
Computational Prediction of the Protonation Sites of Ac-Lys-(Ala)n-Lys-NH2 Peptides through Conceptual DFT Descriptors



Abstract
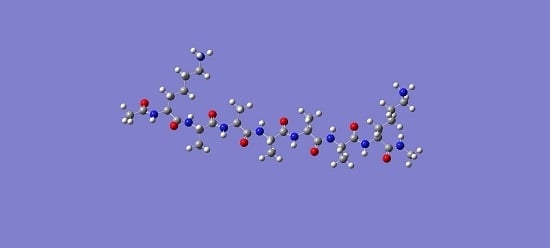
:
1. Introduction
2. Theoretical Background
3. Settings and Computational Methods
4. Results and Discussion
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| DFT | Density Functional Theory |
| KS | Kohn-Sham |
| E | Electronic Energy |
| I | Ionization Potential |
| A | Electron Affinity |
| N | Number of Electrons |
| AGE | Advanced Glycation Endproducts |
| MEDT | Molecular Electron Density Theory |
| SCF | Self Consistent Field |
| KID | Koopmans in DFT |
| HOMO | Higher Ocuppied Molecular Orbital |
| LUMO | Lower Unocuppied Molecular Orbital |
| IEF-PCM | Integral Equation Formalism–Polarized Continuum Model |
| SMD | Solvation Model Density |
References
- Parr, R.; Yang, W. Density-Functional Theory of Atoms and Molecules; Oxford University Press: New York, NY, USA, 1989. [Google Scholar]
- Geerlings, P.; de Proft, F.; Langenaeker, W. Conceptual Density Functional Theory. Chem. Rev. 2003, 103, 1793–1873. [Google Scholar] [CrossRef] [PubMed]
- Toro-Labbé, A. Theoretical Aspects of Chemical Reactivity; Elsevier Science: Amsterdam, The Netherlands, 2007; Volume 19. [Google Scholar]
- Chattaraj, P. Chemical Reactivity Theory—A Density Functional View; CRC Press, Taylor & Francis Group: Boca Raton, FL, USA, 2009. [Google Scholar]
- Domingo, L.R. Molecular Electron Density Theory: A Modern View of Reactivity in Organic Chemistry. Molecules 2016, 21, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Domingo, L.R.; Ríos-Gutiérrez, M.; Pérez, P. Applications of the Conceptual Density Functional Theory Indices to Organic Chemistry Reactivity. Molecules 2016, 21, 1–22. [Google Scholar] [CrossRef] [PubMed]
- Jasinski, R. Searching for Zwitterionic Intermediates in Hetero Diels-Alder Reactions between Methyl α,p-dinitrocinnamate and Vinyl-Alkyl Ethers. Comput. Theor. Chem. 2014, 1046, 93–98. [Google Scholar] [CrossRef]
- Jasiński, R.; Mróz, K.; Kacka, A. Experimental and Theoretical DFT Study on Synthesis of Sterically Crowded 2,3,3,(4)5-Tetrasubstituted-4-nitroisoxazolidines via 1,3-Dipolar Cycloaddition Reactions Between Ketonitrones and Conjugated Nitroalkenes. J. Heterocycl. Chem. 2016, 53, 1424–1429. [Google Scholar] [CrossRef]
- Jasiński, R.; Jasińska, E.; Dresler, E. A DFT Computational Study of the Molecular Mechanism of [3+2] Cycloaddition Reactions between Nitroethene and Benzonitrile N-oxides. J. Mol. Mod. 2017, 23, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, N. Advanced Glycation Endproducts—Role in Pathology of Diabetic Complications. Diabetes Res. Clin. Pract. 2005, 67, 3–21. [Google Scholar] [CrossRef] [PubMed]
- Makowska, J.; Bagińska, K.; Liwo, A.; Chmurzynski, L.; Scheraga, H. Acidic-Basic Properties of Three Alanine-Based Peptides Containing Acidic and Basic Side Chains: Comparison between Theory and Experiment. Biopolymers 2008, 90, 724–732. [Google Scholar] [CrossRef] [PubMed]
- Makowska, J.; Liwo, A.; Chmurzynski, L.; Scheraga, H.A. Influence of the Length of the Alanine Spacer on the Acidic-Basic Properties of the Ac-Lys-(Ala)n-Lys-NH2 Peptides (n = 0, 1, 2, …, 5). J. Solut. Chem. 2012, 41, 1738–1746. [Google Scholar] [CrossRef] [PubMed]
- Huzinaga, S.; Andzelm, J.; Klobukowski, M.; Radzio-Audzelm, E.; Sakai, Y.; Tatewaki, H. Gaussian Basis Sets for Molecular Calculations; Elsevier: Amsterdam, The Netherlands, 1984. [Google Scholar]
- Easton, R.; Giesen, D.; Welch, A.; Cramer, C.; Truhlar, D. The MIDI! Basis Set for Quantum Mechanical Calculations of Molecular Geometries and Partial Charges. Theor. Chem. Acc. 1996, 93, 281–301. [Google Scholar] [CrossRef]
- Lewars, E. Computational Chemistry—Introduction to the Theory and Applications of Molecular and Quantum Mechanics; Kluwer Academic Publishers: Dordrecht, The Netherlands, 2003. [Google Scholar]
- Young, D. Computational Chemistry—A Practical Guide for Applying Techniques to Real-World Problems; John Wiley & Sons: New York, NY, USA, 2001. [Google Scholar]
- Jensen, F. Introduction to Computational Chemistry, 2nd ed.; John Wiley & Sons: Chichester, UK, 2007. [Google Scholar]
- Cramer, C. Essentials of Computational Chemistry—Theories and Models, 2nd ed.; John Wiley & Sons: Chichester, UK, 2004. [Google Scholar]
- Peverati, R.; Truhlar, D.G. Quest for a Universal Density Functional: The Accuracy of Density Functionals Across a Broad Spectrum of Databases in Chemistry and Physics. Philos. Trans. R. Soc. A 2014, 372, 1–52. [Google Scholar] [CrossRef] [PubMed]
- Glossman-Mitnik, D. A Comparison of the Chemical Reactivity of Naringenin Calculated with M06 Family of Density Functionals. Chem. Cent. J. 2013, 7, 155–161. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Araya, J.I.; Salgado-Morán, G.; Glossman-Mitnik, D. Computational Nanochemistry Report on the Oxicams—Conceptual DFT and Chemical Reactivity. J. Phys. Chem. B 2013, 117, 6639–6651. [Google Scholar] [CrossRef] [PubMed]
- Glossman-Mitnik, D. Computational Nanochemistry Study of the Chemical Reactivity Properties of the Rhodamine B Molecule. Procedia Comput. Sci. 2013, 18, 816–825. [Google Scholar] [CrossRef]
- Martínez-Araya, J.I.; Salgado-Morán, G.; Glossman-Mitnik, D. Computational Nutraceutics: Chemical Reactivity Properties of the Flavonoid Naringin by Means of Conceptual DFT. J. Chem. 2013. [Google Scholar] [CrossRef]
- Glossman-Mitnik, D. Chemical Reactivity Theory within DFT Applied to the Study of the Prunin Flavonoid. Eur. Int. J. Sci. Technol. 2014, 3, 195–207. [Google Scholar]
- Glossman-Mitnik, D. Computational Chemistry of Natural Products: A Comparison of the Chemical Reactivity of Isonaringin Calculated with the M06 Family of Density Functionals. J. Mol. Mod. 2014, 20, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Frau, J.; Muñoz, F.; Glossman-Mitnik, D. A Molecular Electron Density Theory Study of the Chemical Reactivity of Cis- and Trans-Resveratrol. Molecules 2016, 21, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Frau, J.; Glossman-Mitnik, D. Chemical Reactivity Theory Study of Advanced Glycation Endproduct Inhibitors. Molecules 2017, 22, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Parr, R.; Yang, W. Density Functional Approach to the Frontier-Electron Theory of Chemical Reactivity. J. Am. Chem. Soc. 1984, 106, 4049–4050. [Google Scholar] [CrossRef]
- Parr, R.; Szentpaly, L.; Liu, S. Electrophilicity Index. J. Am. Chem. Soc. 1999, 121, 1922–1924. [Google Scholar] [CrossRef]
- Morell, C.; Grand, A.; Toro-Labbé, A. New Dual Descriptor for Chemical Reactivity. J. Phys. Chem. A 2005, 109, 205–212. [Google Scholar] [CrossRef] [PubMed]
- Morell, C.; Grand, A.; Toro-Labbé, A. Theoretical Support for Using the Δf(r) Descriptor. Chem. Phys. Lett. 2006, 425, 342–346. [Google Scholar] [CrossRef]
- Cárdenas, C.; Rabi, N.; Ayers, P.; Morell, C.; Jaramillo, P.; Fuentealba, P. Chemical Reactivity Descriptors for Ambiphilic Reagents: Dual Descriptor, Local Hypersoftness, and Electrostatic Potential. J. Phys. Chem. A 2009, 113, 8660–8667. [Google Scholar] [CrossRef] [PubMed]
- Ayers, P.; Morell, C.; de Proft, F.; Geerlings, P. Understanding the Woodward-Hoffmann Rules by Using Changes in Electron Density. Chem. Eur. J. 2007, 13, 8240–8247. [Google Scholar] [CrossRef] [PubMed]
- Morell, C.; Ayers, P.; Grand, A.; Gutiérrez-Oliva, S.; Toro-Labbé, A. Rationalization of the Diels-Alder Reactions through the Use of the Dual Reactivity Descriptor Δf(r). Phys. Chem. Chem. Phys. 2008, 10, 7239–7246. [Google Scholar] [CrossRef] [PubMed]
- Morell, C.; Hocquet, A.; Grand, A.; Jamart-Grégoire, B. A Conceptual DFT Study of Hydrazino Peptides: Assessment of the Nucleophilicity of the Nitrogen Atoms by Means of the Dual Descriptor Δf(r). J. Mol. Struct. THEOCHEM 2008, 849, 46–51. [Google Scholar] [CrossRef]
- Martínez-Araya, J.I. Why is the Dual Descriptor a More Accurate Local Reactivity Descriptor than Fukui Functions? J. Math. Chem. 2015, 53, 451–465. [Google Scholar] [CrossRef]
- Domingo, L.R.; Pérez, P.; Sáez, J. Understanding the Local Reactivity in Polar Organic Reactions through Electrophilic and Nucleophilic Parr Functions. RSC Adv. 2013, 3, 1486–1494. [Google Scholar] [CrossRef]
- Chamorro, E.; Pérez, P.; Domingo, L.R. On the Nature of Parr Functions to Predict the Most Reactive Sites along Organic Polar Reactions. Chem. Phys. Lett. 2013, 582, 141–143. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09 Revision D.01; Gaussian Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Weigend, F.; Ahlrichs, R. Balanced Basis Sets of Split Valence, Triple Zeta Valence and Quadruple Zeta Valence Quality for H to Rn: Design and Assessment of Accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. [Google Scholar] [CrossRef] [PubMed]
- Weigend, F. Accurate Coulomb-fitting Basis Sets for H to R. Phys. Chem. Chem. Phys. 2006, 8, 1057–1065. [Google Scholar] [CrossRef] [PubMed]
- Peverati, R.; Truhlar, D.G. Improving the Accuracy of Hybrid Meta-GGA Density Functionals by Range Separation. J. Phys. Chem. Lett. 2011, 2, 2810–2817. [Google Scholar] [CrossRef]
- Peverati, R.; Truhlar, D.G. M11-L: A Local Density Functional That Provides Improved Accuracy for Electronic Structure Calculations in Chemistry and Physics. J. Phys. Chem. Lett. 2012, 3, 117–124. [Google Scholar] [CrossRef]
- Peverati, R.; Truhlar, D.G. An Improved and Broadly Accurate Local Approximation to the Exchange-Correlation Density Functional: the MN12-L Functional for Electronic Structure Calculations in Chemistry and Physics. Phys. Chem. Chem. Phys. 2012, 14, 13171–13174. [Google Scholar] [CrossRef] [PubMed]
- Peverati, R.; Truhlar, D.G. Screened-Exchange Density Functionals with Broad Accuracy for Chemistry and Solid-State Physics. Phys. Chem. Chem. Phys. 2012, 14, 16187–16191. [Google Scholar] [CrossRef] [PubMed]
- Peverati, R.; Truhlar, D.G. Exchange-Correlation Functional with Good Accuracy for Both Structural and Energetic Properties while Depending Only on the Density and Its Gradient. J. Chem. Theory Comput. 2012, 8, 2310–2319. [Google Scholar] [CrossRef] [PubMed]
- Marenich, A.; Cramer, C.; Truhlar, D. Universal Solvation Model Based on Solute Electron Density and a Continuum Model of the Solvent Defined by the Bulk Dielectric Constant and Atomic Surface Tensions. J. Phys. Chem. B 2009, 113, 6378–6396. [Google Scholar] [CrossRef] [PubMed]
- Avogadro: An Open-Source Molecular Builder and Visualization Tool—Version 1.2.0. Available online: http://avogadro.openmolecules.net (accessed on 6 October 2016).
- Hanweel, M.; Curtis, D.E.; Lonie, D.; Vandermeersch, T.; Zurek, E.; Hutchison, G. Avogadro: An Advanced Semantic Chemical Editor, Visualization, and Analysis Platform. J. Cheminf. 2012, 4, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and Testing of a General AMBER Force Field. J. Comp. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef] [PubMed]
- Gorelsky, S. AOMix Program for Molecular Orbital Analysis—Version 6.5; University of Ottawa: Ottawa, ON, Canada, 2011. [Google Scholar]
- Gorelsky, S.; Lever, A. Electronic Structure and Spectra of Ruthenium Diimine Complexes by Density Functional Theory and INDO/S - Comparison of the Two Methods. J. Organomet. Chem. 2001, 635, 187–196. [Google Scholar] [CrossRef]
- Sample Availability: Not available.


{kind=link}
{kind=link}
| J | J | J | J | J | J | J | |
|---|---|---|---|---|---|---|---|
| M11 | 2.79 | 2.38 | 3.67 | 0.20 | 5.37 | 0.37 | 5.19 |
| M11L | 1.66 | 0.07 | 1.67 | 0.80 | 1.73 | 0.17 | 1.91 |
| MN12L | 1.87 | 0.14 | 1.88 | 0.87 | 2.01 | 0.8 | 2.20 |
| MN12SX | 0.13 | 0.08 | 0.15 | 0.09 | 0.06 | 0.05 | 0.14 |
| N12 | 1.40 | 0.22 | 1.44 | 0.59 | 1.62 | 0.10 | 1.74 |
| N12SX | 0.00 | 0.07 | 0.07 | 0.03 | 0.07 | 0.03 | 0.08 |
| MN12SX | N12SX | |||||
|---|---|---|---|---|---|---|
| P(mpa) | P(hpa) | P(mpa) | P(hpa) | |||
| KK | −0.63 | 0.76 | 0.74 | −0.69 | 0.76 | 0.73 |
| KAK | −0.62 | 0.76 | 0.73 | −0.69 | 0.76 | 0.73 |
| KA2K | −0.71 | 0.81 | 0.78 | −0.74 | 0.78 | 0.73 |
| KA3K | −0.63 | 0.76 | 0.74 | −0.69 | 0.76 | 0.73 |
| KA4K | −0.71 | 0.81 | 0.78 | −0.74 | 0.81 | 0.78 |
| KA5K | −0.71 | 0.81 | 0.78 | −0.74 | 0.81 | 0.78 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sastre, S.; Frau, J.; Glossman-Mitnik, D. Computational Prediction of the Protonation Sites of Ac-Lys-(Ala)n-Lys-NH2 Peptides through Conceptual DFT Descriptors. Molecules 2017, 22, 458. https://doi.org/10.3390/molecules22030458
Sastre S, Frau J, Glossman-Mitnik D. Computational Prediction of the Protonation Sites of Ac-Lys-(Ala)n-Lys-NH2 Peptides through Conceptual DFT Descriptors. Molecules. 2017; 22(3):458. https://doi.org/10.3390/molecules22030458
Chicago/Turabian StyleSastre, Sebastián, Juan Frau, and Daniel Glossman-Mitnik. 2017. "Computational Prediction of the Protonation Sites of Ac-Lys-(Ala)n-Lys-NH2 Peptides through Conceptual DFT Descriptors" Molecules 22, no. 3: 458. https://doi.org/10.3390/molecules22030458