1. Introduction
Angiogenesis is an important facilitator for the development of solid cancer by sprouting new blood vessels from pre-existing endothelium to transport growth factors, oxygen, and various nutrients to tumors. The process of angiogenesis is complex requiring many inducers and receptors; among those, the vascular endothelial growth factor (VEGF) family members are key signal mediators that control endothelial cells in many aspects, particularly, in supporting tumor invasion and metastasis [
1]. The VEGF subtype A (VEGFA) supersedes other family members (VEGFB, VEGFC, VEGFD) in the field of antiangiogenesis therapy because the first generation of drugs available in the market was targeting VEGF (commonly referred to VEGFA) and VEGF receptors (VEGFR).
VEGF specifically binds to VEGFR1 (Flt-1) or VEGFR2 (Flk-1/KDR), presented on the cell surface of endothelial cells. However, VEGFR2 is the main signal transducer responsible for angiogenesis because it has a strong intrinsic kinase activity and plays a decisive role in the regulation of tumor angiogenesis [
2]. The interaction of VEGF with VEGFR2 causes cell proliferation, survival, migration, capillary-tube formation and permeability increase that occurs in rapidly growing tumors [
3]. VEGF/VEGFR2 signaling pathway is initiated by the dimerization of VEGFR2 and subsequent phosphorylations of several tyrosine residues, e.g., Y951, Y1175, and Y1214. Among these residues, VEGFR2-Tyr1175 is the major autophosphorylation site following VEGF binding that controls angiogenic responses in vascular endothelial cells [
4,
5]. Phosphorylation of VEGFR2-Tyr1175 regulates downstream signaling through phospho-Akt at Ser473 promoting endothelial cell survival and migration [
6,
7]. The Akt activation is an almost indispensible signal transduction for regulating endothelial cell survival and migration through the PI3K/AKT and eNOS pathways [
4,
8]. The endothelial cell motility activated by VEGFR2 also associates with focal adhesion molecule (FAK) via the interaction of VEGFR2 and Shb adaptor protein. Particularly, VEGF-induced FAK phosphorylation at Tyr 397 in endothelial cells is a highly conserved autophosphorylation site by transmitting different signals to their targets. It can sustain an increased formation of actin stress fibers and new focal adhesion resulting in several biological processes, including endothelial cell adhesion, migration, invasion, tube formation and survival [
6,
9,
10,
11].
The first antiangiogenesis drug bevacizumab (a monoclonal antibody for VEGFA) was launched in 2004 and many small molecules that inhibit the activity and signaling cascade of VEGF receptors (VEGFRs) have been developed and approved by the US FDA for the treatment of various types of cancer [
12]. Currently, the antiangiogenesis drugs against VEGF are mostly effective in clinical practice when combined in chemotherapy [
13,
14,
15,
16]. The small molecule VEGFR2 tyrosine kinase inhibitors (TKIs, e.g., sorafenib, sunitinib, pazofinib) usually have urea derivatives in their chemical structures which are related to their biological effects [
17]. Sorafenib, an orally available angiogenesis drug for multi-tyrosine kinase inhibition (VEGFR2, VEGFR3, PDGFR-beta and c-kit), is a representative compound that comprises a biaryl urea in its structure [
18]. It was approved by US FDA for advanced renal cancer in 2005. Thus, the development of novel VEGFR2 inhibitors using structure-based drug design targeting high affinity for VEGFR2 is of interest. We have recently reported synthesis of a small molecule VEGFR2 tyrosine kinase inhibitor VH02 or 1-((1-(1
H-indazol-6-yl)-1
H-1,2,3-triazol-4-yl)methyl)-3-(3-chloromethylphenyl)urea by using a back to front approach from in silico drug design [
19]. VH02 is a type II kinase inhibitor which is more selective to VEGFR2 than type I kinase inhibitors. The core motif of VH02 contains 1-((3-chloromethyl) phenyl)-3-(2-propynyl)urea that is buried in the back pocket of VEGFR2. The design of VH02 allows for strong hydrogen bonding to the kinase domain of VEGFR2 while two bonds interact with the key residues at the ATP binding site (front pocket) namely Glu917 and Cys919. Thus, we aim to confirm and evaluate the antiangiogenic activity of VH02 both in vitro and in vivo by targeting the key signals of VEGF-induced angiogenesis. We explored VEGFR2-Tyr1175 signaling pathway on the activation of Akt, FAK, and p38 in human endothelial cells EA.hy926. The in vivo mouse model of Matrigel plug assay was used to evaluate the effect of VH02 on vascular density and α-SMA coverage of the vessels. In this study, we found that VH02 inhibited angiogenesis upon VEGF stimulation both in vitro and in vivo which has high potential for further lead optimization as a cancer therapy agent.
3. Discussion
Antiangiogenic therapy has added value to traditional chemotherapy for solid tumors due to the suppression of tumor invasion and metastasis. Angiogenesis occurs when there is an imbalance of pro-angiogenic molecules secreted by the tumor mass, including VEGF165 isoform, which is one of most potent proangiogenic factors that sends signals through a surface receptor tyrosine kinase (VEGFR2) on endothelial cells. Binding of VEGF to VEGFR2 is a major mediator causing cascades of signals to initiate new blood vessel formation. Phosphorylation of VEGFR2 in each tyrosine site associates with key events including proliferation, migration, survival and permeation that are necessary for tumor angiogenesis. Suppressing VEGFR2 signaling through the inhibition of receptor tyrosine kinase phosphorylation is now an important target of antiangiogenic therapeutic strategies [
13]. In this study, we explored the antiangiogenic activity of a novel VEGFR2 inhibitor, VH02, both in vitro and in vivo. We found that VH02 inhibited multiple steps of angiogenesis feature including endothelial cell viability, adhesion, migration, and tube formation. The main signaling cascade through VEGFR2 including phosphorylation of VEGFR2 and Akt was also inhibited both in vitro and in vivo. The vascular formation in Matrigel plugs implanted in mice was significantly inhibited along with the abrogation of α-SMA expression. This suggests that the novel antiangiogenic agent VH02 can potentially be a lead compound for further optimization in line of cancer therapeutic agents.
The type II kinase inhibitor VH02 was designed with the core structure of 1-(substituted)-3-prop-2-ynylureas with triazole linker using a back to front approach [
19]. VH02 interacts with the side chains of a hydrophobic part of the back pocket including Ile888, Ile892, Val898, Val899, Leu1019, His1026, Ile1044, Cys1045 and Phe1047. The design of high affinity VEGFR2 inhibitors also extended to various structures including phthalazine derivatives [
20], sulfonyl urea derivatives [
21], guanidinium [
22] and others which have similar side chains interactions. The in vitro cell culture studies revealed that VH02 differentially decreased endothelial cell viability; under VEGF stimulation, the endothelial cells were 100-fold more sensitive to VH02-induced cytotoxicity when compared to vehicle-treated cells. Thus, at low concentrations, this compound selectively inhibited endothelial cell growth in the environment of angiogenic drivers that favors the cytotoxic profile in the normal cell environment where VEGF levels are low. It also appears that VH02 dose-dependently inhibited early steps of angiogenesis, including endothelial cell migration and adhesion.
The endothelial migration processes during angiogenesis involve different stimulators such as chemotaxis (induced by chemoattractants), haptotaxis (activated by immobilized ligands), and mechanotaxis (driven by mechanical force, e.g., sheer stress) [
11]. VEGF is an essential chemotactic factor during angiogenesis that initiates cell migration, cell adhesion, and the interaction of endothelial cells to ECM followed by the tubular network formation. The interaction of cell-matrix during adhesion and the tubular network formation on Matrigel mimicked the in vivo vascular formation. Early steps of endothelial cell migration depend on type IV collagenase activity of MMP-2 and MMP-9 [
23]. These MMPs are major ECM proteolytic enzymes that degrade the basement membrane to facilitate cell invasion, sprouting and migration into the perivascular space. We demonstrated that VH02 effectively inhibited MMP-2 gelatinase activity in a dose-dependent manner, which was strongly associated with lower migratory activity due to the release of pro-angiogenic molecules that induced extracellular matrix degradation [
24]. However, there was no change in MMP-9 activity which also plays an important role in metastasis and angiogenesis regulation. Our result was in agreement with other studies because MMP-9 is not inducible in starved conditions of EA.hy926 [
25,
26]. The inhibitory effect of VH02 was concordant with essential phenomena occurring during angiogenesis, which ultimately resulted in the inhibition of vascular tube formation.
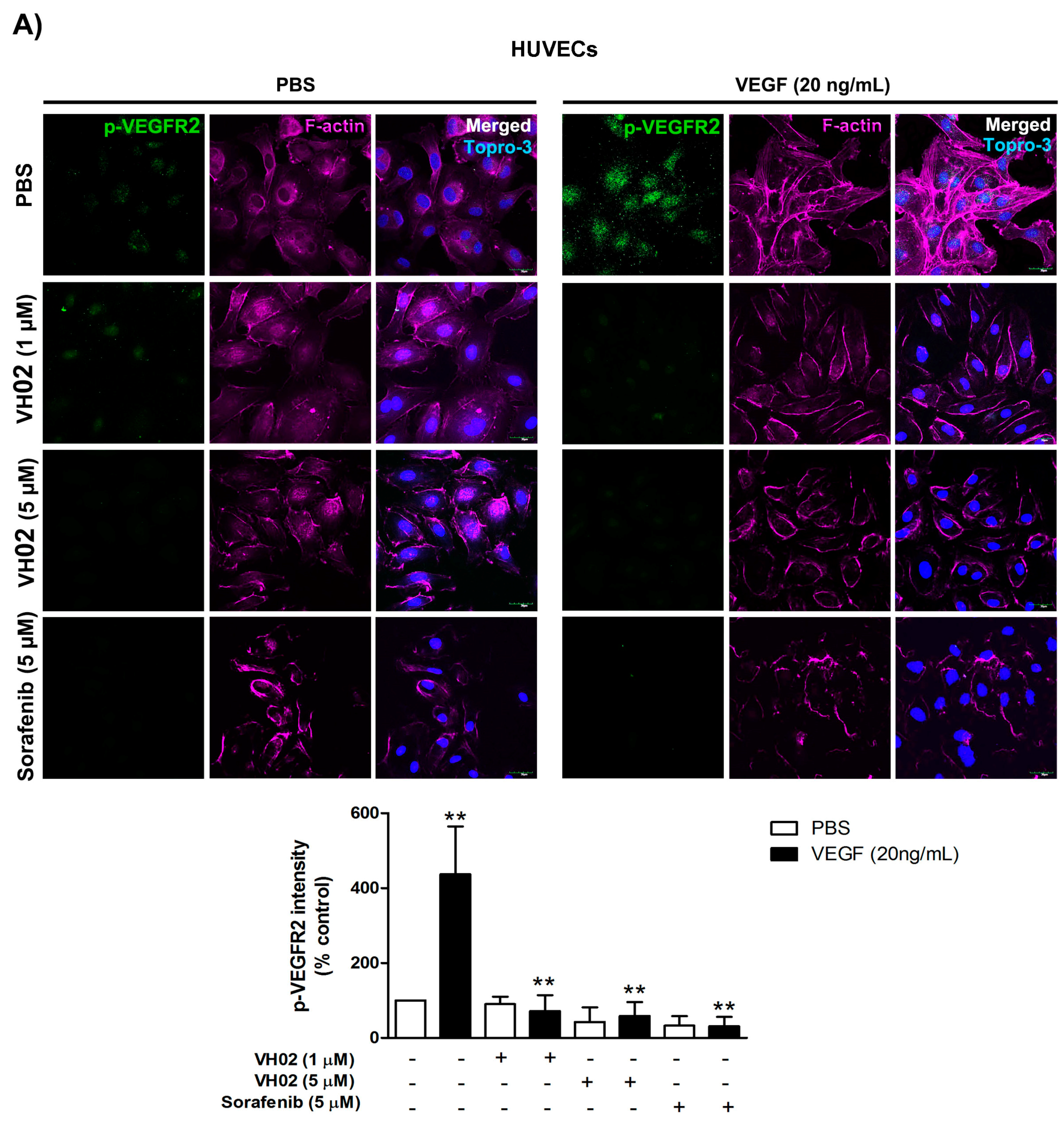
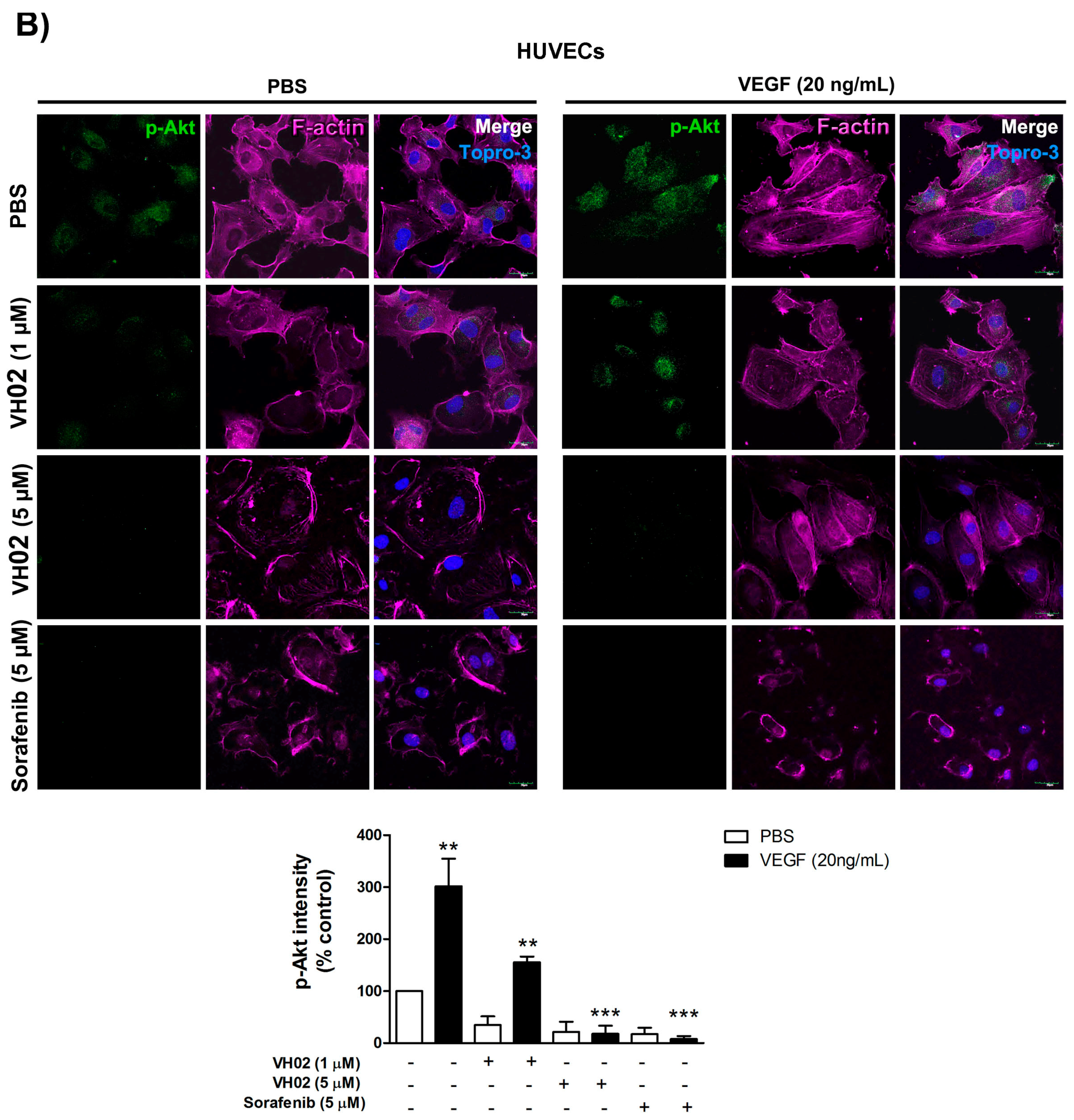
VH02 acts as a small molecule VEGFR2 tyrosine kinase inhibitor; thus, the downstream VEGFR2 signaling pathway was confirmed both in an endothelial cell line and HUVECs. The induction of VEGF/VEGFR2 signaling provides specific binding sites for several signaling protein such as PLC-γ (phospholipase C gamma), Shb (SH2 domain-containing adapter protein B, PI3K (phosphoinositide-3 kinase), Sck (Shc-like protein), or Grb2 (growth factor receptor-bound protein 2). These proteins require specific molecules including Akt, FAK, or P38MAPK, which are all involved in endothelial migration [
6]. Phosphorylation of VEGFR2 Tyr1175 and downstream signaling to Akt and FAK is a major site for regulation of directional endothelial cell migration. We revealed that endothelial cells exposed to VH02 prior to VEGF stimulation inhibited the phosphorylation of VEGFR2 at Tyr1175 confirming the interference of VH02 in the process of VEGF signaling. In addition, VEGF directly mediated both endothelial cell survival and migration through Akt activation on Ser473 [
7]. VH02 apparently decreased p-Akt Ser473, which leads to the inhibition of endothelial cell survival. VEGFR2-1175 activation also induced the FAK phosphorylation on Tyr 397 that is a crucial for control of F-actin fibers associated with cell migration [
6,
10,
11]. As reported previously, downregulation of p-FAK is involved with angiogenic responses by suppressing adhesion molecules and migration via dynamic cytoskeleton organization [
27,
28]. Actin rearrangement plays a crucial role that drives endothelial cell migration upon VEGF induction by forming filopodia, lamellipodia, and stress fibers. VH02 showed inhibitory effect on the phosphorylation of FAK (Y397) with a rapid decay in cytoskeleton integrity. The stimulation of VEGF through VEGFR2 phosphorylation on Tyr1214 leads to SAPK/p28 MAPK activation but it is not a prerequisite of FAK activation [
29]. Despite p38 MAPK kinase also associated in cell migration, we did not find significant changes in p38 MAPK kinase signaling. This suggests that p38 MAPK activation was independent of VEGF-stimulated EA.hy926 migration under growth factor-starved condition. Others also reported that FAK activation through VEGFR2 may be independent of the SAPK2/p38 pathway [
11,
30]. This probably represent that VH02 did not directly affect phosphorylation of p38 MAPK-regulated endothelial cell migration through the VEGF/VEGFR2 complex.
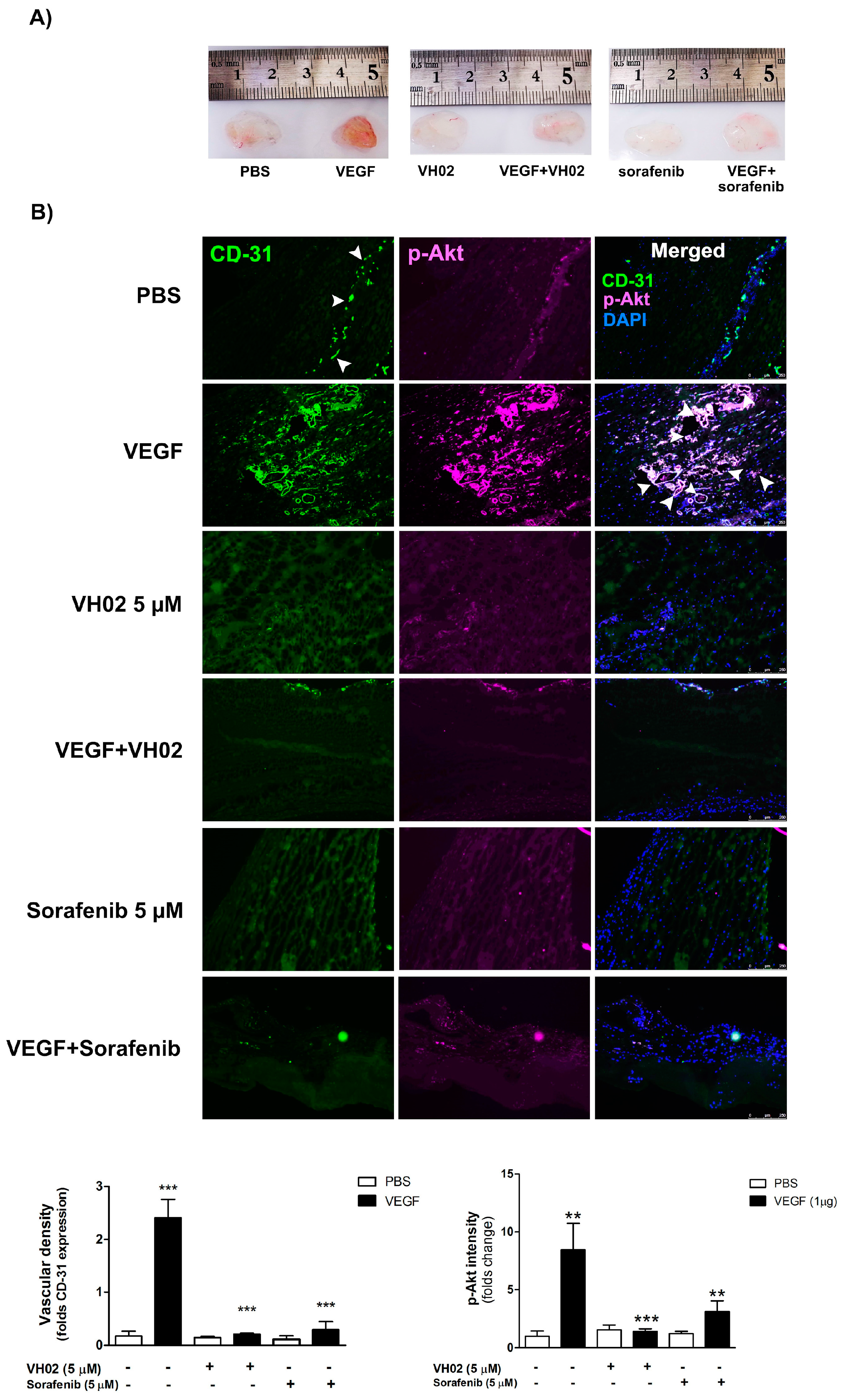
The anti-angiogenic activity of VH02 was confirmed again by in vivo mouse model of vascularization using Matrigel plug assay. A reduction of vascularization in plugs containing VEGF was suppressed by VH02. Immunohistochemical data also revealed that the expression of vascular density and p-Akt intensity were decreased in plugs treated with VH02 or sorafenib (positive drug treatment), which indicated that VH02 significantly diminished angiogenesis in vivo. Besides its angiogenic role, VEGF is also involved in recruitment of pericytes and vascular smooth muscle cells for stabilizing the vessel walls and controlling endothelial cell proliferation [
31]. α-SMA is an actin isoform which is the most frequently used as a marker of pericytes. Many studies used these cells to be a new target of antiangiogenenic therapies by identification of α-SMA coverage of vessels. It is also an important factor for detecting the vasculature which was presented in mouse subcutaneous tissue fibroblast [
32]. Immunohistochemical examination showed that α-SMA coverage of blood vessels in plugs was more sensitive in VEGF stimulation which was correlated with vascular density and p-Akt intensity. The VEGF stimulates the rate of blood vessel formation which is required for pericyte smooth muscle cells to cover new blood vessels while supporting endothelial stabilization and maturation [
31,
33]. However, α-SMA was dramatically decreased after the treatment of VH02 or sorafenib which indicates the suppression of pericyte recruitment and angiogenesis. The overall inhibitory effects of VH02 on all the angiogenesis markers studied were influenced by the interaction of factor 1 (the presence of VEGF) and factor 2 (the presence of VH02 or sorafenib).
4. Materials and Methods
4.1. Reagents and Materials
VH02, 1-((1-(1H-indazol-6-yl)-1H-1,2,3-triazol-4-yl)methyl)-3-(3-chloromethylphenyl)urea was obtained from Kingkarn Sanphanya, The Department of Pharmaceutical Chemistry, Faculty of Pharmacy, Mahidol University (Bangkok, Thailand). Dulbecco’s modified Eagle medium (DMEM), fetal bovine serum (FBS), penicillin/streptomycin, and trypsin-EDTA were all purchased from Gibco BRL (Gaithersburg, MD, USA). VEGF165aa human recombinant protein, monoclonal antibodies against β-actin, VEGFR2, phospho-VEGFR2 (Tyr1175), Akt, phospho-Akt (Ser473), p38 MAPK, phospho-p38 (Thr180/Tyr182), FAK and phospho-FAK (Tyr397), and 4′-6-diamidino-2phenylindole (DAPI) were provided from Cell Signaling Technology (Beverly, MA, USA). Calcein AM fluorescent dye, 3-(4,5-dimethylthiazol-2-yl)-2,5 diphenyltetrazolium bromide (MTT), anti-mouse, and-rabbit IgG-peroxidase antibodies were ordered from Sigma-Aldrich (St. Louis, MO, USA). Matrigel and CD31 mAb were obtained from BD Bioscience (San Jose, CA, USA). Sorafenib was purchased from Selleckchem (Houston, TX, USA). Alexa Fluor 488-conjugated goat anti-rat, Alexa Fluor 555-conjugated goat anti-rabbit, and Topro-3 iodide were obtained from Invitrogen (Eugene, OR, USA). All other chemicals used in this study were analytical grade.
4.2. Cell Culture and Animals
In vitro experiments were first performed in EA.hy926 cells, a human endothelial cell line, purchased from the American Type Culture Collection (ATCC, Rockville, MD, USA). The cells were derived from the fusion of Human umbilical vessel endothelial cells (HUVECs) with the immortal human lung carcinoma cell line A549. EA.hy926 cells were grown in DMEM supplemented with 10% fetal bovine serum (FBS) and 1% penicillin-streptomycin at 37 °C in 5% CO2 atmosphere. EA.hy926 cells were allowed to grow about 80% confluent and subcultured about 1 or 2 times per week.
In separate experiments, human umbilical vein endothelial cells (HUVECs) were used to confirm the activity of VH02 in the endothelial cell line EA.hy926. HUVECs were purchased from Lonza (Walkersvile, MD, USA) and cultured in EGM-2 medium supplemented with 2% FBS, hydrocortisone, hFGFB, VEGF, R3-IGF, ascorbic acid, hEGF, GA-1000, and heparin. Cells were used to evaluate the effects of VH02 on VEGFR and Akt phosphorylation using confocal immunohistochemistry. The in vivo study for the antiagiogenic effect of VH02 was performed in six-week-old male C57BL/6 mice. The experiment procedures complied with Regulations for Animal Experiments and Related Activities at Tohoku University (11th). The experiment protocol was approved by the animal ethic committee of Tohoku University, Japan.
4.3. Cell Viability Assay
EA.hy926 cells were seeded at a density of 1 × 104 cells/well on 96-well plates (Nunc, ThermoFisher Scientific, Waltham, MA, USA). After overnight culture, cells were treated with or without VEGF (25 ng/mL) and various concentrations (0.01–100 μM) of VH02 for 24 h. Then, MTT solution was added for a final concentration 0.25 mg/mL to each well and incubated at 37 °C for 3 h. At the end of incubation period, MTT solution was removed and the formazan crystal product was solubilized with 100 μL of DMSO with constant shaking for 30 min. The optical density was measured using a Multi-detection microplate reader (Synergy, BioTek, Winooski, VT, USA) at a wavelength of 550 nm. Cell viability was expressed as a percentage relative to control. All experiments were performed in triplicate.
4.4. Transwell Migration Assay
The evaluation of endothelial cell migration is the most frequent method for studying chemotaxis in angiogenesis using transwell plates (Costar, #3524, Corning Incorporated, Waltham, NY, USA) with 8 μm diameter pores filters. Briefly, the bottom chambers were filled 600 μL with or without VEGF (25 ng/mL) and various concentrations of VH02. The insert filters were placed into the wells by sterile forceps. Then, cells 1 × 104 cells/well were suspended in 100 μL of media and seeded in the upper chambers. The cultures were allowed to migrate at 37 °C for 16 h. After incubation, the chemoattractant medium in the bottom chambers was removed and replaced with 8 μM Calcein AM fluorescent dye in medium at 37 °C for 45 min. The migrated cells were determined by multi-detection microplate reader at excitation 458 nm and emission at 528 nm (BioTek Instrument, Inc., Winooski, VT, USA).
4.5. Cell Adhesion Assay
Endothelial cells were starved in DMEM containing 2% FBS and incubated at 37 °C overnight. The 96-well plates were coated with 50 μL of Matrigel (0.5 μg/mL) and incubated at 37 °C for 1 h. Wells were blocked with 100 μL of 2% BSA in serum-free media for 1 h and washed with PBS 3 times. Next, 2.5 × 104 cells/well containing 0.1% BSA in serum-free media were adhered in wells co-incubated with 25 ng/mL VEGF and different concentrations of VH02 at 37 °C for 2 h. The cultures were washed with PBS to remove non-adherent cells and filled with MTT solution. The adherent cells were measured at 550 nm using a multi-detection microplate reader (version 4.0.1.10, BioTek Instrument, Inc., Winooski, VT, USA). Percentage of adhesion was expressed using the control group as 100%.
4.6. Capillary-Like Tube Formation Assay
In vitro capillary-like tube formation on a layer of Matrigel was used to evaluate with differentiation of endothelial cells as part of the angiogenesis process. The 24-well plate was coated 250 μL of Matrigel and incubated at 37 °C for 1 h. EA.hy926 cells (6 × 104 cells/well) were suspended in 1 mL DMEM containing 2% FBS, followed by media with or without VEGF (25 ng/mL) and different concentrations of VH02. After 24 h of incubation, endothelial cells forming capillary-like tubes were photographed by an inverted microscope (Olympus DP20-5E, Olympus Corporation, Tokyo, Japan) with 100× magnification and quantifying the tubular length from at least six randomly selected fields by Cell*B imaging software (version 2.7, Olympus DP20:3.0, Münster, Germany). The percentage of tube length was compared to the control group which represents 100%.
4.7. Gelatin Zymography
EA.hy926 cells were starved on 6-well plates, and then DMEM was added to each well containing various concentrations of VH02. VEGF (25 ng/mL) was then added to stimulate cell activity. Cell lysates were collected and the protein samples were separated by 10% SDS-polyacrylamide gels containing 0.1% gelatin. After electrophoresis, the gels were rinsed twice with renaturing buffer (Triton X-100, 2.5% (v/v) in 0.05 M Tris-HCl pH 8.0, 5 mM Ca2Cl) overnight. Then, the gels were incubated twice with developing buffer (0.05 M Tris-HCl pH 8.0, 5 mM Ca2Cl) at 37 °C for overnight. Subsequently, the gels were stained with staining solution (0.1% Coomassie Brilliant Blue R-250 in methanol:acetic acid:water, 4.5:1:4.5, v/v/v), and washed with destaining solution. Finally, the bands indicating MMP-2 (72 kDa) were clearly visible in contrast with the blue background and detected by a high resolution scanner (Canon, Canon USA, Inc., Melville, NY, USA). Intensity of bands were quantitated by Image J software (version 1.47, National Institutes of Health (NIH), Bethesda, MD, USA). The MMP-2 activity was expressed as a percentage relative to the control group.
4.8. Western Blot Analysis
EA.hy926 cells were starved in medium and treated with 1 and 5 μM VH02 (before exposed to VEGF (50 ng/mL). After treatment, cells were lysed with RIPA buffer supplemented with different kinds of inhibitors (1 mM NaVO4, 10 mM NaF, 1% phosphatase inhibitor cocktail mixed with 1% protease inhibitor cocktail). Proteins samples were separated by 10% SDS-polyacrylamide gel electrophoresis (SDS-PAGE) and transferred to PVDF membranes. The membranes were blocked with 5% BSA in TBS-T (5% w/v BSA, 1X TBS, 0.1%Tween-20), and probed with primary monoclonal antibody against VEGFR2, p-VEGFR2 (Tyr1175), Akt, p-Akt (Ser473), p38 MAPK, p-p38 MAPK (Thr180/Tyr182), FAK, p-FAK (Tyr397), and β-actin followed by exposure to a horseradish peroxidase-conjugated anti-mouse or anti-rabbit IgG antibody. The membrane was visualized by enhanced chemiluminescence using ECL plus™ western blotting detection reagents and recorded on Gel documentation (GeneGnome5, Integrated Scientific Solutions, San Diego, CA, USA). Band density was quantified by Image J software.
4.9. Immunofluorescence Microscopy
EA.hy926 cells (7 × 103 cells/well) were plated on 96 well plates and treated as described above. Cells were fixed with 4% formaldehyde for 15 min, permeabilized with 0.1% Triton X-100 in PBS for 5 min, and blocked with 1% BSA for 30 min at room temperature. Then, cells were incubated with anti–actin antibody (1:200) overnight at 4 °C, and covered with anti–rabbit conjugated Alexa Fluor (1:1000), and counterstained with 4′-6-diamidino-2phenylindole (DAPI). The stained cells were visualized under Olympus immunofluorescence microscope (GE Healthcare, Bucks, UK) with 600× magnification.
To ascertain the effects of VH02 on endothelial cells, the assessment of VEGFR2 and Akt phosphorylation, and F-actin staining were performed in HUVECs. Cell staining was analyzed by high content image analysis using confocal microscopy. HUVECs were seeded on collagen type I coated 8-well culture slides (Costar, #354630, Corning Incorporated) overnight, and serum-starved for 4 h in EGM medium. The cells were treated with VH02 for 30 min before 30 min-VEGF stimulation (20 ng/mL). After fixation, the cells were permeabilized with 0.2% Triton X-100 for 10 min and blocked with 2% FBS in PBS for 40 min. The slides were incubated with antibodies against p-VEGFR2-Tyr1175 and p-Akt (1:300), Alexa Fluor 546-conjugated phalloidin (1:500), and Topro-3 iodide (1:500) at 4 °C overnight. Confocal immunofluorescence microscopy photos were taken with an Olympus FV1000 laser scanning biological microscope at 630× magnification. Photographic images were analyzed for the fluorescent intensity according to p-VEGFR2 and p-Akt staining by MetaMorph software (version 7.7.4, Molecular Devices, Sunnyvale, CA, USA). Actin organization was also shown in the photos.
4.10. Matrigel Plug Assay and Immunohistochemistry
Six-week-old male C57BL/6 mice were provided by Prof. Yasufumi Sato in Department of Vascular Biology, Institute of Development, Aging and Cancer (IDAC), Tohoku University, Japan. Mice were subcutaneously injected at opposite iliac regions with 0.6 mL of Matrigel supplemented with 1 μg of VEGF165 with/without 5 μM VH02 or 5 μM sorafenib. After 10 days, the Matrigel plugs were excised and embedded in Tissue Tek OCT (Sakura Finetek Europe B.V., Alphen aan den Rijn, The Netherlands). Cryosections were placed on the glass slides and stained with CD-31, p-Akt, α-SMA mAb, and DAPI. Immunohistochemical staining was performed in Matrigel plugs with Alexa Fluor 488-conjugated goat anti-rat and Alexa Fluor 555-conjugated goat anti-rabbit for 1 h. The vascular expression was photographed by a Leica AF 6500 fluorescence microscope (Wetzlar, Germany) at 250× magnification. Vascular density, p-Akt intensity, and α-SMA coverage on vessel from the histologic sections were quantified in five independent fields of images using the MetaMorph software (version 7.7.4, Molecular Devices, Sunnyvale, CA, USA). The experiment was performed by three mice per groups.
4.11. Statistical Analysis
Each assay was performed in triplicate for at least three independent experiments. All data were expressed as the mean ± SEM. Statistical analysis was accomplished by one-way ANOVA followed by Turkey’s test. In addition, the interaction effects between the conditions (PBS or VEGF) and the drugs (vehicle, VH02 or sorafinib) were detected by two-way ANOVA with Bonferroni posttests for multiple comparisons. The statistical significance was considered when p value was less than 0.05.


 ,
, 

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}








