1. Introduction
Alzheimer’s disease (AD) is a devastating neurodegenerative disorder and the most common cause of dementia [
1]. The complexity of this disease makes drug development efforts to provide effective disease modifying agents a challenging and unmet task since multiple pathogenic factors have been implicated in the development of AD, such as amyloid-β (Aβ) aggregates [
2,
3,
4,
5], oxidative stress, neuroinflammation, and mitochondria dysfunction, among others [
6,
7,
8]. To address this challenge, the multifunctional strategy of small molecule design by employing molecular conjugation or hybridization has recently attracted extensive attention in surmounting the paucity of effective disease-modifying agents in the pipeline of AD therapeutics [
9,
10,
11].
Recently, we developed a novel bivalent ligand strategy to link a multifunctional “war head”, namely curcumin, with a cell membrane/lipid raft (CM/LR) anchor moiety into our molecular design [
12,
13,
14]. Our results demonstrated that this bivalent strategy provided compounds with significantly improved neuroprotection compared to either curcumin or the CM/LR anchor alone, or the combination of these two [
12,
13,
14]. The spacer length between the “war head” and the anchorage moiety proved to be critical for their neuroprotections. Further mechanistic studies employing one of these lead bivalent compounds as a probe (
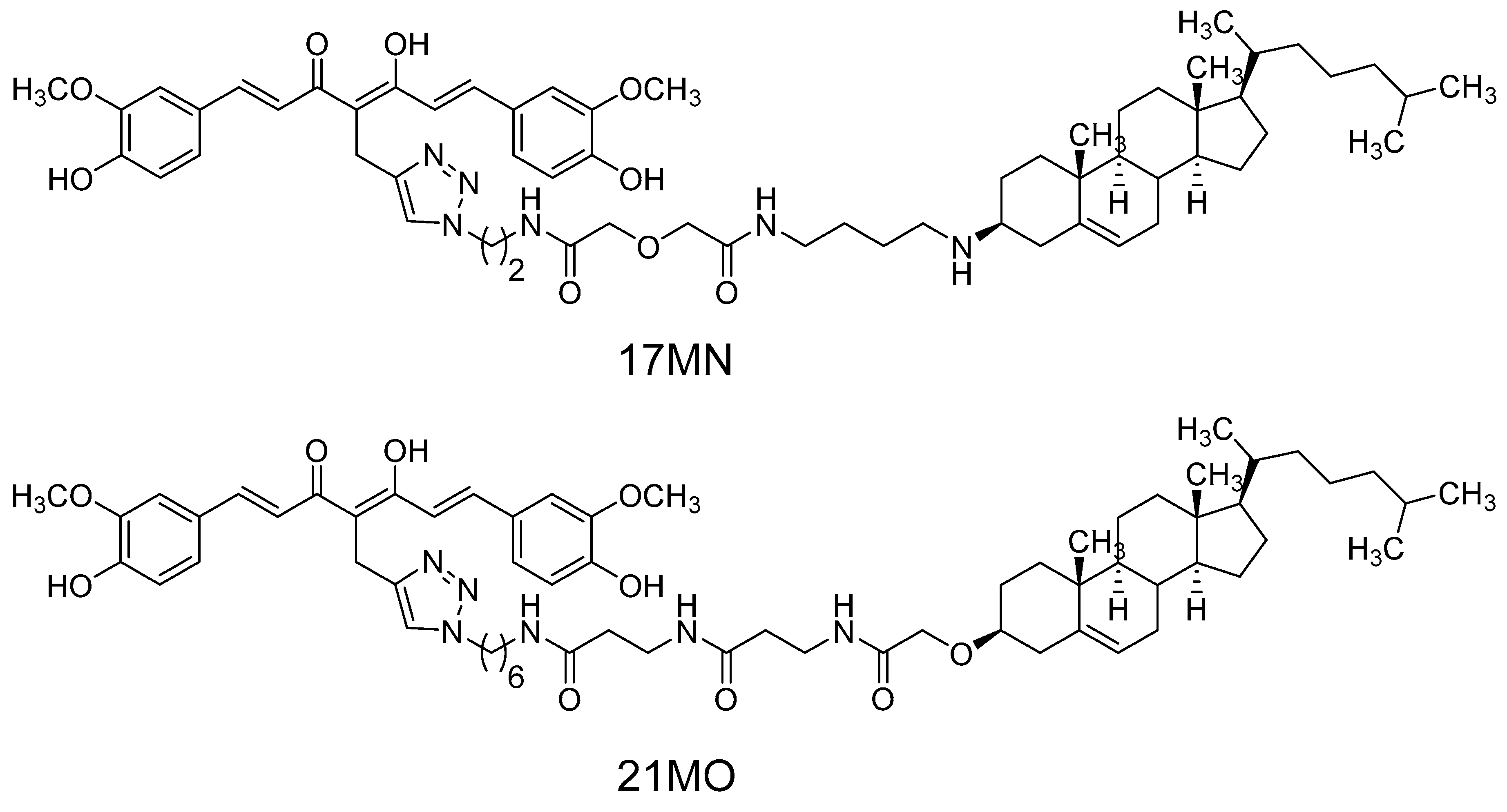
17MN,
Figure 1) demonstrated that our bivalent compound can reverse the change of mitochondrial membrane potential (MMP) and cytosolic Ca
2+ levels induced by the withdrawal of tetracycline (TC) in our MC65 cell model system, thus protecting MC65 cells from TC-removal induced necroptosis [
15]. MC65 is a human neuroblastoma cell line that conditionally expresses a 99-residue carboxyl terminal fragment of Aβ precursor protein (APP) and Aβ after removal of TC. This cell line is widely recognized as one of the cellular models of AD, resulting in intracellular Aβ oligomers (AβOs) formation and oxidative stress. Furthermore, our studies indicated that
17MN interacts with both mitochondria and endoplasmic reticulum (ER), thus suggesting a multiple-site mechanism for the observed neuroprotective activities in MC65 cells for our bivalent compounds. Our studies also noticed that bivalent compounds with varied spacer lengths exhibit differential neuroprotection profile in MC65 cells [
12,
13,
14,
15]. Therefore, it would be interesting to examine how these bivalent compounds with varied spacer lengths behave differentially in the cellular model system. Herein, we report the characterization of another lead bivalent compound,
21MO (
Figure 1), with a longer spacer, in MC65 cells and compare its effects on MMP and Ca
2+ change to our previously reported bivalent compound
17MN.
2. Results and Discussion
Our previous studies have shown that, upon removal of TC, MC65 cells die through necroptosis and bivalent compounds,
17MN protects MC65 cells from TC-removal induced cytotoxicity by engaging target proteins between receptor interacting protein kinase-1 (RIPK1) and Aβ [
15]. Therefore, we initially tested
21MO in MC65 cells to compare whether it functions similarly to
17MN. The results demonstrated that
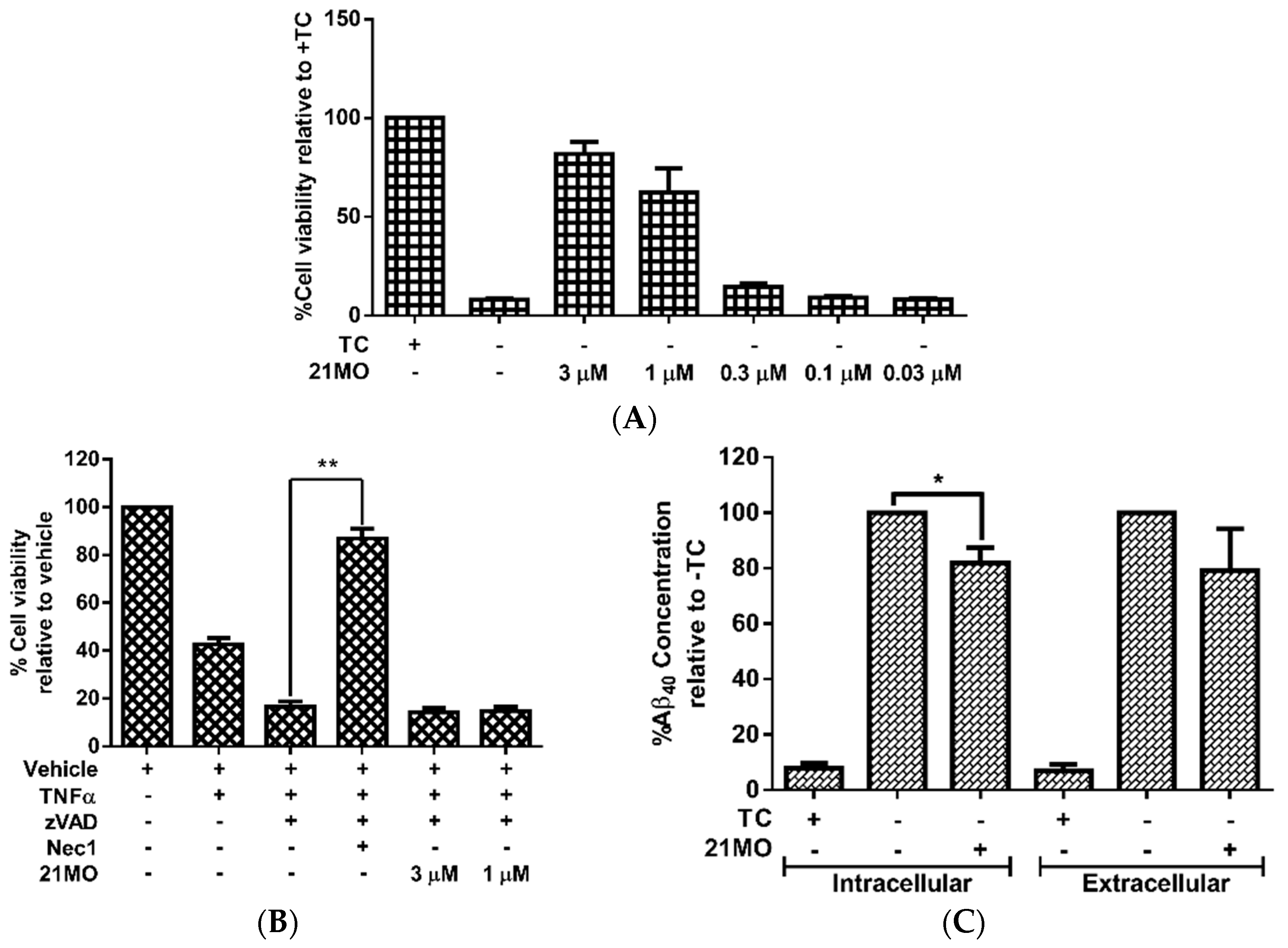
21MO can efficiently rescue MC65 cells from TC-removal induced necroptosis (
Figure 2A) and but could not rescue TNF/zVAD induced necroptosis in U937 cells (
Figure 2B), thus suggesting that
17MN and
21MO may function similarly in MC65 cells under the current experimental conditions. Further studies also demonstrated that
21MO (1 μM) slightly decreased the intracellular level of Aβ
40, but did not interfere with the level of extracellular Aβ
40 (
Figure 2C). Overall,
21MO did not significantly inhibit the total production of Aβ
40, especially when comparing the reduction of intracellular Aβ
40 with the neuroprotective activity of
21MO at this concentration. Our previous studies demonstrated that
21MO exhibits inhibitory effects on the aggregation of small AβOs, but with a much weaker potency compared to its inhibition of MC65 cell death [
12], thus suggesting that the inhibition of Aβ aggregation might only contribute partially towards its overall neuroprotective ability. Taken together, the results suggest that other proteins are involved in the observed neuroprotective activities by these bivalent compounds in MC65 cells.
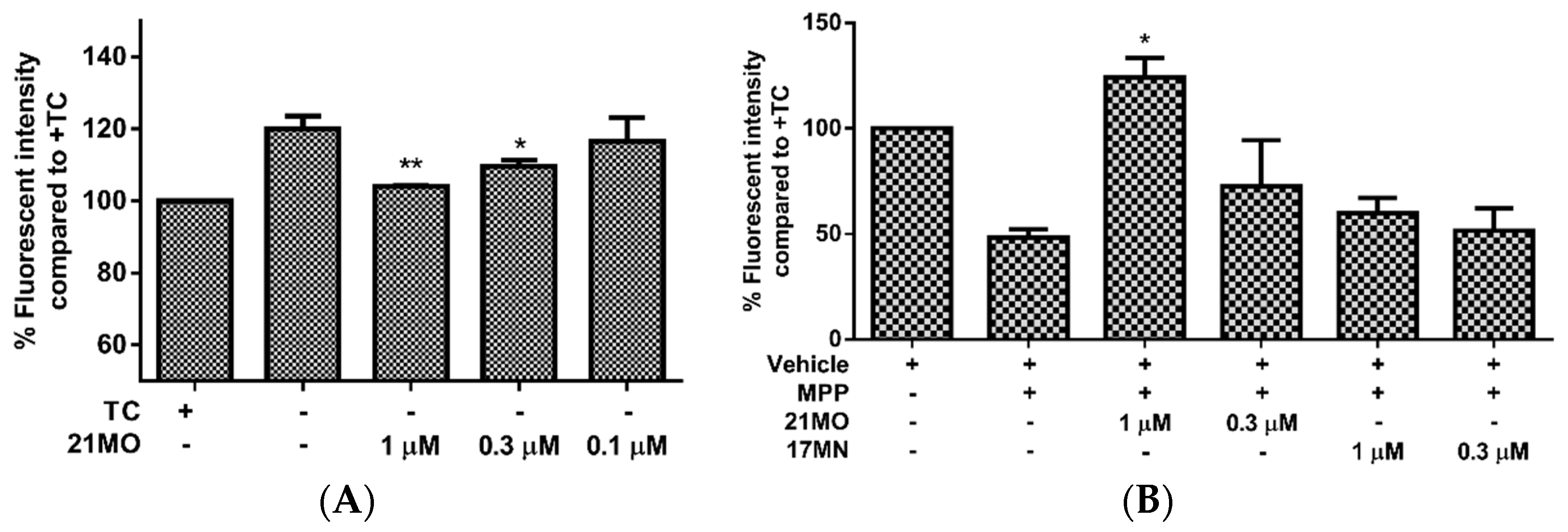
Next, we set out to examine the effects of
21MO on the change of MMP upon the removal of TC in MC65 cells in comparison with
17MN. Consistent with our previous results, the MC65 cells are hyperpolarized (increase in MMP) after the removal of TC and this may indicate the interactions of AβOs with mitochondria caused the change of MMP. Notably,
21MO suppressed the increase of MMP at 0.3 and 1 µM concentrations (
Figure 3A). This may suggest that our bivalent compounds, regardless of spacer lengths, interact with mitochondria to interfere with the effects of AβOs on the mitochondria. Our recent studies have shown that reactive oxygen species (ROS), especially mitochondrial ROS (mitoROS), are involved in the cell death of MC65 cells [
13,
16]. Furthermore, complex I of the electron transport chain is associated with the production of mitoROS [
17]. Then, another cellular model employing neuroblastoma SH-SY5Y cells in the presence of MPP
+, a neurotoxin targeting the complex I of mitochondria, was employed to test the effects of
21MO on the MMP change. Upon addition of MPP
+ the mitochondria of SH-SY5Y cells were depolarized (decrease of MMP) and notably
21MO reversed the MMP change at 1 μM, but
17MN did not, under the same experimental settings (
Figure 3B). Combining the results from MC65 cells, this may indicate that
21MO can reverse the changes induced by either production of AβOs in MC65 cells or MPP
+ in SH-SY5Y cells on mitochondria, but
21MO probably does not affect the MMP by itself.
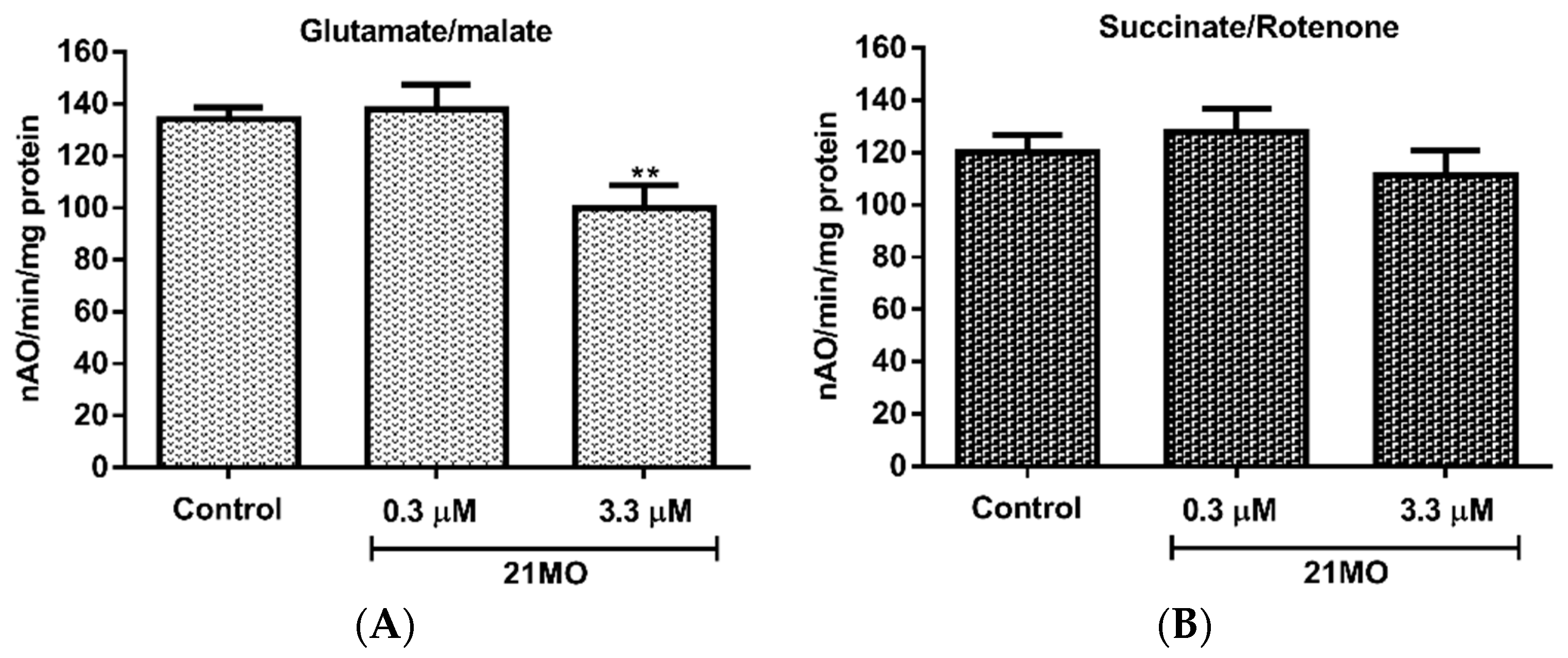
Taken together, the results might suggest that
17MN and
21MO interact differentially with mitochondria with
21MO being more selective to complex I proteins, but these potential mitochondrial proteins all underlie the AβO-induced MMP change. To further confirm this notion and examine whether
17MN and
21MO interact with mitochondria differently, we tested
21MO in the isolated brain mitochondria of mice for its effects on oxy-phosphorylation. Our previous studies have demonstrated that
17MN does not show any significant effects on the oxy-phosphorylation of brain mitochondria [
15]. As shown in
Figure 4, notably,
21MO significantly decreased the oxidation of glutamate (
Figure 4A), the substrate of complex I, but not succinate, the substrate of complex II (
Figure 4B), thus suggesting that
21MO may preferentially target the complex I of brain mitochondria, which is consistent with results from the SH-SY5Y model.
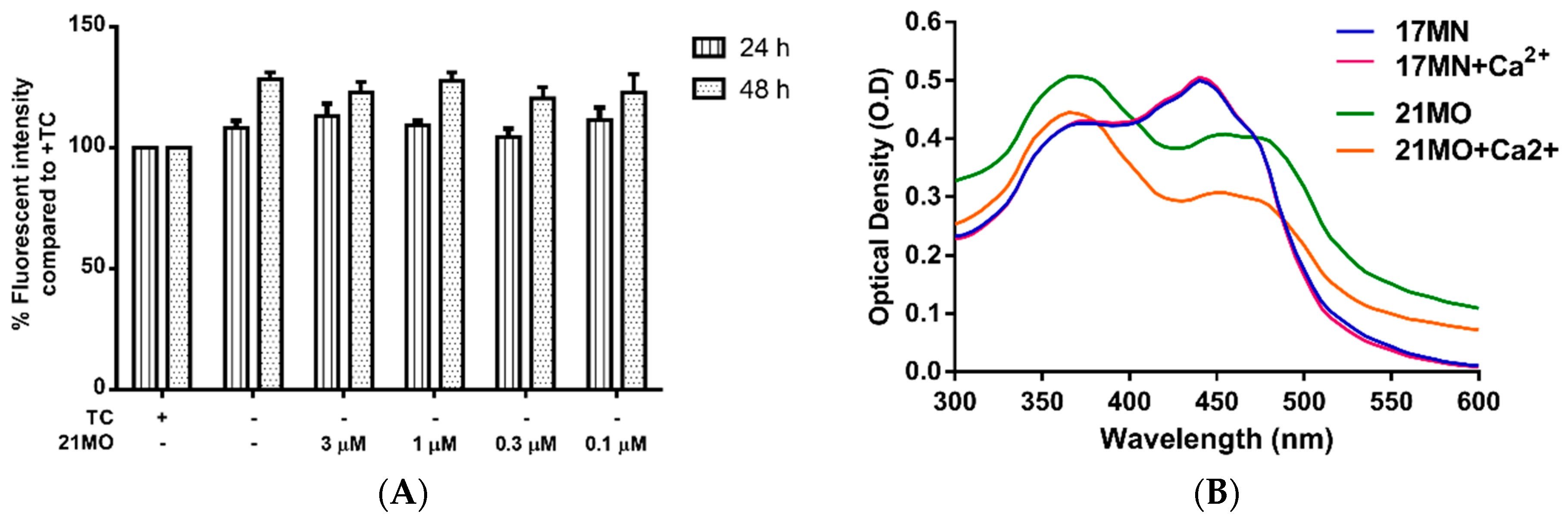
Our previous studies demonstrated that withdrawal of TC resulted in a significant rise of intracellular Ca
2+ levels in MC65 cells and the mobilization of Ca
2+ originated from ER. Furthermore,
17MN can efficiently suppress the increased Ca
2+ level [
15]. Therefore, we decided to compare the effects of
21MO on Ca
2+ levels upon TC-removal in MC65 cells to that of
17MN. Surprisingly,
21MO did not show any significant effects on the increased Ca
2+ level at concentrations up to 3 μM (
Figure 5A) after 24 and 48 h of TC removal. Our previous results have shown that
17MN dose-dependently prevented this increase [
15]. The time dependent change of Ca
2+ upon TC removal is consistent with the production of AβOs [
12], thus suggesting a role of Aβ in the calcium mobilization in this cell model. Further chelating experiments suggested that
21MO does not a form complex with Ca
2+ as evidenced by the unchanged maximum absorption (
Figure 5B), similar to the results of
17MN.
Our previous studies suggested that
17MN can readily pass into MC65 cells and localize into both mitochondria and ER, consistent with the observed effects of
17MN on both MMP and Ca
2+ change. The results of
21MO in MC65 cells may suggest that this bivalent compound with a longer spacer may preferentially interact with mitochondria, but not ER, to exert its neuroprotective activities. The results also implicate that, upon the production of Aβ, especially AβOs, in MC65 cells, multiple pathways are potentially involved to elicit the observed cell death, consistent with the pathogenic complex nature of AD. Our previous studies observed the potency difference of
17MN and
21MO in protecting MC65 cells with
17MN being more potent. This may echo the fact that
17MN can interfere with both mitochondria and ER while
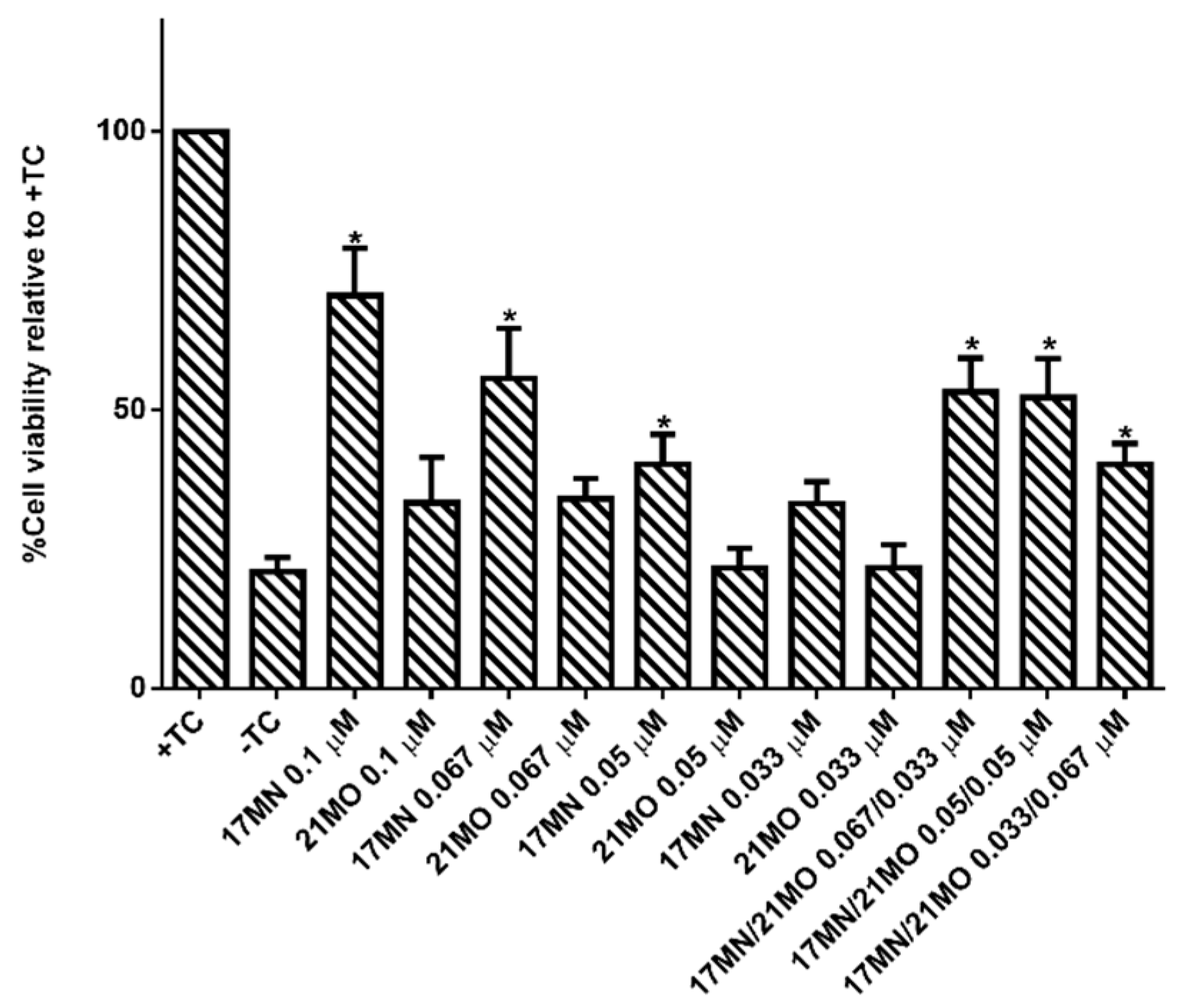
21MO only preferentially interfere with mitochondria in MC65 cells, thus consistent with our original design rationale of developing compounds with multifunctions as effective treatments for AD. To further confirm this, we tested
17MN,
21MO, and the combination of these two for the protective activities under TC-removal conditions in MC65 cells. As shown in
Figure 6,
17MN rescued MC65 cells up to 71% survival at 0.1 μM while
21MO can only slightly increase the survival rate of MC65 cells to 33% at the same concentration, consistent with our previous results [
12,
13]. When examining various combinations of
17MN and
21MO with the total concentration being 0.1 μM, the results showed that the observed protections from these combinations are comparable to that of
17MN alone at corresponding concentrations. The results are consistent with the observed mechanistic study results and our hypothesis that the observed optimal protections of MC65 cells by
17MN are probably due to its dual interactions with both mitochondria and ER, compared to
21MO’s preferential interaction with only mitochondria.
3. Materials and Methods
3.1. Cell Lines and Reagents
MC65 cells (kindly provided by Dr. George M. Martin at the University of Washington, Seattle, WA, USA) were cultured in Dulbecco’s Modified Eagle’s Medium (DMEM) (Life Technologies, Inc., Grand Island, NY, USA) supplemented with 10% of heat-inactivated fetal bovine serum (FBS) (Hyclone, Logan, UT, USA), 1% Penicillin/Streptomycin (P/S) (Invitrogen, Carlsbad, CA, USA), 1 μg/mL Tetracycline (TC) (Sigma Aldrich, St. Louis, MO, USA), and 0.2 mg/mL G418 (Invitrogen). SH-SY5Y cells (ATCC) were cultured in DMEM supplemented with 10% FBS and 1% P/S. All cells were maintained at 37 °C in a fully humidified atmosphere containing 5% CO2.
3.2. MC65 Neuroprotection Assay
MC65 cells were washed twice with PBS, resuspended in Opti-MEM, and seeded in 96-well plates (4 × 104 cells/well). Indicated compounds were then added, and cells were incubated at 37 °C under −TC conditions for 72 h. Then, 10 µL of MTT solution (3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide 5 mg/mL in PBS) were added and the cells were incubated for another 4 h. Cell medium was then removed, and the remaining formazan crystals produced by the cellular reduction of MTT were dissolved in 100 µL of DMSO. Absorbance at 570 nm was immediately recorded using a FlexStation 3 plate reader (Molecular Devices, Sunnyvale, CA, USA). Values were expressed as a percentage relative to those obtained in the +TC controls.
3.3. U937 Necroptosis Assay
U937 cells (2 × 104 cells) were pretreated with compound and caspase pan inhibitor Z-VAD for 1 h. TNF-α was then added and cells were incubated for 72 h. Afterward, 10 μL of MTT solution was added and cells were incubated for an additional 4 h. Cell medium was then removed, and the remaining formazan crystals produced by the cellular reduction of MTT were dissolved in 100 µL of DMSO. Absorbance at 570 nm was immediately recorded using a FlexStation 3 plate reader. Values were expressed as a percentage relative to those obtained in untreated controls.
3.4. Aβ40 ELISA
MC65 cells were collected and washed twice with PBS, resuspended in Opti-MEM, seeded in 6 well plates (1.6 × 106 cells/well), and incubated with compounds at 37 °C under −TC conditions for 48 h. After centrifugation of the plates, 2 mL of medium was carefully collected for analysis of Aβ40 in medium. Cell pellets were lysed by cell extraction buffer (Life Technologies) following the manufacturer’s instructions and the total protein content was quantified by the Bradford method. Samples were analyzed using the Aβ40 Human ELISA Kit (Life Technologies) following the manufacturer’s instructions. The results were normalized by total protein expressed as a percentage relative to those obtained in the −TC control.
3.5. MC65 Mitochondrial Membrane Potential Assay
MC65 cells were collected and washed twice with PBS, resuspended in Opti-MEM, seeded in 6 well plates (1.6 × 106 cells/well), and incubated with compounds at 37 °C under −TC conditions for 48 h. Cells were then collected, washed twice with PBS, and then incubated with 100 nM of tetramethylrhodamine methyl ester (TMRM) in PBS at room temperature for 30 min. Fluorescence was analyzed by flow cytometry.
3.6. SH-SY5Y Mitochondrial Membrane Potential Assay
SH-SY5Y cells (4 × 105 cells) were plated in 12 well plates. After incubation for 24 h, growth medium was removed and cells were treated in DMEM with indicated compounds and MPP+(2.5 mM) for 24 h. TMRM was then added to a final concentration of 100 nM and the cells were further incubated for 30 min. The medium was then collected. The cells were detached by trypsinization, and the medium was then recombined with the cells and centrifuged. After removing the supernatant, the cell pellet was suspended in PBS and the mean fluorescent intensity was recorded by flow cytometry.
3.7. Brain Mitochondrial Isolation and Functional Determination
The Institutional Animal Care and Use Committees (IACUC) of the McGuire VA Medical Center and Virginia Commonwealth University approved this protocol. Brain cortex tissue was collected from C57BL/6 mice after the mouse was anesthetized with sodium pentobarbital (100 mg/kg i.p.) and the heart was removed. Harvested brain tissue was placed into 5 mL MSM buffer (210 mM Mannitol, 70 mM Sucrose, 5.0 mM MOPS, 1.0 mm EDTA, pH 7.4) at 4 °C, and finely minced and incubated with Subtilisin A (1 mg/g tissue) for 1 min. Another 5 mL MSM buffer including 0.2% BSA was added to incubated tissue that was then homogenized by one stroke using a Teflon pestle. The homogenate was centrifuged at 600 g for 10 min at 4 °C. The supernatant was then centrifuged at 5000
g for 10 min at 4 °C to spin down the mitochondria. The mitochondrial pellet was washed once with MSM buffer, then re-suspended in 100–200 µL of MSM buffer. Total protein concentration was measured by the Lowry method using BSA as a standard. Oxygen consumption in mitochondria was measured by a Clark-type oxygen electrode at 30 °C using glutamate + malate (complex I substrates) and succinate + rotenone (complex II substrates) in the presence or absence of compound
17MN [
18].
3.8. MC65 Calcium Level Measurement
MC65 cells were washed twice with PBS, resuspended in Opti-MEM, and seeded in 6-well plates (1.6 × 106 cells/well). Indicated compounds were then added, and cells were incubated at 37 °C under −TC conditions for 48 h. Cells were then harvested, suspended in PBS, and incubated with Fluo-4AM (2 µM) in dark for 30 min. Cells were washed once and then resuspended in PBS. Samples were analyzed by flow cytometry. Values were expressed as a percentage relative to those obtained in −TC controls.
3.9. Biometal Complex Assay
Compound 17MN and 21MO (50 µM) were incubated along with CaCl2 (60 µM) in water (100 µL) were incubated at room temperature for 10 min, then UV absorption was recorded from 300 nm to 600 nm using a FlexStation 3 plate reader.
3.10. Statistical Analyses
The Student’s t-test was used for all statistical analysis. Data are presented as mean ± SEM. The level of significance for all analysis testing was set at * p < 0.05.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}





