On the Reaction Mechanism of the 3,4-Dimethoxybenzaldehyde Formation from 1-(3′,4′-Dimethoxyphenyl)Propene

 and
and
Abstract
:
1. Introduction
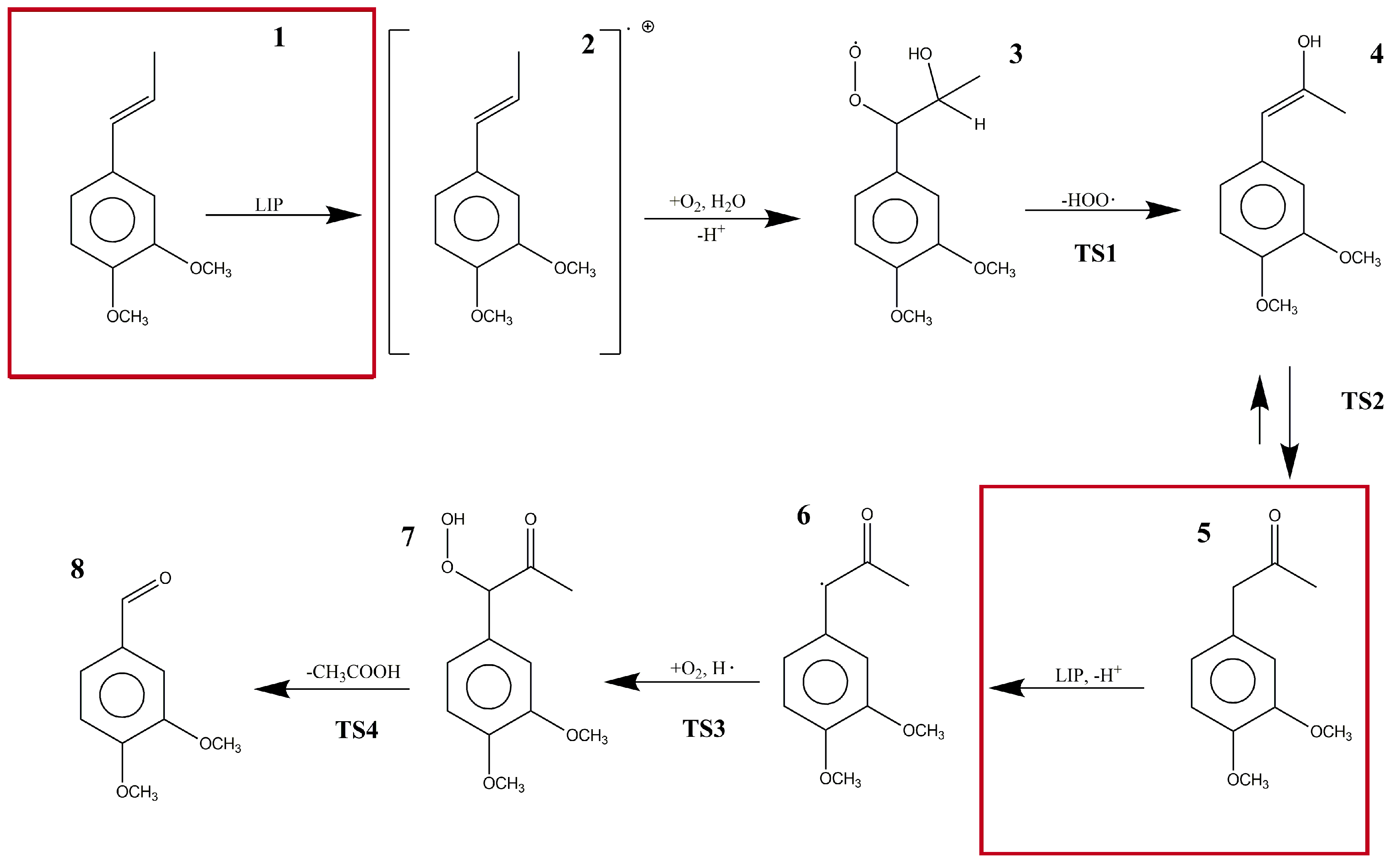
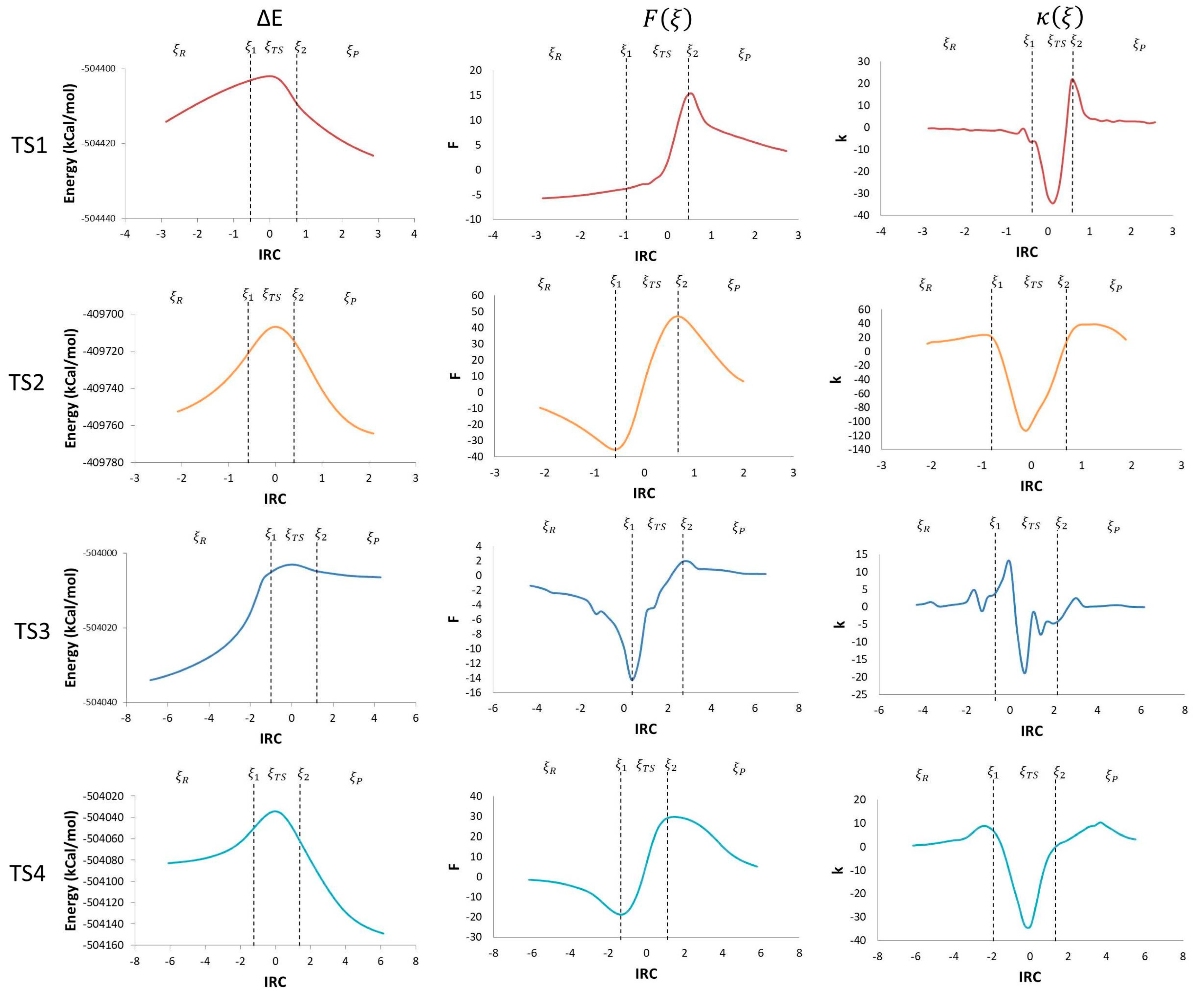
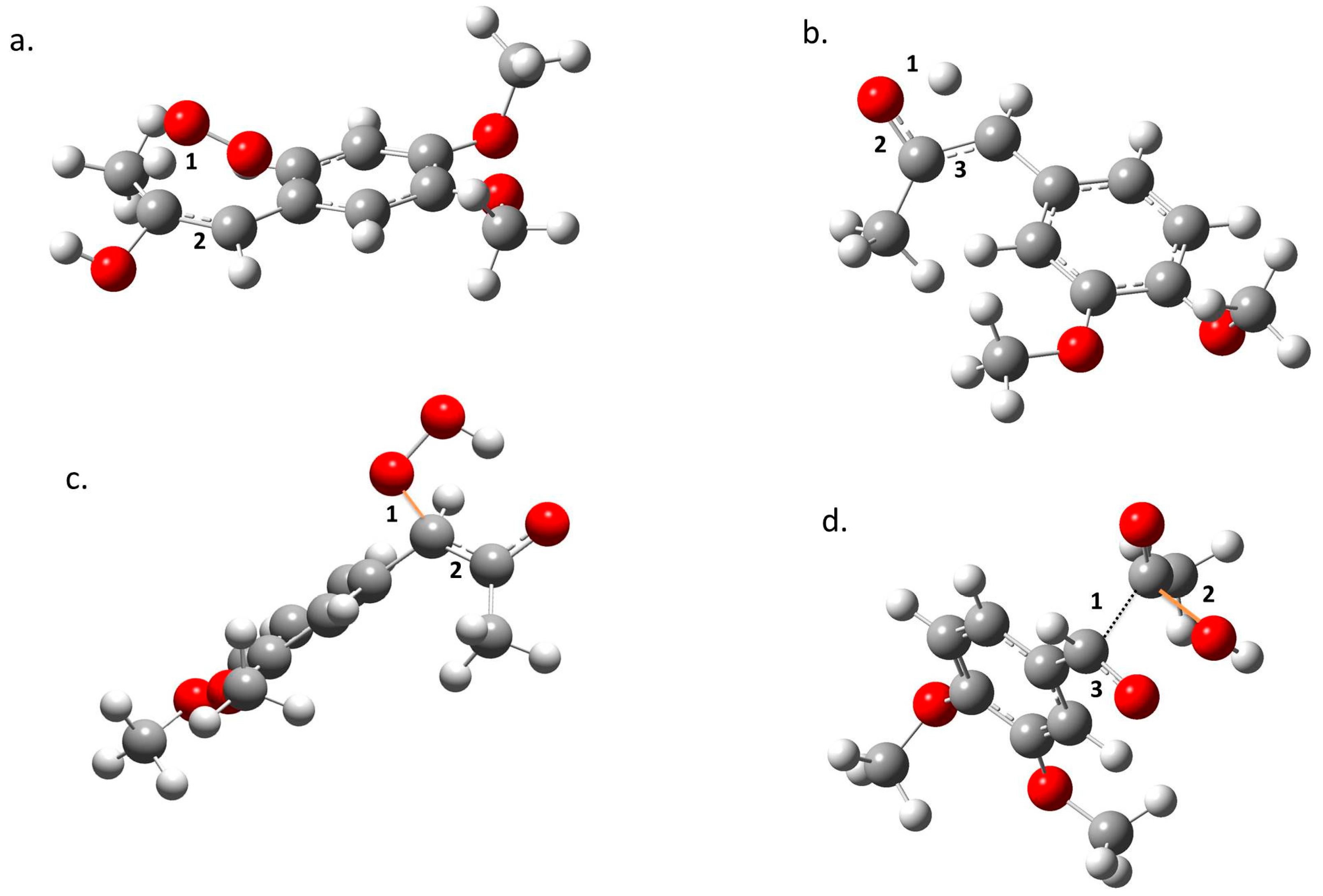
2. Results
3. Discussion
4. Materials and Methods
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Egwim, E.; Kabiru, A.; Tola, A. Partial characterization of lignin peroxidase expressed by bacterial and fungal isolates from termite gut. Biokemistri 2015, 27, 33–38. [Google Scholar]
- Wariishi, H.; Dunford, H.; MacDonald, I.; Gold, M. Manganese Peroxidase from the Lignin-degrading Basidiomycete Phanerochaete chrysosporium. J. Biol. Chem. 1989, 264, 3335–3340. [Google Scholar] [PubMed]
- Semba, Y.; Ishida, M.; Yokobori, S.; Yamagishi, A. Ancestral amino acid substitution improves the thermal stability of recombinant lignin peroxidase from white-rot fungi, Phanerochaete chrysosporium strain UAMH 3641. Protein Eng. Des. Sel. 2015, 28, 221–230. [Google Scholar] [CrossRef] [PubMed]
- MacDonald, J.; Goacher, R.; Abou-Zaid, M.; Master, E. Comparative analysis of lignin peroxidase and manganese peroxidase activity on coniferous and deciduous wood using ToF-SIMS. Appl. Microbiol. Biotechnol. 2016, 100, 8013–8020. [Google Scholar] [CrossRef] [PubMed]
- Kudo, S.; Harada, A.; Kubota, H.; Sasaki, K.; Kaneta, T. Simultaneous Determination of Manganese Peroxidase and Lignin Peroxidase by Capillary Electrophoresis Enzyme Assays. ACS Omega 2017, 2, 7329–7333. [Google Scholar] [CrossRef]
- Rai, N.; Yadav, M.; Yadav, H. Enzymatic Characterisation of Lignin Peroxidase from Luffa aegyptiaca Fruit Juice. Am. J. Plant Sci. 2016, 7, 649–656. [Google Scholar] [CrossRef]
- Blodig, W.; Smith, A.T.; Doyle, W.A.; Piontek, K. Crystal structures of pristine and oxidatively processed lignin peroxidase expressed in Escherichia coli and of the W171F variant that eliminates the redox active tryptophan 171. Implications for the reaction mechanism. J. Mol. Biol. 2001, 305, 851–861. [Google Scholar] [CrossRef] [PubMed]
- Wariishi, H.; Gold, M. Lignin Peroxidase Compound III Mechanism of formation and decomposition. J. Biol. Chem. 1990, 265, 2070–2077. [Google Scholar] [PubMed]
- Harada, A.; Sasaki, K.; Kaneta, T. Direct determination of lignin peroxidase released from Phanerochaete chrysosporium by in-capillary enzyme assay using micellar electrokinetic chromatography. J. Chromatogr. A 2016, 1, 145–149. [Google Scholar] [CrossRef] [PubMed]
- Shah, M.M.; Grover, T.A.; Barr, D.P.; Aust, S.D. On the Mechanism of Inhibition of the Veratryl Alcohol Oxidase Activity of Lignin Peroxidase H2 by EDTA. J. Biol. Chem. 1992, 267, 21564–21569. [Google Scholar] [PubMed]
- Cai, D.; Tien, M. Lignin Peroxidase Catalysis, Oxycomplex, and Heme-Linked Ionization. In Enzymes in Biomass Conversion; Leatham and Himmel; American Chemical Society: Washington, DC, USA, 1991; pp. 180–187, ISBN13:9780841219953. [Google Scholar]
- Higuchi, T. Mechanisms of Lignin Degradation by Lignin Peroxidase and Laccase of White-Rot Fungi. In Plant Cell Wall Polymers; Lewis, N., Paice, M.G., Eds.; American Chemical Society: Washington, DC, USA, 1989; pp. 482–502, ISBN13:9780841216587. [Google Scholar]
- Khindaria, A.; Yamazaki, I.; Aust, S. Veratryl Alcohol Oxidation by Lignin Peroxidase. Biochemistry 1995, 34, 16860–16869. [Google Scholar] [CrossRef] [PubMed]
- Ten Have, R.; Franssen, M.; Field, J. Lignin peroxidase initiates O2-dependent self-propagating chemical reactions which accelerate the consumption of 1-(3′,4′-dimethoxyphenyl)propene. Biochem. J. 2000, 15, 585–591. [Google Scholar] [CrossRef]
- Toro-Labbé, A. Characterization of Chemical Reactions from the Profiles of Energy, Chemical Potential, and Hardness. J. Phys. Chem. A 1999, 103, 4398–4403. [Google Scholar] [CrossRef]
- Jaque, P.; Toro-Labbé, A. Theoretical Study of the Double Proton Transfer in the CHX–XH⋯CHX–XH (X = O, S) Complexes. J. Phys. Chem. A 2000, 104, 995–1003. [Google Scholar] [CrossRef]
- Gutiérrez-Oliva, S.; Herrera, B.; Toro-Labbé, A.; Chermette, H. On the Mechanism of Hydrogen Transfer in the HSCH(O) ⇌ (S)CHOH and HSNO ⇌ SNOH Reactions. J. Phys. Chem. A 2005, 109, 1748–1751. [Google Scholar] [CrossRef] [PubMed]
- Toro-Labbé, A.; Gutiérrez-Oliva, S.; Concha, M.; Murray, J.S.; Politzer, P. Analysis of two intramolecular proton transfer processes in terms of the reaction force. J. Chem. Phys. 2004, 121, 4570–4776. [Google Scholar] [CrossRef] [PubMed]
- Martínez, J.; Toro-Labbé, A. Energy and chemical force profiles from the Marcus equation. Chem. Phys. Lett. 2004, 392, 132–139. [Google Scholar] [CrossRef]
- Politzer, P.; Toro-Labbé, A.; Gutiérrez-Oliva, S.; Herrera, B.; Jaque, P.; Concha, M.; Murray, J.S. The reaction force: Three key points along an intrinsic reaction coordinate. J. Chem. Sci. 2005, 117, 467–472. [Google Scholar] [CrossRef]
- Jaque, P.; Toro-Labbé, A.; Politzer, P.; Geerlings, P. Reaction force constant and projected force constants of vibrational modes along the path of an intramolecular proton transfer reaction. Chem. Phys. Lett. 2008, 456, 135–140. [Google Scholar] [CrossRef]
- Flores-Morales, P.; Gutiérrez-Oliva, S.; Silva, E.; Toro-Labbé, A. The reaction electronic flux: A new descriptor of the electronic activity taking place during a chemical reaction. Application to the characterization of the mechanism of the Schiff’s base formation in the Maillard reaction. J. Mol. Struct. 2010, 943, 121–126. [Google Scholar] [CrossRef]
- Toro-Labbé, A.; Gutiérrez-Oliva, S.; Murray, J.S.; Politzer, P. The reaction force and the transition region of a reaction. J. Mol. Model. 2009, 15, 707–710. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Truhlar, D.G. The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: Two new functionals and systematic testing of four M06-class functionals and 12 other functionals. Theor. Chem. Acc. 2008, 120, 215–241. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision E.01; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- McQuarrie, D.A. Statistical Mechanics; Harper & Row: New York, NY, USA, 1975; pp. 143–151. ISBN 9781891389153. [Google Scholar]
- Marenich, A.V.; Cramer, C.J.; Truhlar, D.G. Performance of SM6, SM8, and SMD on the SAMPL1 test set for the prediction of small-molecule solvation free energies. J. Phys. Chem. B 2009, 113, 4538–4543. [Google Scholar] [CrossRef] [PubMed]
- Marenich, A.V.; Cramer, C.J.; Truhlar, D.G. Universal solvation model based on solute electron density and on a continuum model of the solvent defined by the bulk dielectric constant and atomic surface tensions. J. Phys. Chem. B 2009, 113, 6378–6396. [Google Scholar] [CrossRef] [PubMed]
- Carrol, F.A. Perspectives on Structure and Mechanism in Organic Chemistry; Brooks/Cole Publishing Company: New York, NY, USA, 1998; pp. 322–349. ISBN 9780470276105. [Google Scholar]
- Herrera, B.; Toro-Labbé, A. The Role of Reaction Force and Chemical Potential in Characterizing the Mechanism of Double Proton Transfer in the Adenine−Uracil Complex. J. Phys. Chem. A 2007, 111, 5921–5926. [Google Scholar] [CrossRef] [PubMed]
- Vogt-Geisse, S.; Toro-Labbé, A. The mechanism of the interstellar isomerization reaction HOC+→HCO + HOC+→HCO+ catalyzed by H2H2: New Insights from the reaction electronic flux. J. Chem. Phys. 2009, 130, 244308–244314. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Not Available. |





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
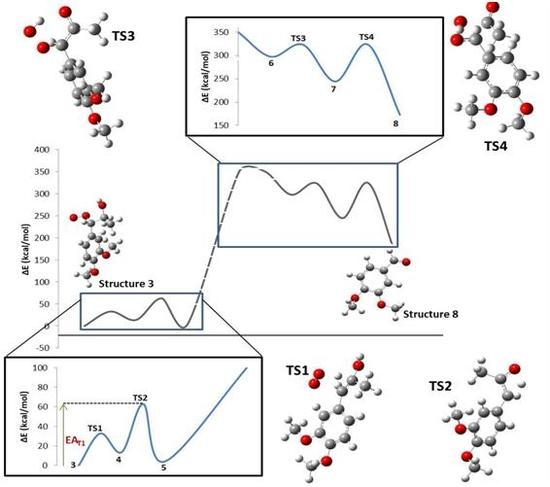
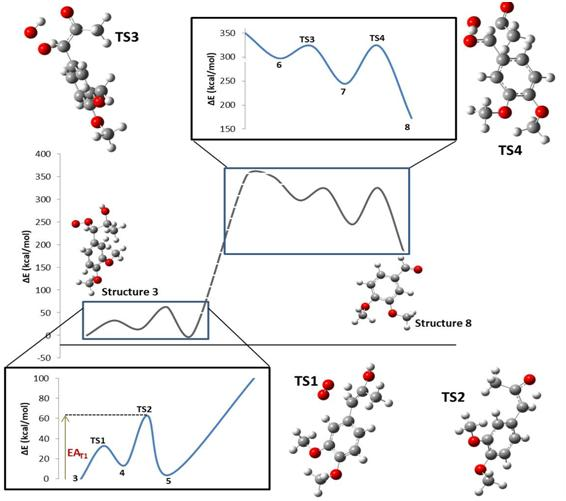
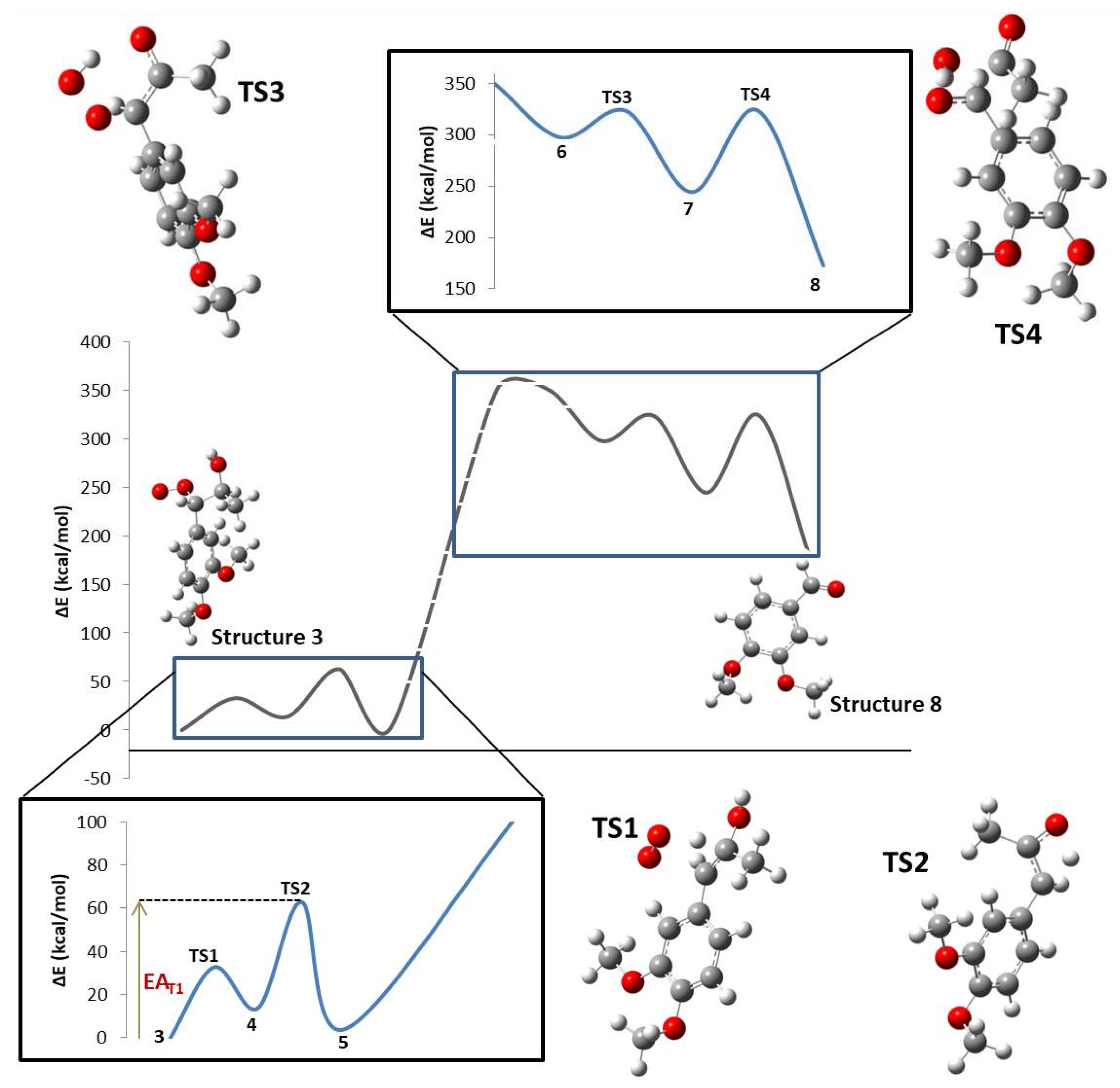
| Step | TS1 | TS2 | TS3 | TS4 |
|---|---|---|---|---|
| ΔE0 (kcal/mol) | 13.81 | −9.56 | −53.17 | −71.50 |
| EA (kcal/mol) | 32.80 | 49.03 | 25.71 | 79.52 |
| ΔH0 (kcal/mol) | 14.40 | −9.56 | −53.76 | −70.91 |
| HA (kcal/mol) | 32.80 | 49.03 | 25.12 | 79.52 |
| ΔG0 (kcal/mol) | 2.15 | −10.01 | −40.58 | −83.06 |
| GA (kcal/mol) | 32.92 | 49.36 | 38.37 | 81.20 |
| ΔS0 (cal/mol.K) | 41.10 | 1.54 | −44.21 | 40.73 |
| SA (cal/mol.K) | −0.42 | −1.10 | −44.45 | −5.63 |
| ΔPV0 (kcal/mol) | 0.59 | 0.00 | −0.59 | 0.59 |
| PVA (kcal/mol) | 0.00 | 0.00 | −0.59 | 0.00 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cuesta, S.; Arias, J.; Gallegos, F.; Alzate-Morales, J.; Meneses, L. On the Reaction Mechanism of the 3,4-Dimethoxybenzaldehyde Formation from 1-(3′,4′-Dimethoxyphenyl)Propene. Molecules 2018, 23, 412. https://doi.org/10.3390/molecules23020412
Cuesta S, Arias J, Gallegos F, Alzate-Morales J, Meneses L. On the Reaction Mechanism of the 3,4-Dimethoxybenzaldehyde Formation from 1-(3′,4′-Dimethoxyphenyl)Propene. Molecules. 2018; 23(2):412. https://doi.org/10.3390/molecules23020412
Chicago/Turabian StyleCuesta, Sebastián, Josefa Arias, Felipe Gallegos, Jans Alzate-Morales, and Lorena Meneses. 2018. "On the Reaction Mechanism of the 3,4-Dimethoxybenzaldehyde Formation from 1-(3′,4′-Dimethoxyphenyl)Propene" Molecules 23, no. 2: 412. https://doi.org/10.3390/molecules23020412