
Computational Approaches for the Discovery of Human Proteasome Inhibitors: An Overview


Abstract
:1. Introduction
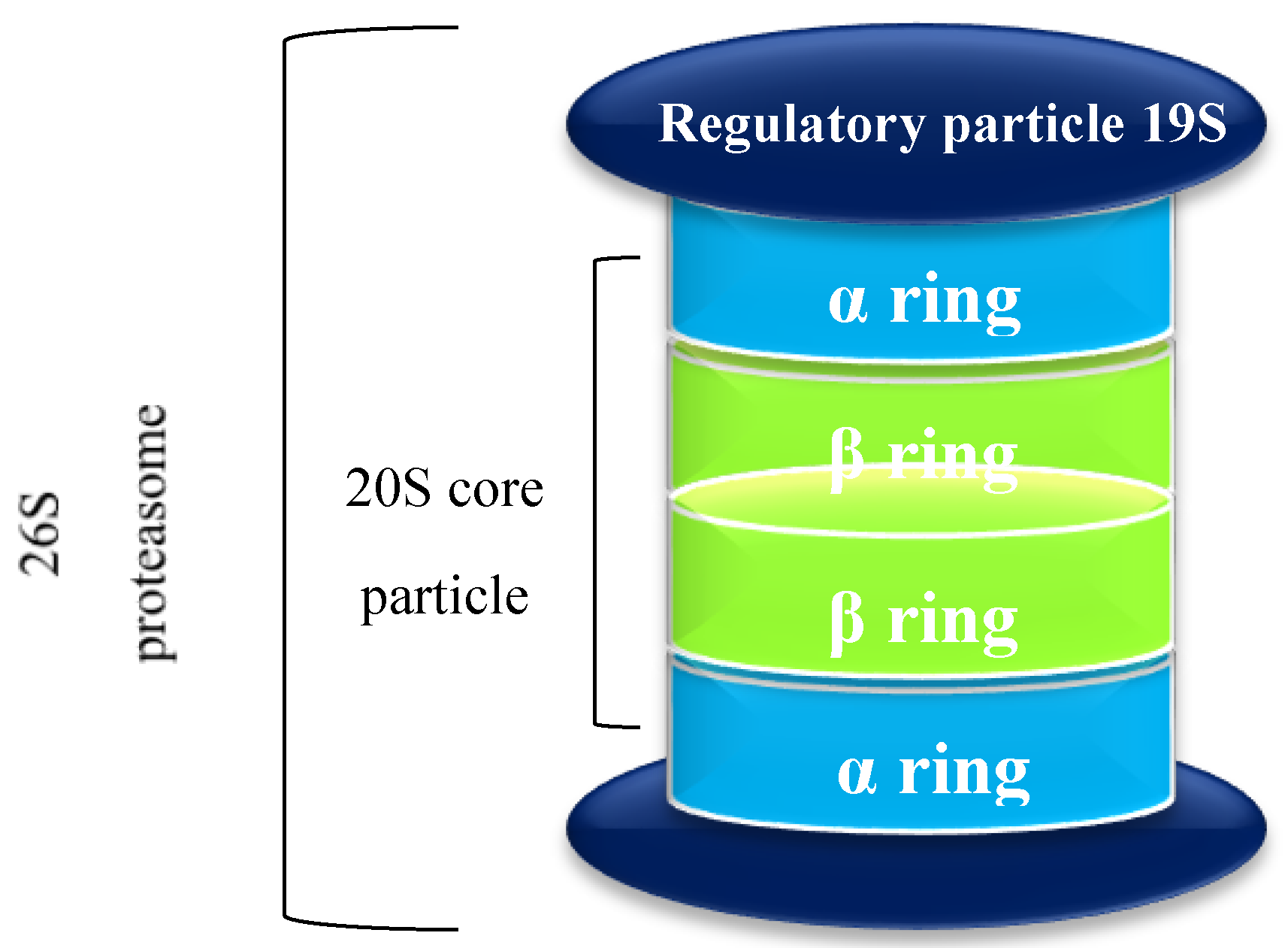
1.1. The Proteasome
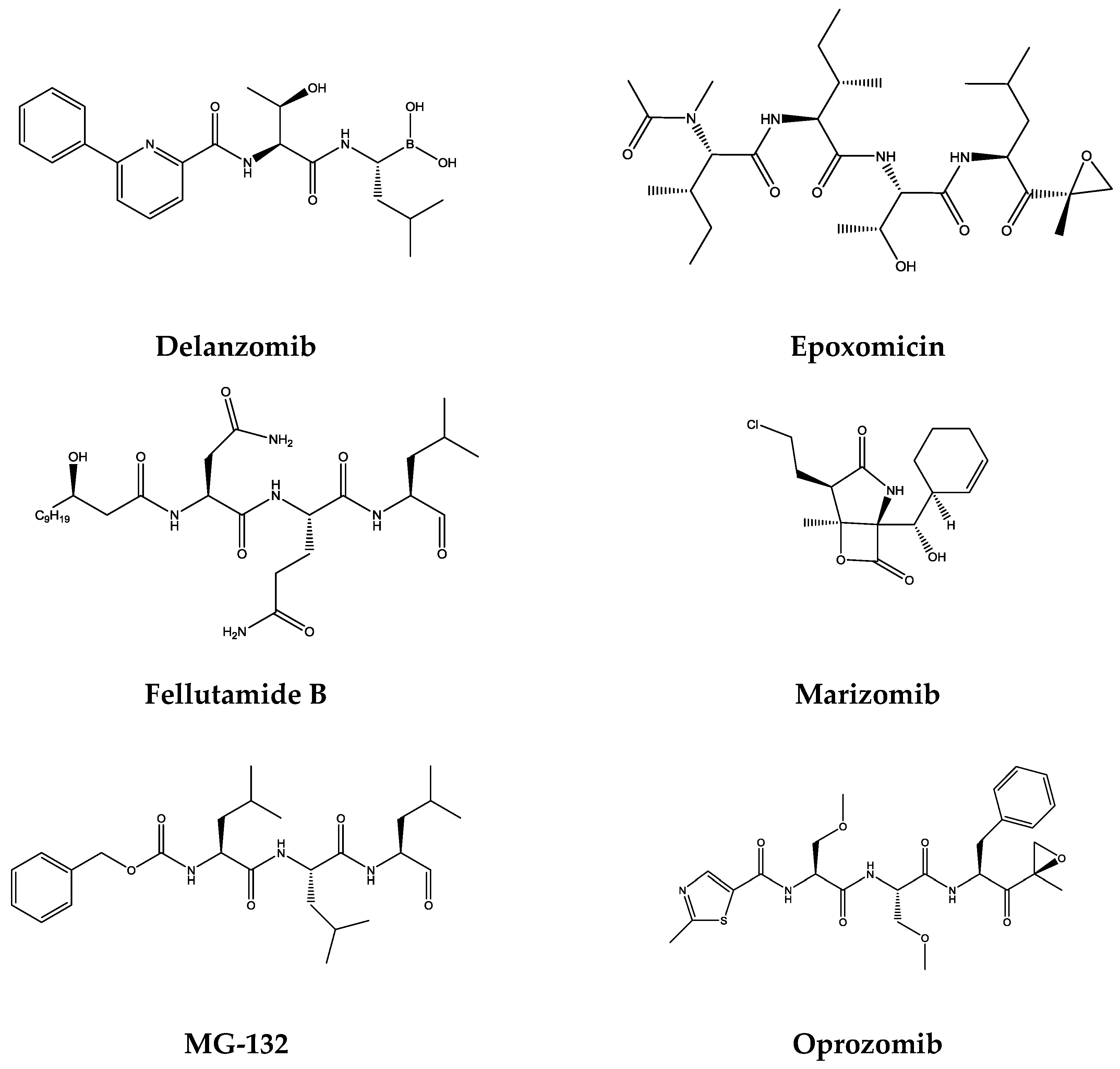
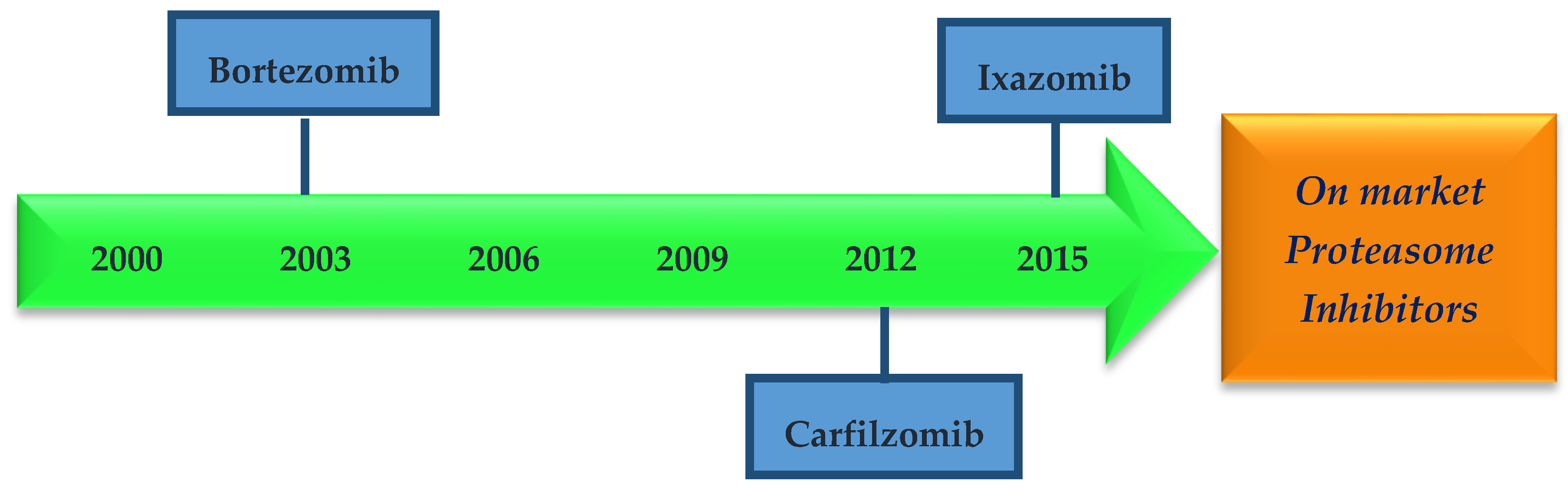
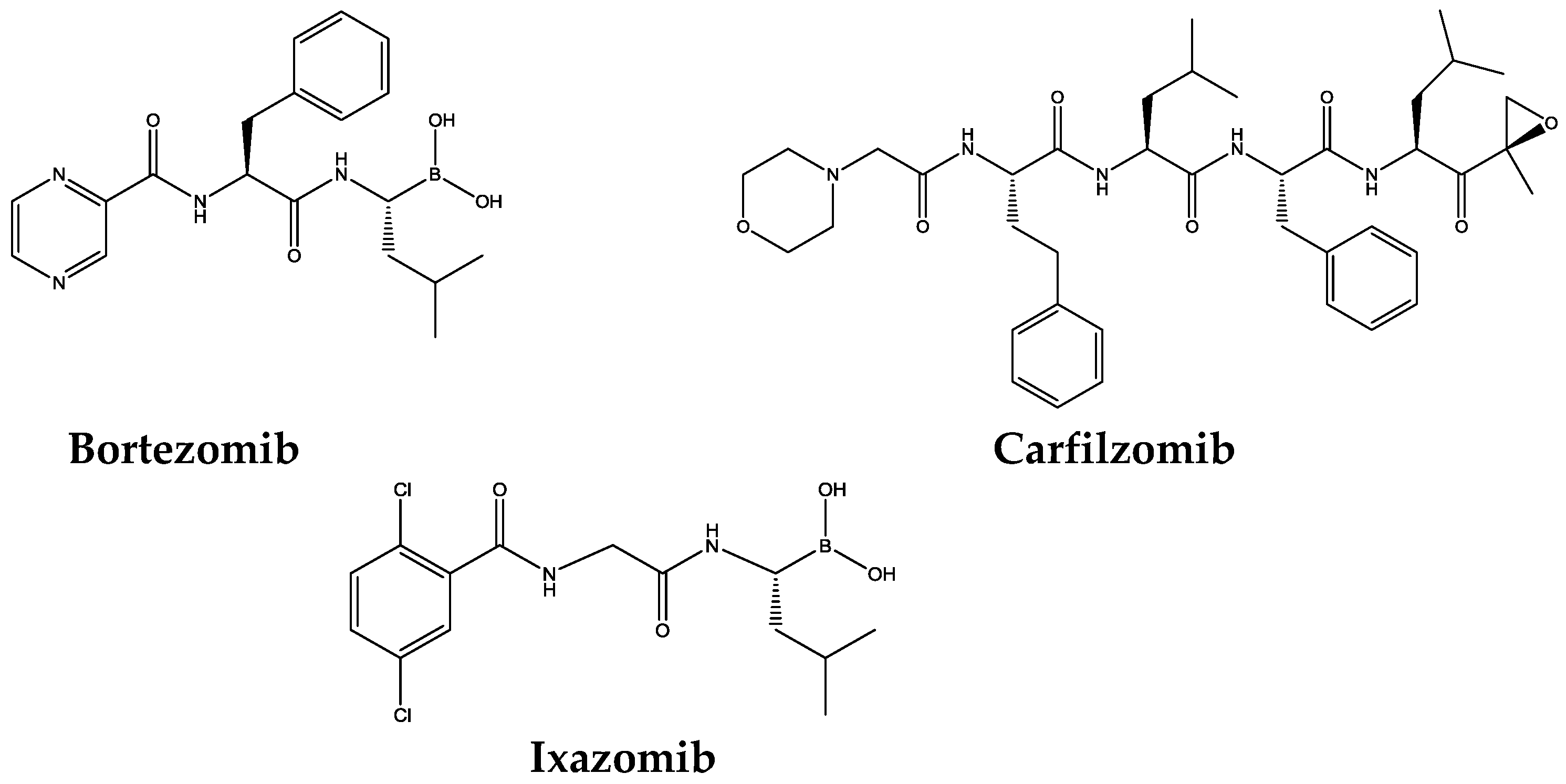
1.2. Proteasome Inhibitors
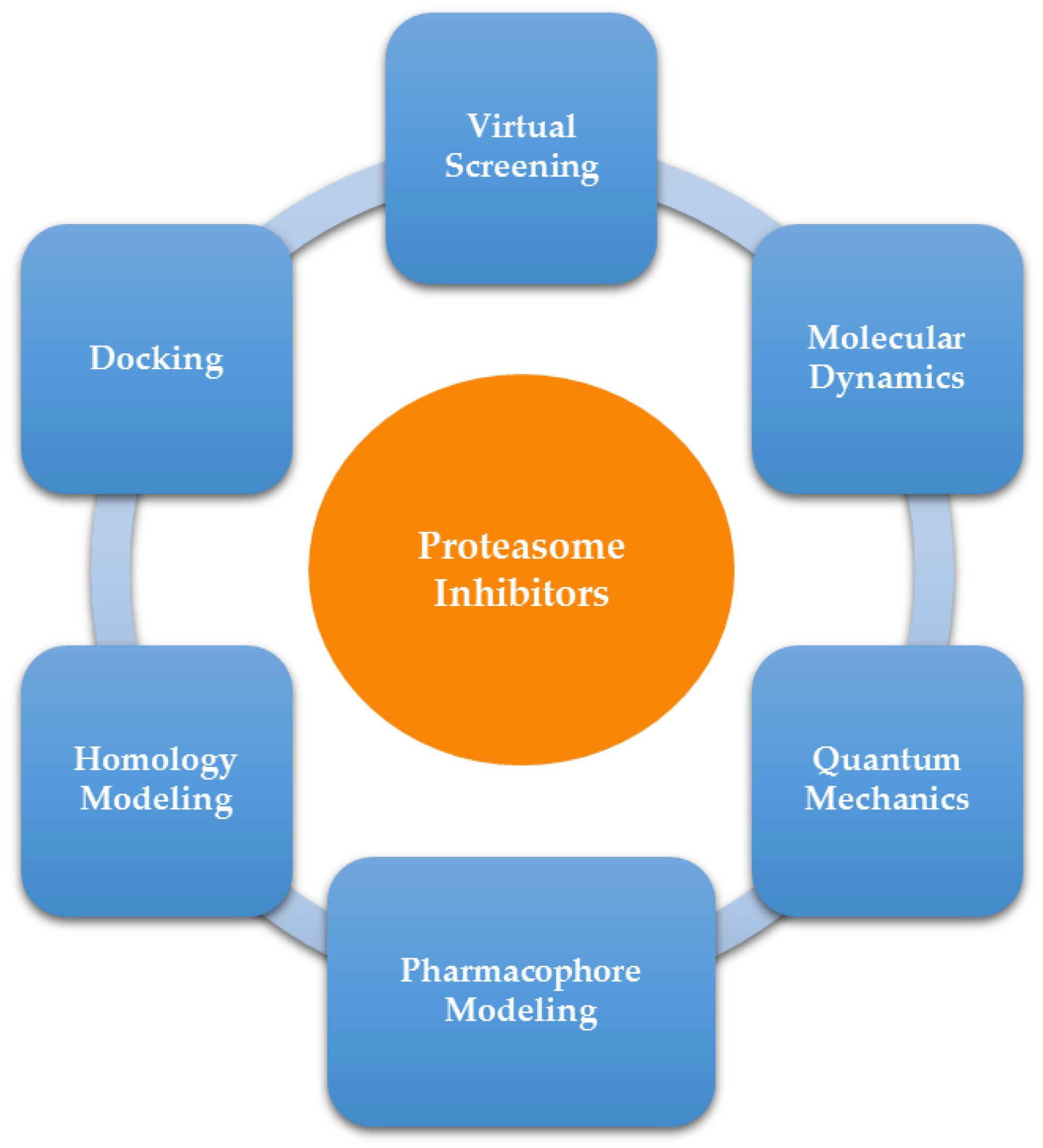
2. Computer-Aided Drug Design
2.1. Homology Modeling
2.2. Pharmacophore Modeling
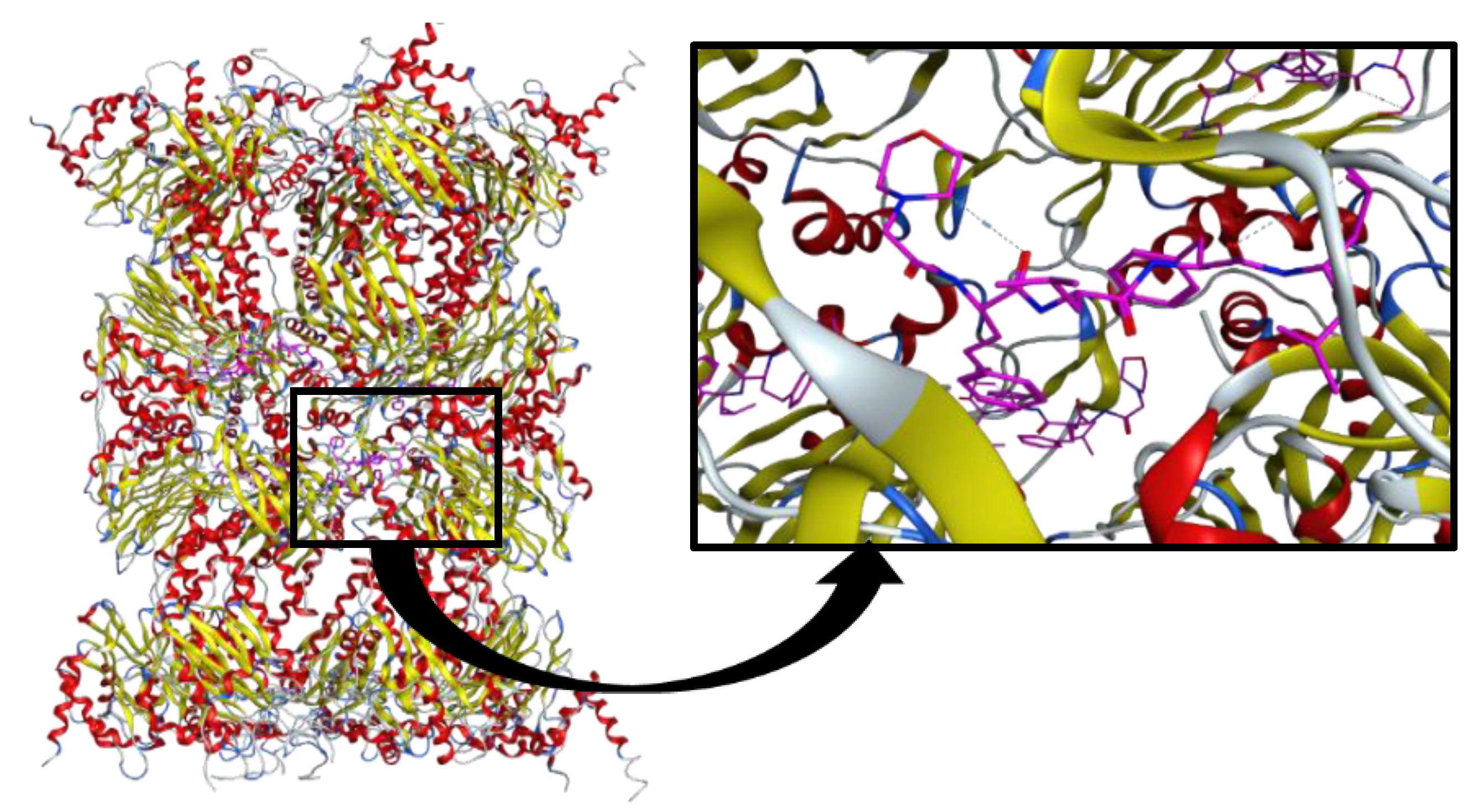
2.3. Molecular Docking
2.4. Virtual Screening
2.5. Combined Methods: Docking, Quantum Mechanics, QM/MM and Molecular Dynamics
3. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Chondrogianni, N.; Voutetakis, K.; Kapetanou, M.; Delitsikou, V.; Papaevgeniou, N.; Sakellari, M.; Lefaki, M.; Filippopoulou, K.; Gonos, E.S. Proteasome activation: An innovative promising approach for delaying aging and retarding age-related diseases. Ageing Res. Rev. 2015, 23, 37–55. [Google Scholar] [CrossRef] [PubMed]
- Jung, T.; Grune, T. Structure of the proteasome. Prog. Mol. Biol. Transl Sci. 2012. [Google Scholar] [CrossRef]
- Stein, M.L.; Groll, M. Applied techniques for mining natural proteasome inhibitors. Biochim. Biophys. Acta 2014, 1843, 26–38. [Google Scholar] [CrossRef] [PubMed]
- Da Fonseca, P.C.A.; He, J.; Morris, E.P. Molecular model of the human 26S proteasome. Mol. Cell. 2012, 46, 54–66. [Google Scholar] [CrossRef] [PubMed]
- Sommer, T.; Wolf, D.H. The ubiquitin-proteasome-system. Biochim. Biophys. Acta. 2014. [Google Scholar] [CrossRef] [PubMed]
- Marastoni, M.; Scotti, A.; Trapella, C.; Ferretti, V.; Sforza, F.; Gavioli, R. Synthesis and activity of isoxazoline vinyl ester pseudopeptides as proteasome inhibitors. J. Pept. Sci. 2014, 20, 258–265. [Google Scholar] [CrossRef] [PubMed]
- Adams, J. The development of proteasome inhibitors as anticancer drugs. Cancer Cell 2004, 5, 417–421. [Google Scholar] [CrossRef]
- Guo, N.; Peng, Z. MG132, a proteasome inhibitor, induces apoptosis in tumor cells. Asia. Pac. J. Clin. Oncol. 2013, 9, 6–11. [Google Scholar] [CrossRef] [PubMed]
- Deshaies, R.J. Proteotoxic crisis, the ubiquitin-proteasome system, and cancer therapy. BMC Biol. 2014. [Google Scholar] [CrossRef] [PubMed]
- Da Fonseca, P.C.A.; Morris, E.P. Structure of the human 26S proteasome: Subunit radial displacements open the gate into the proteolytic core. J. Biol. Chem. 2008, 283, 23305–23314. [Google Scholar] [CrossRef] [PubMed]
- Kisselev, A.F.; Garcia-Calvo, M.; Overkleeft, H.S.; Peterson, E.; Pennington, M.W.; Ploegh, H.L.; Thornberry, N.; Goldberg, A.L. The caspase-like sites of proteasomes, their substrate specificity, new inhibitors and substrates, and allosteric interactions with the trypsin-like sites. J. Biol. Chem. 2003, 278, 35869–25877. [Google Scholar] [CrossRef] [PubMed]
- Teicher, B.A.; Tomaszewski, J.E. Proteasome inhibitors. Biochem. Pharmacol. 2015. [Google Scholar] [CrossRef]
- Gomez, A.M.; Vrolix, K.; Martínez-Martínez, P.; Molenaar, P.C.; Phernambucq, M.; van der Esch, E.; Duimel, H.; Verheyen, F.; Voll, R.E.; Manz, R.A.; et al. Proteasome inhibition with bortezomib depletes plasma cells and autoantibodies in experimental autoimmune myasthenia gravis. J. Immunol. 2011, 186, 2503–2513. [Google Scholar] [CrossRef] [PubMed]
- Fissolo, N.; Kraus, M.; Reich, M.; Ayturan, M.; Overkleeft, H.; Driessen, C.; Weissert, R. Dual inhibition of proteasomal and lysosomal proteolysis ameliorates autoimmune central nervous system inflammation. Eur. J. Immunol. 2008, 38, 2401–2411. [Google Scholar] [CrossRef] [PubMed]
- Ichikawa, H.T.; Conley, T.; Muchamuel, T.; Jiang, J.; Lee, S.; Owen, T.; Barnard, J.; Nevarez, S.; Goldman, B.I.; Kirk, C.J.; et al. Beneficial effect of novel proteasome inhibitors in murine lupus via dual inhibition of type I interferon and autoantibody-secreting cells. Arthritis Rheum. 2012, 64, 493–503. [Google Scholar] [CrossRef] [PubMed]
- Elliott, P.J.; Pien, C.S.; McCormack, T.A.; Chapman, I.D.; Adams, J. Proteasome inhibition: A novel mechanism to combat asthma. J. Allergy Clin. Immunol. 1999, 104, 294–300. [Google Scholar] [CrossRef]
- Elliot, P.J.; Zollner, T.M.; Boehncke, W.-H. Proteasome inhibition: A new anti-inflammatory strategy. J. Mol. Med. 2003, 81, 235–245. [Google Scholar]
- Everly, M.J. A summary of bortezomib use in transplantation across 29 centers. Clin. Transpl. 2009, 323–337. [Google Scholar]
- Czesny, B.; Goshu, S.; Cook, J.L.; Williamson, K.C. The proteasome inhibitor epoxomicin has potent Plasmodium falciparum gametocytocidal activity. Antimicrob. Agents Chemother. 2009, 53, 4080–4085. [Google Scholar] [CrossRef] [PubMed]
- Vale, N.; Pérez, B.; Gomes, A.; Gomes, J.R.B.; Gomes, P. “Recycling” Classical Drugs for Malaria. Chem. Rev. 2014, 114, 11164–11220. [Google Scholar]
- Crunkhorn, S. Novel proteasome inhibitor combats malaria. Nat. Rev. Drug Discov. 2016, 15, 232–233. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; O′Donoghue, A.J.; van der Linden, W.A.; Xie, S.C.; Yoo, E.; Foe, I.T.; Tilley, L.; Craik, C.S.; da Fonseca, P.C.; Bogyo, M. Structure- and function-based design of Plasmodium-selective proteasome inhibitors. Nature 2016, 530, 233–236. [Google Scholar] [CrossRef] [PubMed]
- Kisselev, A.F.; van der Linden, W.A.; Overkleeft, H.S. Proteasome inhibitors: An expanding army attacking a unique target. Chem. Biol. 2012, 19, 99–115. [Google Scholar] [CrossRef] [PubMed]
- Ciechanover, A. Intracellular protein degradation from a vague idea through the lysosome and the ubiquitin-proteasome system and on to human diseases and drug targeting: Nobel Lecture, December 8, 2004. Ann. N. Y. Acad. Sci. 2007. [Google Scholar] [CrossRef] [PubMed]
- Verbrugge, S.E.; Scheper, R.J.; Lems, W.F.; de Gruijl, T.D.; Jansen, G. Proteasome inhibitors as experimental therapeutics of autoimmune diseases. Arthritis Res. Ther. 2015. [Google Scholar] [CrossRef] [PubMed]
- Blackburn, C.; Gigstad, K.M.; Hales, P.; Garcia, K.; Jones, M.; Bruzzese, F.J.; Barrett, C.; Liu, J.X.; Soucy, T.A.; Sappal, D.S.; et al. Characterization of a new series of non-covalent proteasome inhibitors with exquisite potency and selectivity for the 20S beta5-subunit. Biochem. J. 2010, 430, 461–476. [Google Scholar] [CrossRef] [PubMed]
- Finley, D. Recognition and processing of ubiquitin-protein conjugates by the proteasome. Annu. Rev. Biochem. 2009, 78, 477–513. [Google Scholar] [CrossRef] [PubMed]
- Huber, E.; Basler, M.; Schwab, R.; Heinemeyer, W.; Kirk, C.J.; Groettrup, M.; Groll, M. Immuno- and constitutive proteasome crystal structures reveal differences in substrate and inhibitor specificity. Cell 2012, 148, 727–738. [Google Scholar]
- Tanahashi, N.; Murakami, Y.; Minami, Y.; Shimbara, N.; Hendil, K.B.; Tanaka, K. Hybrid proteasomes. Induction by interferon-gamma and contribution to ATP-dependent proteolysis. J. Biol. Chem. 2000, 275, 14336–14345. [Google Scholar] [CrossRef] [PubMed]
- Murata, S.; Sasaki, K.; Kishimoto, T.; Niwa, S.-I.; Hayashi, H.; Takahama, Y.; Tanaka, K. Regulation of CD8+ T cell development by thymus-specific proteasomes. Science 2007, 316, 1349–1353. [Google Scholar] [CrossRef] [PubMed]
- Murata, S.; Takahama, Y.; Tanaka, K. Thymoproteasome: Probable role in generating positively selecting peptides. Curr. Opin. Immunol. 2008, 20, 192–196. [Google Scholar] [CrossRef] [PubMed]
- De Bettignies, G.; Coux, O. Proteasome inhibitors: Dozens of molecules and still counting. Biochimie 2010, 92, 1530–1545. [Google Scholar] [CrossRef] [PubMed]
- Jung, T.; Catalgol, B.; Grune, T. The proteasomal system. Mol. Aspects Med. 2009, 30, 191–296. [Google Scholar] [CrossRef] [PubMed]
- Bedford, L.; Paine, S.; Sheppard, P.W.; Mayer, R.J.; Roelofs, J. Assembly, structure, and function of the 26S proteasome. Trends Cell Biol. 2010, 20, 391–401. [Google Scholar] [CrossRef] [PubMed]
- Lander, G.C.; Estrin, E.; Matyskiela, M.E.; Bashore, C.; Nogales, E.; Martin, A. Complete subunit architecture of the proteasome regulatory particle. Nature 2012, 482, 186–191. [Google Scholar] [CrossRef] [PubMed]
- Goldberg, A. Development of proteasome inhibitors as research tools and cancer drugs. J. Cell Biol. 2012, 199, 583–588. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.M.; Yu, Y.; Cheg, Y. Structure characterization of the 26S proteasome. Biochim. Biophys. Acta 2011, 1809, 67–79. [Google Scholar] [CrossRef] [PubMed]
- Ginodi, I.; Vider-Shalit, T.; Tsaban, L.; Louzoun, Y. Precise score for the prediction of peptides cleaved by the proteasome. Bioinformatics 2008, 24, 477–483. [Google Scholar] [CrossRef] [PubMed]
- Hadeler, K.P.; Kuttler, C.; Nussbaum, A.K. Cleaving proteins for the immune system. Math. Biosci. 2004, 188, 63–79. [Google Scholar] [CrossRef] [PubMed]
- Groll, M.; Heinemeyer, W.; Jäger, S.; Ullrich, T.; Bochtler, M.; Wolf, D.H.; Huber, R. The catalytic sites of 20S proteasomes and their role in subunit maturation: A mutational and crystallographic study. Proc. Natl. Acad. Sci. 1999, 96, 10976–10983. [Google Scholar] [CrossRef] [PubMed]
- Kisselev, A.F.; Akopian, T.N.; Woo, K.M.; Goldberg, A.L. The sizes of peptides generated from protein by mammalian 26 and 20 S proteasomes. J. Biol. Chem. 1999, 274, 3363–3371. [Google Scholar]
- Nussbaum, A.K.; Dick, T.P.; Keilholz, W.; Schirle, M.; Stevanović, S.; Dietz, K.; Heinemeyer, W.; Groll, M.; Wolf, D.H.; Huber, R.; et al. Cleavage motifs of the yeast 20S proteasome β subunits deduced from digests of enolase 1. Proc. Natl. Acad. Sci. 1998, 95, 12504–12509. [Google Scholar] [CrossRef] [PubMed]
- Diez-Rivero, C.M.; Lafuente, E.M.; Reche, P.A. Computational analysis and modeling of cleavage by the immunoproteasome and the constitutive proteasome. BMC Bioinformatics 2010. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, Y.; Zhao, X.; Zhu, X.; Wu, G.; Li, Y.; Ma, Y.; Yuan, Y.; Yang, J.; Hu, Y.; Ai, L.; et al. Design, synthesis, biological evaluation, and structure-activity relationship (SAR) discussion of dipeptidyl boronate proteasome inhibitors, part I: Comprehensive understanding of the SAR of alpha-amino acid boronates. J. Med. Chem. 2009, 52, 4192–4199. [Google Scholar] [CrossRef] [PubMed]
- Beck, P.; Dubiella, C.; Groll, M. Covalent and non-covalent reversible proteasome inhibition. Biol. Chem. 2012, 393, 1101–1120. [Google Scholar] [CrossRef] [PubMed]
- Borissenko, L.; Groll, M. 20S proteasome and its inhibitors: Crystallographic knowledge for drug development. Chem. Rev. 2007, 107, 687–717. [Google Scholar] [CrossRef] [PubMed]
- Harshbarger, W.; Miller, C.; Diedrich, C.; Sacchettini, J. Crystal Structure of the Human 20S Proteasome in Complex with Carfilzomib. Structure 2015, 23, 418–424. [Google Scholar] [CrossRef] [PubMed]
- Kaffy, J.; Bernadat, G.; Ongeri, S. Non-covalent proteasome inhibitors. Curr. Pharm. Des. 2013, 19, 4115–4130. [Google Scholar] [CrossRef] [PubMed]
- Demo, S.D.; Kirk, C.J.; Aujay, M.A.; Buchholz, T.J.; Dajee, M.; Ho, M.N.; Jiang, J.; Laidig, G.J.; Lewis, E.R.; Parlati, F.; et al. Antitumor activity of PR-171, a novel irreversible inhibitor of the proteasome. Cancer Res. 2007, 67, 6383–6391. [Google Scholar] [CrossRef] [PubMed]
- Piva, R.; Ruggeri, B.; Williams, M.; Costa, G.; Tamagno, I.; Ferrero, D.; Giai, V.; Coscia, M.; Peola, S.; Massaia, M.; et al. CEP-18770: A novel, orally active proteasome inhibitor with a tumor-selective pharmacologic profile competitive with bortezomib. Blood 2008, 111, 2765–2775. [Google Scholar] [CrossRef] [PubMed]
- Hines, J.; Groll, M.; Fahnestock, M.; Crews, C.M. Proteasome inhibition by fellutamide B induces nerve growth factor synthesis. Chem. Biol. 2008, 15, 501–512. [Google Scholar] [CrossRef] [PubMed]
- Kupperman, E.; Lee, E.C.; Cao, Y.; Bannerman, B.; Fitzgerald, M.; Berger, A.; Yu, J.; Yang, Y.; Hales, P.; Bruzzese, F.; et al. Evaluation of the Proteasome Inhibitor MLN9708 in Preclinical Models of Human Cancer. Cancer Res. 2010, 70, 1970–1980. [Google Scholar] [CrossRef] [PubMed]
- Groll, M.; Potts, B.C. Proteasome structure, function, and lessons learned from beta-lactone inhibitors. Curr. Top Med. Chem. 2011, 11, 2850–2878. [Google Scholar] [CrossRef] [PubMed]
- Momose, I.; Umezawa, Y.; Hirosawa, S.; Iijima, M.; Iinuma, H.; Ikeda, D. Synthesis and activity of tyropeptin A derivatives as potent and selective inhibitors of mammalian 20S proteasome. Biosci. Biotechnol. Biochem. 2005, 69, 1733–1742. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.J.; Aujay, M.A.; Bennett, M.K.; Dajee, M.; Demo, S.D.; Fang, Y.; Ho, M.N.; Jiang, J.; Kirk, C.J.; Laidig, G.J.; et al. Design and synthesis of an orally bioavailable and selective peptide epoxyketone proteasome inhibitor (PR-047). J. Med. Chem. 2009, 52, 3028–3038. [Google Scholar] [CrossRef] [PubMed]
- Kisselev, A.F.; Groettrup, M. Subunit specific inhibitors of proteasomes and their potential for immunomodulation. Curr. Opin. Chem. Biol. 2014, 23, 16–22. [Google Scholar] [CrossRef] [PubMed]
- Smith, B.R.; Eastman, C.M.; Njardarson, J.T. Beyond C, H, O, and N! Analysis of the Elemental Composition of U.S. FDA Approved Drug Architectures. J. Med. Chem. 2014, 57, 9764–9773. [Google Scholar] [CrossRef] [PubMed]
- Furukawa, Y.; Kikuchi, J. Molecular pathogenesis of multiple myeloma. Int. J. Clin. Oncol. 2015, 29, 413–422. [Google Scholar] [CrossRef] [PubMed]
- Torimoto, Y.; Shindo, M.; Ikuta, K.; Kohgo, Y. Current therapeutic strategies for multiple myeloma. Int. J. Clin. Oncol. 2015, 20, 423–430. [Google Scholar] [CrossRef] [PubMed]
- European Medicines Agency Find medicine Velcade. Available online: http://www.ema.europa.eu/ema/index.jsp?curl=pages/medicines/human/medicines/000539/human_med_001130.jsp&mid=WC0b01ac058001d124 (accessed on 3 March 2016).
- Sánchez-Serrano, I. Success in translational research: Lessons from the development of bortezomib. Nat. Rev. Drug Discov. 2006, 5, 107–114. [Google Scholar] [CrossRef] [PubMed]
- Kortuem, K.; Stewart, K. Carfilzomib. Blood 2013, 121, 893–897. [Google Scholar] [CrossRef] [PubMed]
- Merin, N.M.; Kelly, K.R. Clinical use of proteasome inhibitors in the treatment of multiple myeloma. Pharmaceuticals (Basel) 2015. [Google Scholar] [CrossRef] [PubMed]
- U.S. Food and Drug Administration. Research, C. for D. E. and Approved Drugs–Ixazomib 2015. Available online: http://www.fda.gov/Drugs/InformationOnDrugs/ApprovedDrugs/ucm473804.htm (accessed on 28 April 2016).
- Shirley, M. Ixazomib: First Global Approval. Drugs 2015, 75, 1935–1940. [Google Scholar] [CrossRef] [PubMed]
- Lusher, S.; Azevedo, R.; McGuire, R.; Vlieg, J. Computer-Aided Drug Design. In International Computational Drug Discovery Course; Computational Chemistry List: Nijmegen, The Netherlands, 2007. [Google Scholar]
- Liao, C. Software and resources for computational medicinal chemistry. Future Med Chem. 2011, 3, 1057–1085. [Google Scholar] [CrossRef] [PubMed]
- Devi, R.V.; Sathya, S.S.; Coumar, M.S. Evolutionary algorithms for de novo drug design—A survey. Appl. Soft Comput. 2015, 27, 543–552. [Google Scholar] [CrossRef]
- Kazi, A.; Lawrence, H.; Guida, W.C.; Mclaughlin, M.L.; Springett, G.M.; Berndt, N.; Yip, R.M.; Sebti, S.M. Discovery of a novel proteasome inhibitor selective for cancer cells over non-transformed cells. Cell Cycle 2009, 8, 1940–1951. [Google Scholar] [CrossRef] [PubMed]
- Gräwert, M.A.; Gallastegui, N.; Stein, M.; Schmidt, B.; Kloetzel, P.M.; Huber, R.; Groll, M. Elucidation of the α-keto-aldehyde binding mechanism: A lead structure motif for proteasome inhibition. Angew. Chemie. Int. Ed. 2011, 50, 542–544. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.-Y. Computational virtual screening towards designing novel anticancer drugs. In Virtual Screening; Taha, M., Ed.; InTech: Rijeka, Croat, 2012; pp. 91–100. [Google Scholar]
- Triggle, D.J. Comprehensive Medicinal Chemistry II: Computer-Assisted Drug Design; Taylor, J.B., Triggle, D.J., Eds.; Elsevier Science: Amsterdam, The Netherlands, 2006; Volume 4, pp. 119–281. [Google Scholar]
- Zhu, M.; Li, M. Revisiting the homology modeling of G-protein coupled receptors: β1-adrenoceptor as an example. Mol. Biosyst. 2012. [Google Scholar] [CrossRef] [PubMed]
- Groll, M.; Ditzel, L.; Löwe, J.; Stock, D.; Bochtler, M.; Bartunik, H.D.; Huber, R. Structure of 20S proteasome from yeast at 2.4 A resolution. Nature 1997, 386, 463–471. [Google Scholar] [PubMed]
- Kopp, F.; Hendil, K.B.; Dahlmann, B.; Kristensen, P.; Sobek, A.; Uerkvitz, W. Subunit arrangement in the human 20S proteasome. Proc. Natl. Acad. Sci. 1997, 94, 2939–2944. [Google Scholar] [CrossRef] [PubMed]
- Haass, C.; Kloetzel, P.M. The drosophila proteasome undergoes changes in its subunit pattern during development. Exp. Cell Res. 1989, 180, 243–252. [Google Scholar] [CrossRef]
- Morimoto, Y.; Mizushima, T.; Yagi, A.; Tanahashi, N.; Tanaka, K.; Ichihara, A.; Tsukihara, T. Ordered Structure of the Crystallized Bovine 20S Proteasome. J. Biochem. 1995, 471–474. [Google Scholar]
- Vyas, V.K.; Ukawala, R.D.; Ghate, M.; Chintha, C. Homology modeling a fast tool for drug discovery: Current perspectives. Indian J. Pharm. Sci. 2012. [Google Scholar] [CrossRef] [PubMed]
- Mozzicafreddo, M.; Cuccioloni, M.; Cecarini, V.; Eleuteri, A.M.; Angeletti, M. Homology modeling and docking analysis of the interaction between polyphenols and mammalian 20S proteasomes. J. Chem. Inf. Model. 2009, 49, 401–409. [Google Scholar] [CrossRef] [PubMed]
- LaFranzo, N.A.; Strulson, M.K.; Yanker, D.M.; Dang, L.T.; Maurer, J.A. Sequence or structure: Using bioinformatics and homology modeling to understand functional relationships in cAMP/cGMP binding domains. Mol. Biosyst. 2010, 6, 894–901. [Google Scholar] [CrossRef] [PubMed]
- Lei, M.; Liu, Y.; Zhu, Y.; Liu, Z. Progress of computer-aided drug design (CADD) of proteasome inhibitors. Curr. Top. Med. Chem. 2011, 11, 2931–2944. [Google Scholar] [CrossRef] [PubMed]
- Basic Local Alignment Search Tool (BLAST). The National Center for Biotechnology Information, Bethesda (USA). Available online: http://blast.ncbi.nlm.nih.gov/BLAST.cgi (accessed on 6 June 2016).
- The Uniprot Consortium The Universal Protein Resource (UniProt). Available online: http://www.uniprot.org (accessed on 6 June 2016).
- Molecular Operating Environment (MOE), Chemical Computing Group Inc.: Montreal, QC, Canada, 2016.
- Furet, P.; Imbach, P.; Noorani, M.; Koeppler, J.; Laumen, K.; Lang, M.; Guagnano, V.; Fuerst, P.; Roesel, J.; Zimmermann, J.; et al. Entry into a new class of potent proteasome inhibitors having high antiproliferative activity by structure-based design. Society 2004, 47, 4810–4813. [Google Scholar] [CrossRef] [PubMed]
- Loizidou, E.Z.; Zeinalipour-Yazdi, C.D. Computational inhibition studies of the human proteasome by argyrin-based analogues with subunit specificity. Chem. Biol. Drug Des. 2014, 84, 99–107. [Google Scholar] [CrossRef] [PubMed]
- Ehrlich, P. Über den jetzigen stand der chemotherapie. Berichte der Dtsch. Chem. Gesellschaft 1909, 42, 17–47. [Google Scholar] [CrossRef]
- Gund, P. Three-dimensional pharmacophoric pattern searching. Prog. Mol. Subcell. Bio. 1977, 5, 117–143. [Google Scholar]
- International Union of Pure and Applied Chemistry. Glossary of terms used in medicinal chemistry. Available online: https://fenix.tecnico.ulisboa.pt/downloadFile/3779571244449/Pure_Appl.%20Chem_Gloss%C3%83%C2%A1rio.pdf (accessed on 21 April 2016).
- Hein, M.; Zilian, D.; Sotriffer, C. Docking compared to 3D-pharmacophores: The scoring function challenge. Drug Discov. Today Technol. 2010, 7, e229–e236. [Google Scholar] [CrossRef]
- Gao, Q.; Yang, L.; Zhu, Y. Pharmacophore based drug design approach as a practical process in drug discovery. Curr. Comput. Aided. Drug Des. 2010, 6, 37–49. [Google Scholar] [CrossRef] [PubMed]
- Gallastegui, N.; Beck, P.; Arciniega, M.; Huber, R.; Hillebrand, S.; Groll, M. Hydroxyureas as noncovalent proteasome inhibitors. Angew. Chem. Int. Ed. Engl. 2012, 51, 247–249. [Google Scholar] [CrossRef] [PubMed]
- Pautasso, C.; Troia, R.; Genuardi, M.; Palumbo, A. Pharmacophore modeling technique applied for the discovery of proteasome inhibitors. Expert Opin. Drug Discov. 2014, 9, 931–943. [Google Scholar] [CrossRef] [PubMed]
- DS Catalyst–Accelrys. Available online: http://accelrys.com/ (accessed on 4 April 2016).
- Phase–Schrödinger. Available online: http://www.schrodinger.com/Phase/ (accessed on 4 April 2016).
- Lei, M.; Zhao, X.; Wang, Z.; Zhu, Y. Pharmacophore modeling, docking studies, and synthesis of novel dipeptide proteasome inhibitors containing boron atoms. J. Chem. Inf. Model 2009, 49, 2092–2100. [Google Scholar] [CrossRef] [PubMed]
- Li, A.; Sun, H.; Du, L.; Wu, X.; Cao, J.; You, Q.; Li, Y. Discovery of novel covalent proteasome inhibitors through a combination of pharmacophore screening, covalent docking, and molecular dynamics simulations. J. Mol. Model. 2014. [Google Scholar] [CrossRef] [PubMed]
- Guo, L.; Yan, Z.; Zheng, X.; Hu, L.; Yang, Y.; Wang, J. A comparison of various optimization algorithms of protein-ligand docking programs by fitness accuracy. J. Mol. Model. 2014, 20, 17–21. [Google Scholar] [CrossRef] [PubMed]
- Kumalo, H.M.; Bhakat, S.; Soliman, M.E.S. Theory and applications of covalent docking in drug discovery: Merits and pitfalls. Molecules 2015, 20, 1984–2000. [Google Scholar] [CrossRef] [PubMed]
- Singh, J.; Petter, R.C.; Baillie, T.A.; Whitty, A. The resurgence of covalent drugs. Nat. Rev. Drug Discov. 2011, 10, 307–317. [Google Scholar] [CrossRef] [PubMed]
- Genetic optimisation for ligand docking (GOLD). The Cambridge Crystallographic Data Centre. Available online: http://www.ccdc.cam.ac.uk/Solutions/GoldSuite/Pages/GOLD.aspx (accessed on 26 April 2016).
- Muegge, I.; Enyede, I. Docking and scoring. In Computational Medicinal Chemistry for Drug Discovery; Bultinck, P., Winter, H.D., Langenaeker, W., Tollenaere, J., Eds.; Marcel Dekker: New York, NY, USA, 2004. [Google Scholar]
- Shin, W.H.; Seok, C. GalaxyDock: Protein-ligand docking with flexible protein side-chains. J. Chem. Inf. Model. 2012, 52, 3225–3232. [Google Scholar] [CrossRef] [PubMed]
- Novikov, F.; Chilov, G. Molecular docking: Theoretical background, practical applications and perspectives. Mendeleev Commun. 2009, 19, 237–242. [Google Scholar] [CrossRef]
- Kitchen, D.B.; Decornez, H.; Furr, J.R.; Bajorath, J. Docking and scoring in virtual screening for drug discovery: Methods and applications. Nat. Rev. Drug Discov. 2004, 3, 935–949. [Google Scholar] [CrossRef] [PubMed]
- Yuriev, E.; Holien, J.; Ramsland, P.A. Improvements, trends, and new ideas in molecular docking: 2012–2013 in review. J. Mol. Recognit. 2015, 581–604. [Google Scholar] [CrossRef] [PubMed]
- Molecular Graphics Laboratory, The Scripps Research Institute. AutoDock. Available online: http://autodock.scripps.edu/ (accessed on 9 March 2016).
- UCSF Molecular Design Institute. DOCK. Available online: http://dock.compbio.ucsf.edu/Contact_Info/index.htm (accessed on 9 March 2016).
- BioSolveIT. FlexX. Available online: http://www.biosolveit.de/flexx/index.html?ct=1 (accessed on 24 March 2016).
- Tripos International St. Louis, U. Surflex-Dock. Available online: http://www.tripos.com/index.php?family=modules,SimplePage,,,&page=surflex_dock&s= 0 (accessed on 24 March 2016).
- Glide–Schrodinger. Available online: http://www.schrodinger.com/productpage/14/5/ (accessed on 24 March 2016).
- Thomsen, R.; Christensen, M.H. MolDock: A New Technique for High-Accuracy Molecular Docking. J. Med. Chem. 2006, 49, 3315–3321. [Google Scholar] [CrossRef] [PubMed]
- OpenEye Scientific Software, FRED. Available online: https://docs.eyesopen.com/oedocking/fred.html#fred (accessed on 24 April 2016).
- Molsoft L.L.C. ICM. Available online: http://www.molsoft.com/docking.html (accessed on 24 April 2016).
- Sousa, S.F.; Ribeiro, A.J.M.; Coimbra, J.T.S.; Neves, R.P.P.; Martins, S.A.; Moorthy, N.S.H.N.; Fernandes, P.A.; Ramos, M.J. Protein-ligand docking in the new millennium—A retrospective of 10 years in the field. Curr. Med. Chem. 2013, 20, 2296–2314. [Google Scholar] [CrossRef] [PubMed]
- Carlson, H.A.; Smith, R.D.; Damm-Ganamet, K.L.; Stuckey, J.A.; Ahmed, A.; Convery, M.A.; Somers, D.O.; Kranz, M.; Elkins, P.A.; Cui, G.; et al. CSAR 2014: A benchmark exercise using unpublished data from pharma. J. Chem. Inf. Model. 2016. [Google Scholar] [CrossRef] [PubMed]
- Hartshorn, M.J.; Verdonk, M.L.; Chessari, G.; Brewerton, S.C.; Mooij, W.T.M.; Mortenson, P.N.; Murray, C.W. Diverse, high-quality test set for the validation of protein-ligand docking performance. J. Med. Chem. 2007, 50, 726–741. [Google Scholar] [CrossRef] [PubMed]
- Liebeschuetz, J.W.; Cole, J.C.; Korb, O. Pose prediction and virtual screening performance of GOLD scoring functions in a standardized test. J. Comput. Aided. Mol. Des. 2012, 26, 737–748. [Google Scholar] [CrossRef] [PubMed]
- Kazi, A.; Daniel, K.G.; Smith, D.M.; Kumar, N.B.; Dou, Q.P. Inhibition of the proteasome activity, a novel mechanism associated with the tumor cell apoptosis-inducing ability of genistein. Biochem. Pharmacol. 2003, 66, 965–976. [Google Scholar] [CrossRef]
- Smith, D.M.; Daniel, K.G.; Wang, Z.; Guida, W.C.; Chan, T.-H.; Dou, Q.P. Docking studies and model development of tea polyphenol proteasome inhibitors: Applications to rational drug design. Proteins 2004, 54, 58–70. [Google Scholar] [CrossRef] [PubMed]
- Rydzewski, R.M.; Burrill, L.; Mendonca, R.; Palmer, J.T.; Rice, M.; Tahilramani, R.; Bass, K.E.; Leung, L.; Gjerstad, E.; Janc, J.W.; et al. Optimization of subsite binding to the beta5 subunit of the human 20S proteasome using vinyl sulfones and 2-keto-1,3,4-oxadiazoles: Syntheses and cellular properties of potent, selective proteasome inhibitors. J. Med. Chem. 2006, 49, 2953–2968. [Google Scholar] [CrossRef] [PubMed]
- Nam, S.; Smith, D.M.; Dou, Q.P. Ester bond-containing tea polyphenols potently inhibit proteasome activity in vitro and in vivo. J. Biol. Chem. 2001, 276, 13322–13330. [Google Scholar] [CrossRef] [PubMed]
- Milacic, V.; Banerjee, S.; Landis-Piwowar, K.R.; Sarkar, F.H.; Majumdar, A.P.N.; Dou, Q.P. Curcumin inhibits the proteasome activity in human colon cancer cells in vitro and in vivo. Cancer Res. 2008, 68, 7283–7292. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Landis-Piwowar, K.R.; Lu, D.; Yuan, P.; Li, L.; Prem-Veer Reddy, G.; Yuan, X.; Dou, Q.P. Pristimerin induces apoptosis by targeting the proteasome in prostate cancer cells. J. Cell. Biochem. 2008, 103, 234–244. [Google Scholar] [CrossRef] [PubMed]
- Leban, J.; Blisse, M.; Krauss, B.; Rath, S.; Baumgartner, R.; Seifert, M.H.J. Proteasome inhibition by peptide-semicarbazones. Bioorganic Med. Chem. 2008, 16, 4579–4588. [Google Scholar] [CrossRef] [PubMed]
- Seifert, M.H.J.; Schmitt, F.; Herz, T.; Kramer, B. ProPose: A docking engine based on a fully configurable protein-ligand interaction model. J. Mol. Model. 2004, 10, 342–357. [Google Scholar] [CrossRef] [PubMed]
- Seifert, M.H.J. ProPose: Steered virtual screening by simultaneous protein-ligand docking and ligand-ligand alignment. J. Chem. Inf. Model. 2005, 45, 449–460. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Shi, Y.; Jin, H.; Liu, Z.; Zhang, L.; Zhang, L. Covalent complexes of proteasome model with peptide aldehyde inhibitors MG132 and MG101: Docking and molecular dynamics study. J. Mol. Model. 2009, 15, 1481–1490. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Zhu, X.; Wu, G.; Ma, Y.; Li, Y.; Zhao, X.; Yuan, Y.; Yang, J.; Yu, S.; Shao, F.; et al. Synthesis, in vitro and in vivo biological evaluation, docking studies, and structure-activity relationship (SAR) discussion of dipeptidyl boronic acid proteasome inhibitors composed of β-amino acids. J. Med. Chem. 2010, 53, 1990–1999. [Google Scholar] [CrossRef] [PubMed]
- Smoum, R.; Rubinstein, A.; Dembitsky, V.M.; Srebnik, M. Boron containing compounds as protease inhibitors. Chem. Rev. 2012, 112, 4156–4220. [Google Scholar] [CrossRef] [PubMed]
- Kanwar, J.; Moahammad, I.; Yang, H.; Huo, C.; Chan, T.H.; Dou, Q.P. Computational modeling of the potential interactions of the proteasome β5 subunit and catechol-O-methyltransferase-resistant EGCG analogs. Int. J. Mol. Med. 2010, 26, 837–843. [Google Scholar]
- Shi, G.; Sun, Q.; Yang, H.; Dou, Q.; Deng, Q.; Wang, H.; Zhong, G. Molecular modeling for the interaction between proteasome beta 5 subunit and organotin compounds. Sci. China Chem. 2010, 53, 2387–2393. [Google Scholar] [CrossRef]
- Bonfili, L.; Cuccioloni, M.; Mozzicafreddo, M.; Cecarini, V.; Angeletti, M.; Eleuteri, A.M. Identification of an EGCG oxidation derivative with proteasome modulatory activity. Biochimie 2011, 93, 931–940. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Xu, B.; Fang, Y.; Yang, Z.; Cui, J.; Zhang, L.; Zhang, L. Synthesis and SAR study of novel peptide aldehydes as inhibitors of 20S proteasome. Molecules 2011, 16, 7551–7564. [Google Scholar] [CrossRef] [PubMed]
- Xu, K.; Xiao, Z.; Tang, Y.B.; Huang, L.; Chen, C.-H.; Ohkoshi, E.; Lee, K.-H. Design and synthesis of naphthoquinone derivatives as antiproliferative agents and 20S proteasome inhibitors. Bioorg. Med. Chem. Lett. 2012, 22, 2772–2774. [Google Scholar] [CrossRef] [PubMed]
- Pham, T.H.; Hovhannisyan, A.; Bouvier, D.; Tian, L.; Reboud-Ravaux, M.; Melikyan, G.; Bouvier-Durand, M. A new series of N5 derivatives of the 1,1,5-trimethyl furo[3,4-c]pyridine-3,4-dione (cerpegin) selectively inhibits the post-acid activity of mammalian 20S proteasomes. Bioorg. Med. Chem. Lett. 2012, 22, 3822–3827. [Google Scholar] [CrossRef] [PubMed]
- Santoro, A.M.; Lo Giudice, M.C.; D’Urso, A.; Lauceri, R.; Purrello, R.; Milardi, D. Cationic porphyrins are reversible proteasome inhibitors. J. Am. Chem. Soc. 2012, 134, 10451–10457. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hovhannisyan, A.; Pham, T.H.; Bouvier, D.; Qin, L.; Melikyan, G.; Reboud-Ravaux, M.; Bouvier-Durand, M. C1 and N5 derivatives of cerpegin: Synthesis of a new series based on structure-activity relationships to optimize their inhibitory effect on 20S proteasome. Bioorganic Med. Chem. Lett. 2013, 23, 2696–2703. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.; Sun, J.; Xu, Q.; Liu, Y.; Wei, J.; Young, C.Y.F.; Yuan, H.; Lou, H. Marchantin M: A novel inhibitor of proteasome induces autophagic cell death in prostate cancer cells. Cell Death Dis. 2013. [Google Scholar] [CrossRef] [PubMed]
- Orabi, K.Y.; Abaza, M.S.; El Sayed, K.A.; Elnagar, A.Y.; Al-Attiyah, R.; Guleri, R.P. Selective growth inhibition of human malignant melanoma cells by syringic acid-derived proteasome inhibitors. Cancer Cell Int. 2013. [Google Scholar] [CrossRef] [PubMed]
- Zuo, J.; Caifeng, B.; Yuhua, F.; Daniela, B.; Chiara, N.; Kenyon, G.D.; Ping, D.Q. Cellular and computational studies of proteasome inhibition and apoptosis induction in human cancer cells by amino acid Schiff base-copper complexes. J. Inorg. Biochem. 2013, 118, 83–93. [Google Scholar] [CrossRef] [PubMed]
- Bordessa, A.; Keita, M.; Maréchal, X.; Formicola, L.; Lagarde, N.; Rodrigo, J.; Bernadat, G.; Bauvais, C.; Soulier, J.-L.; Dufau, L.; et al. α- and β-hydrazino acid-based pseudopeptides inhibit the chymotrypsin-like activity of the eukaryotic 20S proteasome. Eur. J. Med. Chem. 2013, 70, 505–524. [Google Scholar] [CrossRef] [PubMed]
- Kawamura, S.; Unno, Y.; Tanaka, M.; Sasaki, T.; Yamano, A.; Hirokawa, T.; Kameda, T.; Asai, A.; Arisawa, M.; Shuto, S. Investigation of the noncovalent binding mode of covalent proteasome inhibitors around the transition state by combined use of cyclopropylic strain-based conformational restriction and computational modeling. J. Med. Chem. 2013. [Google Scholar] [CrossRef] [PubMed]
- Hasegawa, M.; Yasuda, Y.; Tanaka, M.; Nakata, K.; Umeda, E.; Wang, Y.; Watanabe, C.; Uetake, S.; Kunoh, T.; Shionyu, M.; et al. A novel tamoxifen derivative, ridaifen-F, is a nonpeptidic small-molecule proteasome inhibitor. Eur. J. Med. Chem. 2014, 71, 290–305. [Google Scholar] [CrossRef] [PubMed]
- Troiano, V.; Scarbaci, K.; Ettari, R.; Micale, N.; Cerchia, C.; Pinto, A.; Schirmeister, T.; Novellino, E.; Grasso, S.; Lavecchia, A.; et al. Optimization of peptidomimetic boronates bearing a P3 bicyclic scaffold as proteasome inhibitors. Eur. J. Med. Chem. 2014. [Google Scholar] [CrossRef] [PubMed]
- Scarbaci, K.; Troiano, V.; Ettari, R.; Pinto, A.; Micale, N.; di Giovanni, C.; Cerchia, C.; Schirmeister, T.; Novellino, E.; Lavecchia, A.; et al. Development of novel selective peptidomimetics containing a boronic acid moiety, targeting the 20 s proteasome as anticancer agents. ChemMedChem 2014, 9, 1801–1816. [Google Scholar] [CrossRef] [PubMed]
- Voss, C.; Scholz, C.; Knorr, S.; Beck, P.; Stein, M.L.; Zall, A.; Kuckelkorn, U.; Kloetzel, P.M.; Groll, M.; Hamacher, K.; et al. α-Keto phenylamides as P1′-extended proteasome inhibitors. ChemMedChem 2014, 2557–2564. [Google Scholar] [CrossRef] [PubMed]
- Scotti, A.; Trapella, C.; Ferretti, V.; Gallerani, E.; Gavioli, R.; Marastoni, M. Studies of C-terminal naphthoquinone dipeptides as 20S proteasome inhibitors. J. Enzyme Inhib. Med. Chem. 2015. [Google Scholar] [CrossRef] [PubMed]
- Xu, K.; Wang, K.; Yang, Y.; Yan, D.-A.; Huang, L.; Chen, C.-H.; Xiao, Z. Discovery of novel non-covalent inhibitors selective to the β5-subunit of the human 20S proteasome. Eur. J. Med. Chem. 2015, 98, 61–68. [Google Scholar] [CrossRef] [PubMed]
- Krüger, D.M.; Evers, A. Comparison of structure- and ligand-based virtual screening protocols considering hit list complementarity and enrichment factors. ChemMedChem 2010, 5, 148–158. [Google Scholar] [CrossRef] [PubMed]
- Wermuth, C.G. The Practice of Medicinal Chemistry, 3rd ed.; Elsevier: Amsterdam, The Netherlands, 2008. [Google Scholar]
- Schuster, D.; Laggner, C.; Steindl, T.M.; Langer, T. Development and validation of an in silico P450 profiler based on pharmacophore models. Curr. Drug Discov. Technol. 2006. [Google Scholar] [CrossRef]
- Basse, N.; Montes, M.; Maréchal, X.; Qin, L.; Bouvier-Durand, M.; Genin, E.; Vidal, J.; Villoutreix, B.O.; Reboud-Ravaux, M. Novel organic proteasome inhibitors identified by virtual and in vitro screening. J. Med. Chem. 2010, 53, 509–513. [Google Scholar] [CrossRef] [PubMed]
- Maréchal, X.; Genin, E.; Qin, L.; Sperandio, O.; Montes, M.; Basse, N.; Richy, N.; Miteva, M.A.; Vidal, J.; Villoutreix, B.O. 1,2,4-Oxadiazoles identified by virtual screening and their non-covalent inhibition of the human 20S Proteasome. Curr. Med. Chem. 2013. [Google Scholar] [CrossRef]
- Miller, Z.; Kim, K.-S.; Lee, D.-M.; Kasam, V.; Baek, S.E.; Lee, K.H.; Zhang, Y.-Y.; Ao, L.; Carmony, K.; Lee, N.-R.; et al. Proteasome inhibitors with pyrazole scaffolds from structure-based virtual screening. J. Med. Chem. 2015. [Google Scholar] [CrossRef] [PubMed]
- Kasam, V.; Lee, N.; Kim, K.; Zhan, C. Selective immunoproteasome inhibitors with non-peptide scaffolds identified from structure-based virtual screening. Bioorg. Med. Chem. Lett. 2014, 24, 3614–3617. [Google Scholar] [CrossRef] [PubMed]
- Pundir, S.; Vu, H.-Y.; Solomon, V.R.; McClure, R.; Lee, H. VR23: A quinoline-sulfonyl hybrid proteasome inhibitor that selectively kills cancer via cyclin E-mediated centrosome amplification. Cancer Res. 2015, 75, 7164–7175. [Google Scholar] [CrossRef] [PubMed]
- Wei, D.; Lei, B.; Tang, M.; Zhan, C.-G. Fundamental reaction pathway and free energy profile for inhibition of proteasome by epoxomicin. J. Am. Chem. Soc. 2012, 134, 10436–10450. [Google Scholar] [CrossRef] [PubMed]
- Wei, D.; Lei, B.; Tang, M.; Zhan, C.G. Fundamental reaction pathway and free energy profile for inhibition of proteasome by syringolin A (SylA). J. Am. Chem. Soc. 2015, 13, 6857–6865. [Google Scholar] [CrossRef] [PubMed]
- Sun, Q.; Xu, B.; Niu, Y.; Xu, F.; Liang, L.; Wang, C.; Yu, J.; Yan, G.; Wang, W.; Jin, H.; et al. Synthesis, bioactivity, docking and molecular dynamics studies of furan-based peptides as 20S proteasome inhibitors. ChemMedChem 2015, 10, 498–510. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Proteasome Inhibitors | Structural Class | Catalytic Subunit | |||
|---|---|---|---|---|---|
| β1 | β2 | β5 | Reference | ||
| Bortezomib | Boronates | 74 nM | 4200 nM | 7 nM | [49] |
| Carfilzomib | Epoxyketones | 2400 nM | 3600 nM | 6 nM | [49] |
| Delanzomib | Boronates | <100 nM | >100 nM | 3.8 nM | [50] |
| Epoxomicin | Epoxyketones | − | − | 5.7 nM | [51] |
| Fellutamide B | Aldehydes | 1200 nM | 2000 nM | 9.4 nM | [51] |
| Ixazomib | Boronates | 31 nM | 3500 nM | 3.4 nM | [52] |
| Marizomib | β-Lactones | 330 nM | 26 nM | 2.5 nM | [53] |
| MG-132 | Aldehydes | 1400 nM | 4500 nM | 68 nM | [54] |
| Oprozomib | Epoxyketones | − | − | 36 nM | [55] |
| Organism | PDB ID (Resolution) | Percentage Identity | ||
|---|---|---|---|---|
| β1 | β2 | β5 | ||
| Homo sapiens | 4R3O (2.60 Å) | − | − | − |
| 4R67 (2.89 Å) | ||||
| Bos taurus | 1IRU (2.75 Å) | 94 | 99 | 96 |
| Mus musculus | 3UNB (2.90 Å) | 94 | 97 | 95 |
| 3UNE (3.20 Å) | ||||
| Saccharomyces cerevisia | 3GPT (2.41 Å) | 55 | 19 | 67 |
| 5CZ4 (2.30 Å) | ||||
| 4NNN (2.50 Å) | ||||
| 3D29 (2.60 Å) | ||||
| 3MG0 (2.68 Å) | ||||
| 3UN8 (2.70 Å) | ||||
| 4INR (2.70 Å) | ||||
| 2F16 (2.80 Å) | ||||
| 1JD2 (3.00 Å) | ||||
| Reference | Software | Training Set | Test Set | PDB | PM Features |
|---|---|---|---|---|---|
| Lei et al. [96] | Catalyst | 24 dipeptide inhibitors | 26 molecules | − | 2 HBA 1, 2HBD 2, 2 Hyd 3, 2 PI 4 |
| Li et al. [97] | LigandScout | − | 3 molecules | 3 UNB | 2 HBA 1, 2 HBD 2, 1 Hyd 3, several excluded volumes |
© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Guedes, R.A.; Serra, P.; Salvador, J.A.R.; Guedes, R.C. Computational Approaches for the Discovery of Human Proteasome Inhibitors: An Overview. Molecules 2016, 21, 927. https://doi.org/10.3390/molecules21070927
Guedes RA, Serra P, Salvador JAR, Guedes RC. Computational Approaches for the Discovery of Human Proteasome Inhibitors: An Overview. Molecules. 2016; 21(7):927. https://doi.org/10.3390/molecules21070927
Chicago/Turabian StyleGuedes, Romina A., Patrícia Serra, Jorge A. R. Salvador, and Rita C. Guedes. 2016. "Computational Approaches for the Discovery of Human Proteasome Inhibitors: An Overview" Molecules 21, no. 7: 927. https://doi.org/10.3390/molecules21070927