
5-Phenyl-10,15,20-Tris(4-sulfonatophenyl)porphyrin: Synthesis, Catalysis, and Structural Studies


Abstract
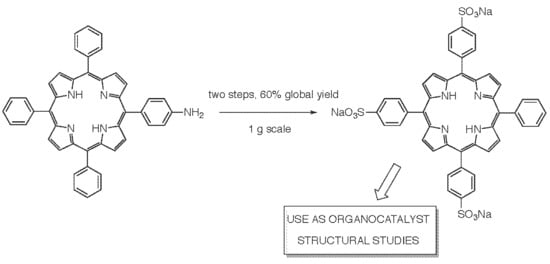
:
1. Introduction
2. Results and Discussion
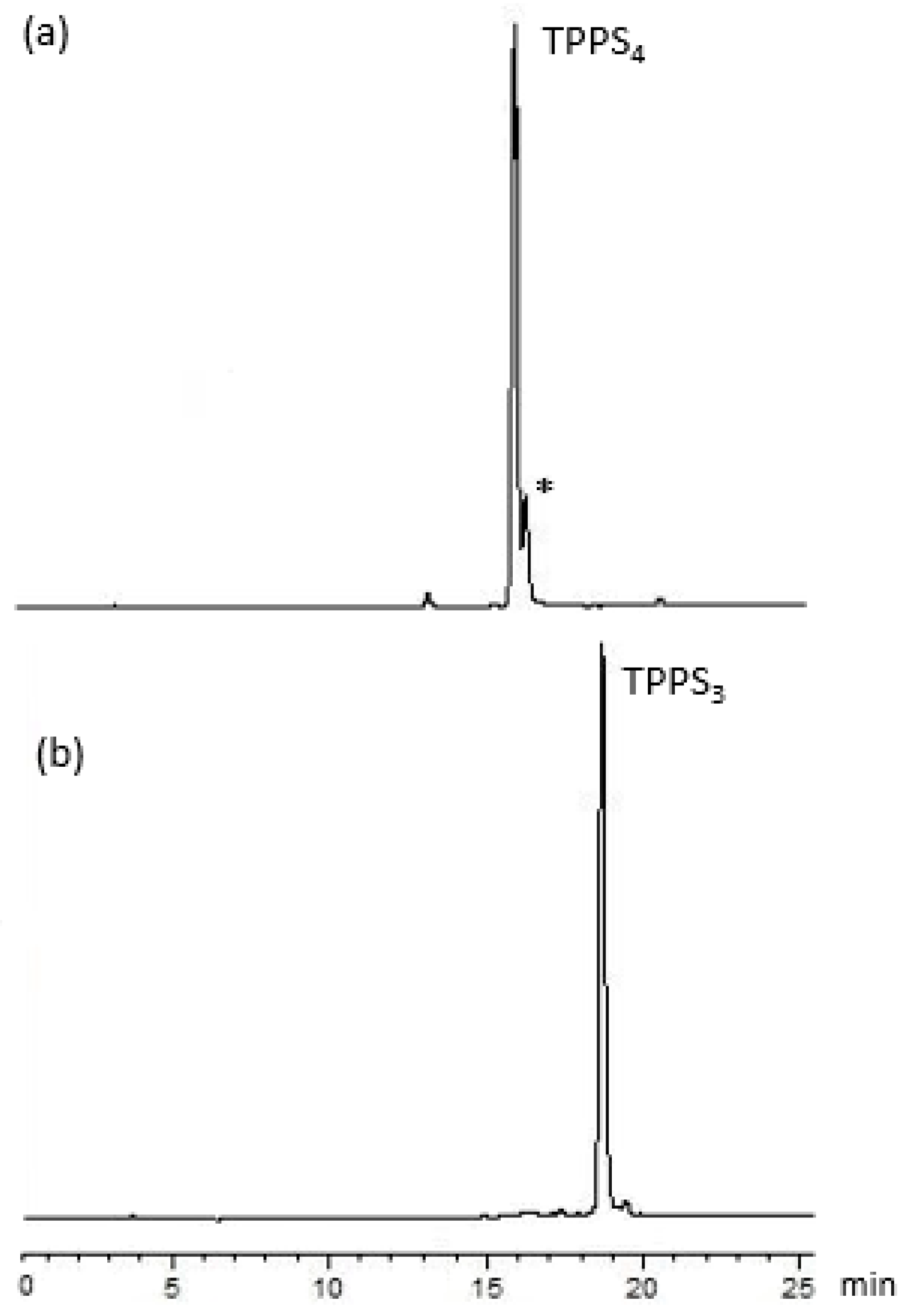
2.1. Reductive Deamination Route for the Gram-Scale Synthesis of Porphyrin 1
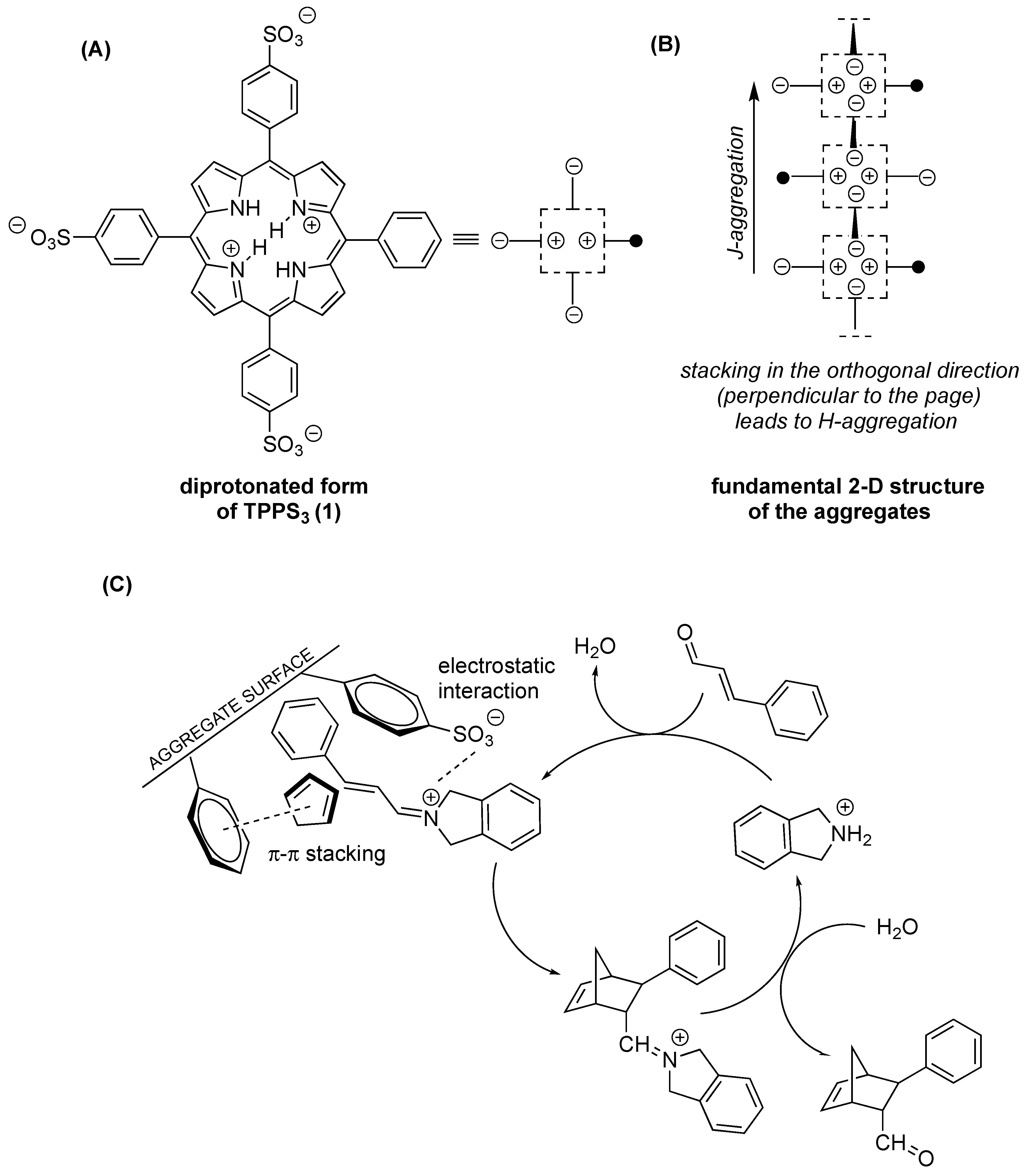
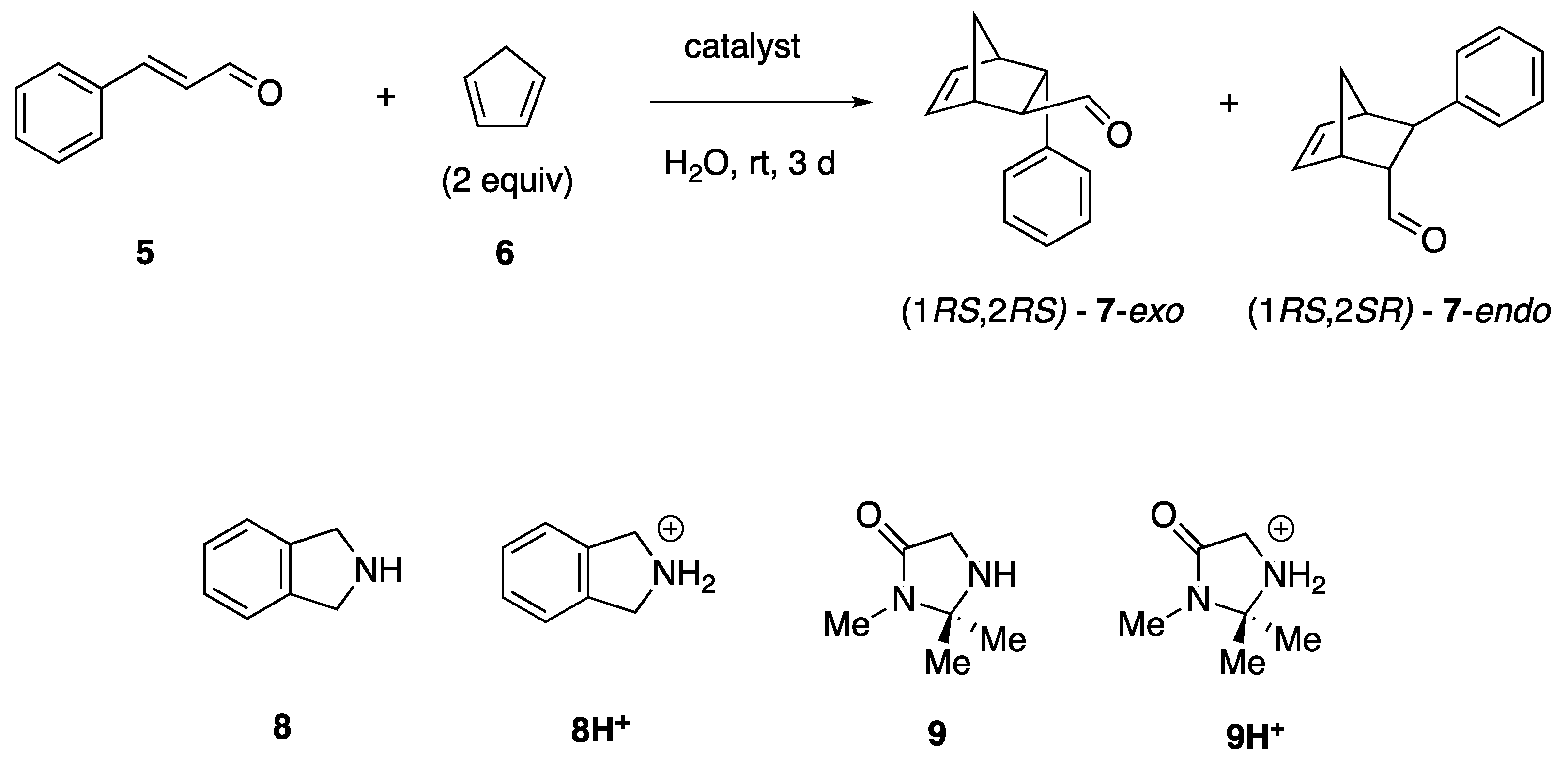
2.2. Heterogeneous Supramolecular Catalysis of the Aqueous Diels–Alder Reaction by TPPS3 (1) Aggregates
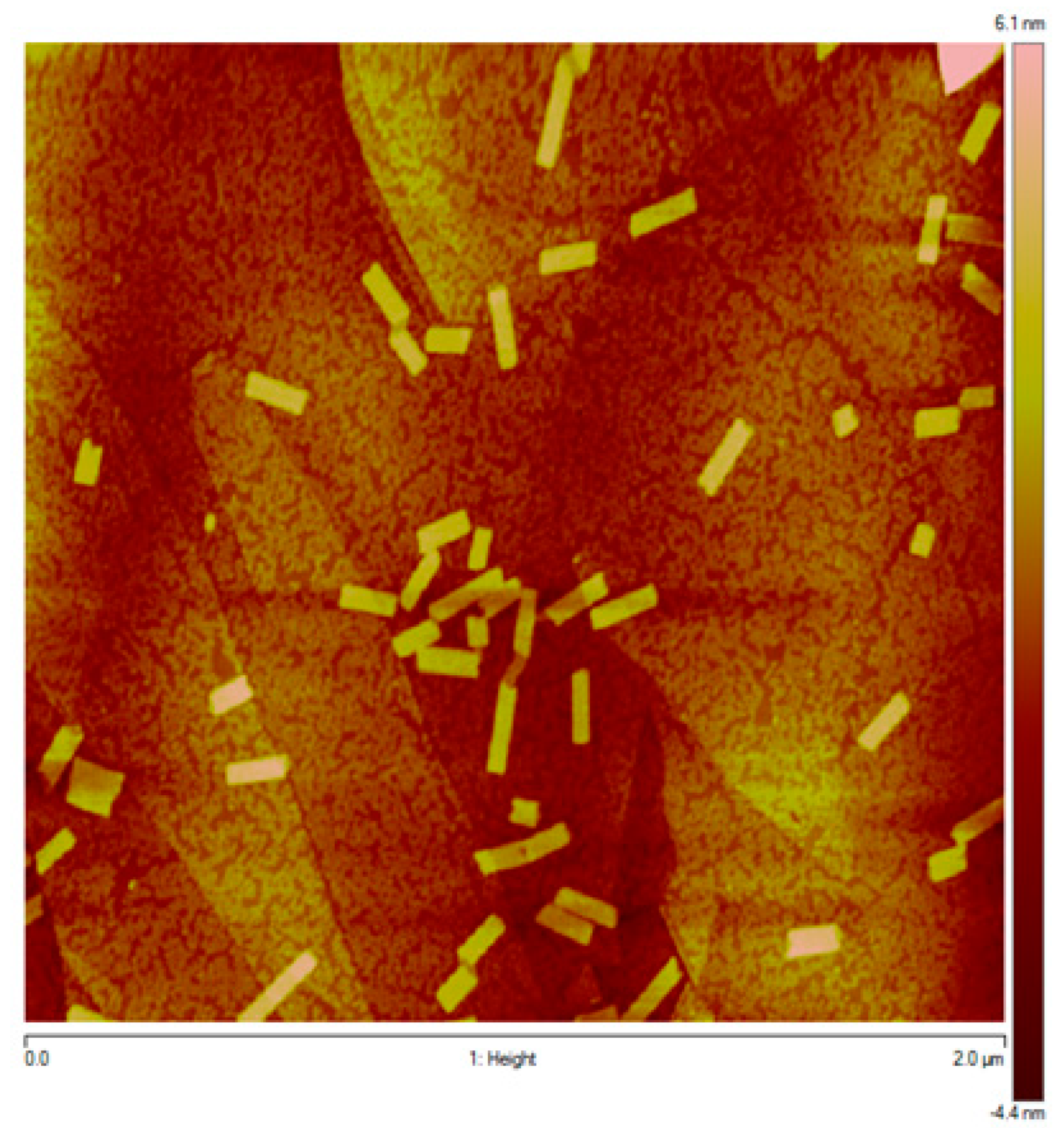
2.3. Atomic Force and Transmission Electron Microscopy Study of the Morphology of TPPS3 (1) Aggregates
3. Materials and Methods
3.1. General Methods
3.2. Synthetic Procedures and Product Characterization
3.3. Representative Procedure for the Catalysis of the Diels-Alder Reaction by the [zw-TPPS3·(amine)] Heteroaggreates
3.4. Procedure for the Preparation of Solutions TPPS3 (1) Nanotubes for PFM and Cryo-TEM Imaging
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Hambright, P. Chemistry of Water-Soluble Porphyrins. In The Porphyrin Handbook; Kadish, K.M., Smith, K.M., Guilard, R., Eds.; Academic Press: San Diego, CA, USA, 2000; Volume 3, pp. 130–210. ISBN 0-12-393203-3. [Google Scholar]
- Miyajima, K.; Komatsu, H.; Inoue, K.; Handa, T.; Nakagaki, M. The protective effects of cyclodextrins against the oxidation of methyl orange by singlet oxygen. Bull. Chem. Soc. Jpn. 1990, 63, 6–10. [Google Scholar] [CrossRef]
- Sailer, R.; Strauss, W.S.L.; Emmert, H.; Stock, K.; Steiner, R.; Schneckenburger, H. Plasma membrane associated location of sulfonated meso-tetraphenylporphyrins of different hydrophilicity probed by total internal reflection fluorescence spectroscopy. Photochem. Photobiol. 2000, 71, 460–465. [Google Scholar] [CrossRef]
- Kano, K.; Sato, T.; Yamada, S.; Ogawa, T. Fluorescence quenching of water-soluble porphyrins. A novel fluorescence quenching of anionic porphyrin by anionic anthraquinone. J. Phys. Chem. 1983, 87, 566–569. [Google Scholar] [CrossRef]
- Yeats, A.L.; Schwab, A.D.; Massare, B.; Johnston, D.E.; Johnson, A.T.; de Paula, J.C.; Smith, W.F. Photoconductivity of Self-Assembled Nanotapes Made from meso-Tris(4-sulfonatophenyl) monophenylporphine. J. Phys. Chem. C 2008, 112, 2170–2176. [Google Scholar] [CrossRef]
- Kitagawa, Y.; Segawa, H.; Ishii, K. Magneto-chiral dichroism of organic compounds. Angew. Chem. Int. Ed. 2011, 50, 9133–9136. [Google Scholar] [CrossRef]
- Zhang, Y.H.; Chen, D.-M.; He, F.; Liu, F.-C. Raman and infrared spectral study of meso-sulfonatophenylsubstituted porphyrins (TPPSn, n = 1, 2A, 2O, 3, 4). Spectrochim. Acta Part A 2003, 59, 87–101. [Google Scholar] [CrossRef]
- Kobayashi, T. J-Aggregates; World Scientific Publishing: Singapore, 2012; Volume 2, ISBN 978-981-4365-74-1. [Google Scholar]
- Würthner, F.; Kaiser, T.E.; Saha-Möller, C.R. J-Aggregates: From Serendipitous Discovery to Supramolecular Engineering of Functional Dye Materials. Angew. Chem. Int. Ed. 2011, 50, 3376–3410. [Google Scholar] [CrossRef]
- Ribó, J.M.; Crusats, J.; Sagués, F.; Claret, J.; Rubires, R. Chiral sign induction by vortices during the formation of mesophases in stirred solutions. Science 2001, 292, 2063–2066. [Google Scholar] [CrossRef]
- Micali, N.; Engelkamp, H.; van Rhee, P.G.; Christianen, P.C.M.; Scolaro, L.M.; Maan, J.C. Selection of supramolecular chirality by application of rotational and magnetic forces. Nature Chem. 2012, 4, 201–207. [Google Scholar] [CrossRef]
- Escudero, C.; Crusats, J.; Díez-Pérez, I.; El-Hachemi, Z.; Ribó, J.M. Folding and Hydrodynamic Forces in J-Aggregates of 5-Phenyl-10,15,20-tris-(4-sulfophenyl)porphyrin. Angew. Chem. Int. Ed. 2006, 45, 8032–8035. [Google Scholar] [CrossRef]
- Tantussi, F.; Fuso, F.; Allegrini, M.; Micali, N.; Occhiuto, I.G.; Scolaro, L.M.; Patanè, S. Linear and circular dichroism in porphyrin J-aggregates probed by polarization modulated scanning near-field optical microscopy. Nanoscale 2014, 6, 10874. [Google Scholar] [CrossRef] [PubMed]
- Crusats, J.; Claret, J.; Díez-Pérez, I.; El-Hachemi, Z.; García-Ortega, H.; Rubires, R.; Sagués, F.; Ribó, J.M. Chiral shape and enantioselective growth of colloidal particles of self-assembled meso-tetra(phenyl and 4-sulfonatophenyl)porphyrins. Chem. Commun. 2003, 1588–1589. [Google Scholar] [CrossRef]
- El-Hachemi, Z.; Arteaga, O.; Canillas, A.; Crusats, J.; Escudero, C.; Kuroda, R.; Harada, T.; Rosa, M.; Ribó, J.M. On the Mechano-Chiral Effect of Vortical Flows on the Dichroic Spectra of 5-Phenyl-10,15,20-tris(4-sulfonatophenyl)porphyrin J-Aggregates. Chem. Eur. J. 2008, 14, 6438–6443. [Google Scholar] [CrossRef] [PubMed]
- Winkelman, J. The distribution of tetraphenylporphinesulfonate in the tumor-bearing rat. J. Cancer. Res. 1962, 22, 589–596. [Google Scholar]
- Pasternack, R.F.; Huber, P.R.; Boyd, P.; Engasser, G.; Francesconi, L.; Gibbs, E.; Fasella, P.; Venturo, G.C.; Hinds, L.C. On the aggregation of meso-substituted water-soluble porphyrins. J. Am. Chem. Soc. 1972, 94, 4511–4517. [Google Scholar] [CrossRef] [PubMed]
- Sutter, T.P.G.; Rahimi, R.; Hambright, P.; Bommer, J.C.; Kumar, M.; Neta, P.J. Steric and inductive effects on the basicity of porphyrins and on the site of protonation of porphyrin dianions: Radiolytic reduction of porphyrins and metalloporphyrins to chlorins or phlorins. J. Chem Soc. Faraday Trans. 1993, 89, 495–502. [Google Scholar] [CrossRef]
- Rubires, R.; Crusats, J.; El-Hachemi, Z.; Jaramillo, T.; Lopez, M.; Valls, E.; Farrera, J.A.; Ribó, J.M. Self-assembly in water of the sodium salts of meso-sulfonatophenyl substituted porphyrins. New J. Chem. 1999, 189–198. [Google Scholar] [CrossRef]
- Lindsey, J.S.; Schreiman, I.C.; Hsu, H.C.; Kearney, P.C.; Marguerettaz, A.M. Rothemund and Longo Reactions Revisited: Synthesis of Tetraphenylporphyrins under Equilibrium Conditions. J. Org. Chem. 1987, 52, 827–836. [Google Scholar] [CrossRef]
- Ye, B.-H.; Naruta, Y. A novel method for the synthesis of regiospecifically sulfonated porphyrin monomers and dimers. Tetrahedron 2003, 59, 3593–3601. [Google Scholar] [CrossRef]
- Hosomizu, K.; Oodoi, M.; Umeyama, T.; Matano, Y.; Yoshida, K.; Isoda, S.; Isosomppi, M.; Tkachenko, N.V.; Lemmetyinen, H.; Imahori, H. Substituent Effects of Porphyrins on Structures and Photophysical Properties of Amphiphilic Porphyrin Aggregates. J. Phys. Chem. B 2008, 112, 16517–16524. [Google Scholar] [CrossRef]
- Moyano, A.; Arlegui, A.; Soler, B.; Crusats, J.; El-Hachemi, Z.; Ribó, J.M. Absolute Asymmetric Synthesis via Top-Bottom Chirality Transfer. In Proceedings of the XXVII RSEQ Biennal Meeting in Organic Chemistry, Santiago de Compostela, Spain, 20–22 June 2018. [Google Scholar]
- Kruper, W.J., Jr.; Chamberlin, T.A.; Kochanny, M. Regiospecific aryl nitration of meso-substituted tetraarylporphyrins: A simple route to bifunctional porphyrins. J. Org. Chem. 1989, 54, 2753–2756. [Google Scholar] [CrossRef]
- Kornblum, N. Replacement of the aromatic primary amino group by hydrogen. Org. React. 1944, 2, 262–340. [Google Scholar]
- Geoffroy, O.T.; Morinelli, T.A.; Meier, G.P. Chemoselective one-pot deamination of aryl amines. Tetrahedron Lett. 2001, 42, 5367–5369. [Google Scholar] [CrossRef]
- Zurita, A.; Duran, A.; Ribó, J.M.; El-Hachemi, Z.; Crusats, J. Hyperporphyrin effects extended into a J-aggregate supramolecular structure in water. RSC Adv. 2017, 7, 3353–3357. [Google Scholar] [CrossRef]
- Cabrer, A.; Ribó, J.M.; El-Hachemi, Z.; Crusats, J. 5,10,15,20-Tetrasulfonatophenylporphyrin regioisomers: How the location of the sulfonato groups determines the formation of their supramolecular aggregates. J. Porphyrins Phthalocyanines 2015, 19, 852–857. [Google Scholar] [CrossRef]
- Barona-Castaño, J.C.; Carmona-Vargas, C.C.; Brocksom, T.J.; de Oliveira, K.T. Porphyrins as Catalysts in Scalable Organic Reactions. Molecules 2016, 21, 310. [Google Scholar] [CrossRef] [PubMed]
- Simonneaux, G.; Le Maux, P.; Ferrand, Y.; Rault-Berthelot, J. Asymmetric heterogeneous catalysis by metalloporphyrins. Coord. Chem. Rev. 2006, 250, 2212–2221. [Google Scholar] [CrossRef]
- Nakagaki, S.; Baio Ferreira, G.K.; Ucoski, G.M.; Dias de Freitas Castro, K.A. Chemical Reactions Catalyzed by Metalloporphyrin-Based Metal-Organic Frameworks. Molecules 2013, 18, 7279–7308. [Google Scholar] [CrossRef] [Green Version]
- Rybicka-Jasinska, K.; Shan, W.; Zawada, K.; Kadish, K.M.; Gryko, D. Porphyrins as Photoredox Catalysts: Experimental and Theoretical Studies. J. Am. Chem. Soc. 2016, 138, 15451–15458. [Google Scholar] [CrossRef]
- El-Hachemi, Z.; Escudero, C.; Acosta-Reyes, F.; Casas, M.T.; Altoe, V.; Aloni, S.; Oncins, G.; Sorrenti, A.; Crusats, J.; Campos, J.L.; Ribó, J.M. Structure vs. properties—chirality, optics and shapes—in amphiphilic porphyrin J-aggregates. J. Mater. Chem. C 2013, 1, 3337–3346. [Google Scholar] [CrossRef]
- Hayashi, Y.; Samanta, S.; Gotoh, H.; Ishikawa, H. Asymmetric Diels–Alder Reactions of α,β-Unsaturated Aldehydes Catalyzed by a Diarylprolinol Silyl Ether Salt in the Presence of Water. Angew. Chem. Int. Ed. 2008, 47, 6634–6637. [Google Scholar] [CrossRef] [PubMed]
- Breslow, R. Hydrophobic Effects on Simple Organic Reactions in Water. Acc. Chem. Res. 1991, 24, 159–164. [Google Scholar] [CrossRef]
- Sun, Z.; She, Y.; Cao, M.; Zhou, Q.; Lu, X.; Zhang, S. Synthesis of meso-tetraphenylporphyrins in mixed solvent systems. Arkivoc 2013, 389–400. [Google Scholar] [CrossRef]
Sample Availability: Samples of compound 1 are available from the authors. |








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Catalyst (mol%) | Yield b | 7-exo:7-endo b |
|---|---|---|---|
| 1 | none | <2% | 52:48 |
| 2 | 8H+ TsO− (30 mol%) | 75% | 70:30 |
| 3 | 9H+ TsO− (30 mol%) | 50% | 57:43 |
| 4 c | [8H+ TPPS3−]x (15 mol%) | 19% | 60:40 |
| 5 c | [9H+ TPPS3−]x (15 mol%) | 7% | 56:44 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Arlegui, A.; El-Hachemi, Z.; Crusats, J.; Moyano, A. 5-Phenyl-10,15,20-Tris(4-sulfonatophenyl)porphyrin: Synthesis, Catalysis, and Structural Studies. Molecules 2018, 23, 3363. https://doi.org/10.3390/molecules23123363
Arlegui A, El-Hachemi Z, Crusats J, Moyano A. 5-Phenyl-10,15,20-Tris(4-sulfonatophenyl)porphyrin: Synthesis, Catalysis, and Structural Studies. Molecules. 2018; 23(12):3363. https://doi.org/10.3390/molecules23123363
Chicago/Turabian StyleArlegui, Aitor, Zoubir El-Hachemi, Joaquim Crusats, and Albert Moyano. 2018. "5-Phenyl-10,15,20-Tris(4-sulfonatophenyl)porphyrin: Synthesis, Catalysis, and Structural Studies" Molecules 23, no. 12: 3363. https://doi.org/10.3390/molecules23123363