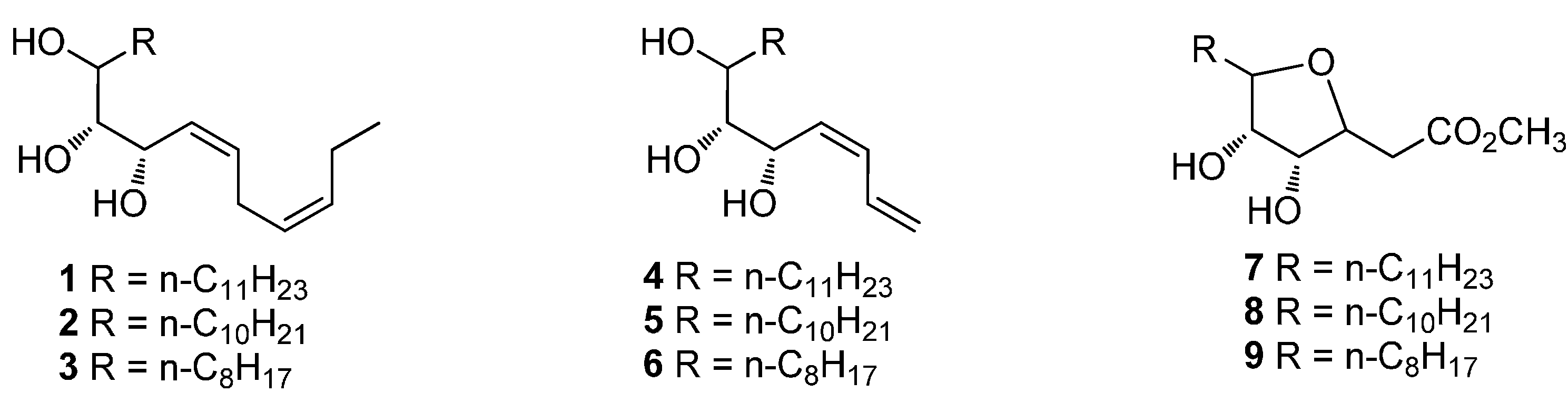
3.2. Synthesis of Compounds 1–9
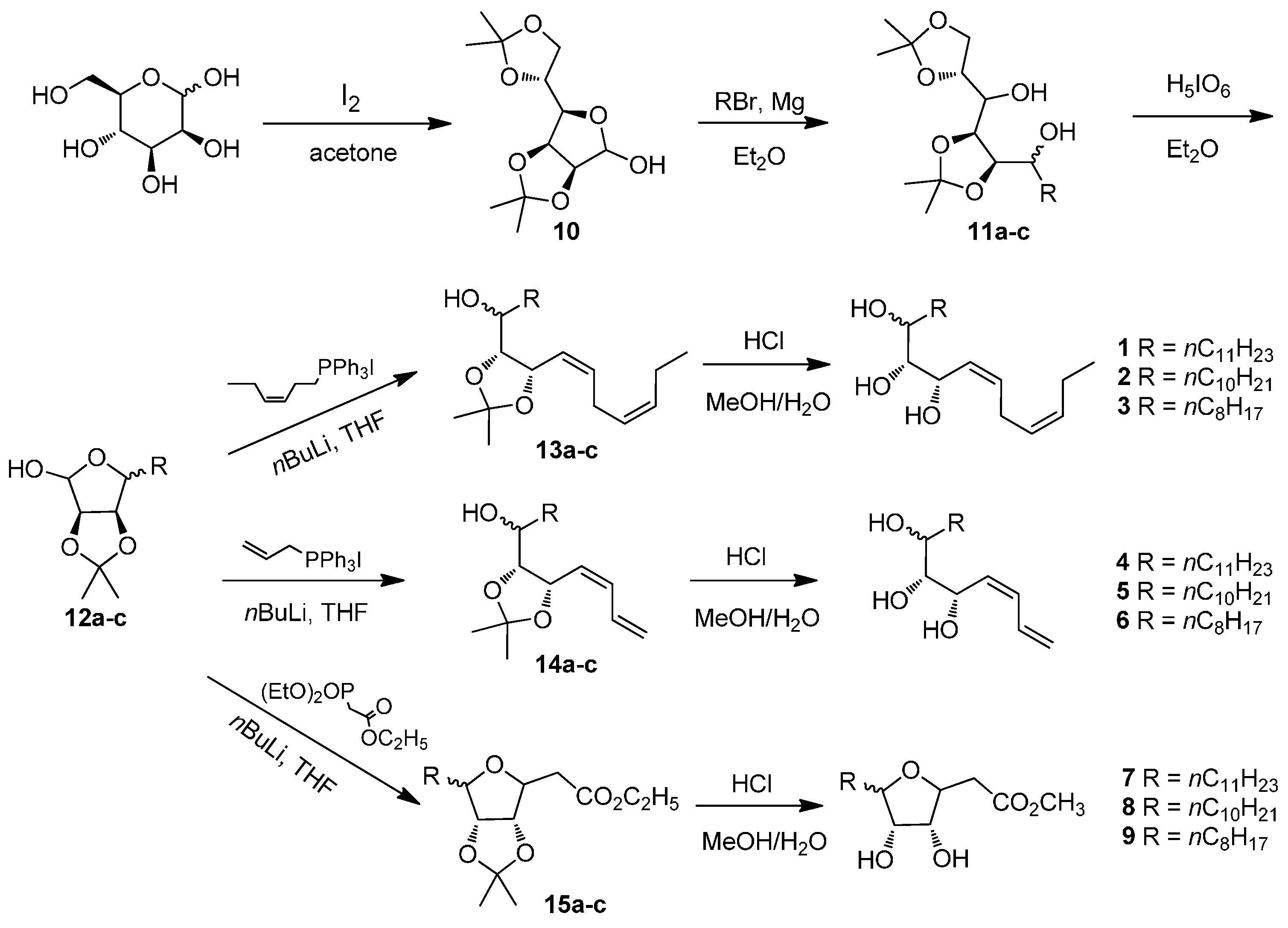
(2R,4S,5R)-1,2:4,5-Diisopropylideneundecyl-1,2,3,4,5,6-hexaol (11a). To a suspension of magnesium turnings (3.65 g, 0.15 mol) in 100 mL of anhydrous ether, 1-bromoundecane (4.7 g, 0.02 mol) was added. The reaction was initiated by addition of 0.1 mL dibromoethane. Then 1-bromoundecane (18.8 g, 0.08 mol) was added dropwise to keep gently refluxing. After 3 h, the mixture was cooled to room temperature and then −10 °C, which was followed by addition of compound 10 (2.6 g, 0.01 mol). After completion of addition, the mixture was warmed to room temperature and stirred for 2 h. Ice water was added to quench the reaction and the resulting suspension was filtered through Celite. The filtrate was concentrated and purified by column chromatography on silica gel (ethyl acetate/petroleum ether = 1:5) to give compound 11a as a colorless oil (3.54 g, 85%). 1H-NMR (400 MHz, CDCl3) δ: 0.87 (t, J = 6.8 Hz, 3H, CH3), 1.23–1.53 (m containing 4 s, 32H, 4CH3, 10CH2), 3.59–3.62 (m, 1H, CH), 3.77–3.88 (m, 1H, CH), 3.97–4.14 (m, 4H, 2CH, CH2), 4.34–4.41 (m, 1H, CH); HRMS (ESI) m/z 439.3036 (calcd. for C24H44O6Na [M + Na]+, 439.3036).
(3R,4S)-3,4-Isopropylidene-5-undecyltetrahydrofuran-2,3,4-triol (12a). A mixture of periodic acid (3.84 g, 16.95 mmol) and compound 11a (2.35 g, 5.65 mmol) in 200 mL of anhydrous ether was stirred for 4 h at room temperature. The suspension was filtered to remove the extra periodic acid. The filtrate was washed with saturated NaHCO3 solution to neutral and then washed with brine. The organic phase was dried over Na2SO4, filtered and concentrated. The residue was purified by column chromatography on silica gel (ethyl acetate/petroleum ether = 1:10) to give compound 12a as a colorless oil (1.55 g, 87%). 1H-NMR (400 MHz, CDCl3) δ: 0.86 (t, J = 6.8 Hz, 3H, CH3), 1.24–1.46 (m containing 2 s, 24H, 2CH3, 9CH2), 1.68 (dd, J = 14.7, 7.2 Hz, 2H, CH2), 3.31(s, 1H, OH), 4.10 (dt, J = 6.8, 3.6 Hz, 1H, CH), 4.56–4.57 (m, 1H, CH), 4.60–4.64 (m, 1H, CH), 5.32 (d, J = 1.6 Hz, 1H, CH); HRMS (ESI) m/z 337.2355 (calcd. for C18H34O6Na [M + Na]+, 337.2354).
(3Z,6Z,8S,9R)-8,9-Isopropylidene-heneicosa-3, 6-diene-8, 9, 10-triol (13a). n-Butyl lithium (9.88 mL, 1.6 M) was added dropwise to a solution of (Z)-hex-3-ene triphenylphosphonium iodine (5.81 g, 12.3 mmol) in 70 mL of THF at −30 °C. After 0.5 h, a solution of compound 12a (1.10 g, 3.5 mmol) in 5 mL of THF was added to the mixture, which was then allowed to warm to 0 °C. The reaction was continued for another 6 h. The resulting mixture was filtered. The filtrate was dried over Na2SO4, filtered and concentrated. The residue was purified by column chromatography on silica gel (ethyl acetate/petroleum ether = 1:20) to give compound 13a as a colorless oil (0.65 g, 49%). 1H-NMR (400 MHz, CDCl3) δ: 0.88 (t, J = 6.8 Hz, 3H, CH3), 0.97 (t, J = 7.4 Hz, 3H, CH3), 1.26–1.51 (m containing 2 s, 26H, 2CH3, 10CH2), 2.02–2.10 (m, 2H, CH2), 2.80–2.86 (m, 2H, CH2), 3.56–3.59 (m 1H, CH), 3.97–4.00 (m, 1H, CH), 4.98 (dd, J = 8.6, 6.8 Hz, 1H, CH), 5.24–5.74 (m, 4H, 2CH=CH); HRMS (ESI) m/z 403.3191 (calcd. for C24H44O3Na [M + Na]+, 403.3188).
(3Z,6Z,8S,9R,10R)-Henicosa-3,6-diene-8,9,10-triol (1). Compound 13a (0.46 g, 1.2 mmol) was added to a solution of concentrated HCl (11 mL) in water/methanol (75 mL, v/v = 1:9). The mixture was stirred for 3 h at room temperature and then neutralized with NaOH solution. The resulting mixture was concentrated to remove methanol. The residue was diluted with EtOAc and washed with water. The organic phase was dried over Na2SO4, filtered and concentrated. The residue was purified by column chromatography on silica gel (ethyl acetate/petroleum ether = 1:2) to give compound 1 as a colorless oil (0.32 g, 78%). 1H-NMR (400 MHz, CDCl3) δ: 0.87 (t, J = 6.8 Hz, 3H, CH3), 0.97 (t, J = 7.5 Hz, 3H, CH3), 1.25–1.51 (m, 20H, 10CH2), 2.02–2.08 (m, 2H, CH2), 2.15 (brs, 3H, 3OH), 2.79–2.89 (m, 2H, CH2), 3.28–3.48 (m, 1H, CH), 3.64–3.85 (m, 1H, CH), 4.22–4.67 (m, 1H, CH), 5.27–5.83 (m, 4H, CH=CH); HRMS (ESI) m/z 363.2875 (calcd. for C21H40O3Na [M + Na]+, 363.2875).
(3Z,6Z,8S,9R,10R)-Icosa-3,6-diene-8,9,10-triol (2). To a suspension of magnesium turnings (4.86 g, 0.2 mol) in 200 mL of anhydrous ether, 1-bromodecane (8.85 g, 0.04 mol) was added. The reaction was initiated by addition of 0.1 mL dibromoethane. Then 1-bromodecane (35.39 g, 0.16 mol) was added dropwise to keep gently refluxing. After 3 h, the mixture was cooled to room temperature and then −10 °C, which was followed by addition of compound 10 (5.21 g, 0.02 mol). After completion of addition, the mixture was warmed to room temperature and stirred for 2 h. Ice water was added to quench the reaction and the resulting suspension was filtered through Celite. The filtrate was concentrated and purified by column chromatography on silica gel (ethyl acetate/petroleum ether = 1:5) to give compound 11b as a colorless oil (7.08 g, 88%). A mixture of periodic acid (11.97 g, 52.5 mmol) and compound 11b (7.04 g, 17.5 mmol) in 250 mL of anhydrous ether was stirred for 4 h at room temperature. The suspension was filtered to remove the extra periodic acid. The filtrate was washed with saturated NaHCO3 solution to neutral and then washed with brine. The organic phase was dried over Na2SO4, filtered and concentrated. The residue was purified by column chromatography on silica gel (ethyl acetate/petroleum ether = 1:10) to give compound 12b as a colorless oil (4.16 g, 79%). n-Butyl lithium (11.8 mL, 1.6 M) was added dropwise to a solution of (Z)-hex-3-ene triphenylphosphonium iodine (7.44 g, 15.75 mmol) in 80 mL of THF at −30 °C. After 0.5 h, a solution of compound 12b (1.35 g, 4.5 mmol) in 5 mL of THF was added to the mixture, which was then allowed to warm to 0 °C. The reaction was continued for another 6 h. The resulting mixture was filtered. The filtrate was dried over Na2SO4, filtered and concentrated. The residue was purified by column chromatography on silica gel (ethyl acetate/petroleum ether = 1:20) to give compound 13b as a colorless oil (0.76 g, 46%). Compound 13b (0.75 g, 2.05 mmol) was added to a solution of concentrated HCl (8.5 mL) in water/methanol (100 mL, v/v = 1:9). The mixture was stirred for 3 h at room temperature and then neutralized with NaOH solution. The resulting mixture was concentrated to remove methanol. The residue was diluted with EtOAc and washed with water. The organic phase was dried over Na2SO4, filtered and concentrated. The residue was purified by column chromatography on silica gel (ethyl acetate/petroleum ether = 1:2) to give compound 2 as a colorless oil (0.48 g, 72%). 1H-NMR (400 MHz, CDCl3) δ: 0.88 (t, J = 6.4 Hz, 3H, CH3), 0.97 (t, J = 7.5 Hz, 3H, CH3), 1.24–1.73 (m, 18H, 9CH2), 2.00–2.10 (m containing brs, 3H, CH2, OH), 2.44 (brs, 1H, OH), 2.63 (brs, 1H, OH), 2.77–2.91 (m, 2H, CH2), 3.39–3.46 (m, 1H, CH), 3.66–3.83 (m, 1H, CH), 4.22–4.30 (m, 0.5H, CH), 4.64–4.66 (m, 0.5H, CH), 5.28–5.83 (m, 4H, 2CH=CH); HRMS (ESI) m/z 349.2713 (calcd. for C20H38O3Na [M + Na]+, 349.2706).
(3Z,6Z,8S,9R,10R)-Octadeca-3,6-diene-8,9,10-triol (3). To a suspension of magnesium turnings (4.86 g, 0.2 mol) in 200 mL of anhydrous ether, 1-bromoctane (7.72 g, 0.04 mol) was added. The reaction was initiated by addition of 0.1 mL dibromoethane. Then 1-bromodecane (30.9 g, 0.16 mol) was added dropwise to keep gently refluxing. After 3 h, the mixture was cooled to room temperature and then −10 °C, which was followed by addition of compound 10 (5.2 g, 0.02 mol). After completion of addition, the mixture was warmed to room temperature and stirred for 2 h. Ice water was added to quench the reaction and the resulting suspension was filtered through Celite. The filtrate was concentrated and purified by column chromatography on silica gel (ethyl acetate/petroleum ether = 1:5) to give compound 11c as a colorless oil (5.91 g, 79%). A mixture of periodic acid (10.77 g, 47.25 mmol) and compound 11c (5.90 g, 15.75 mmol) in 250 mL of anhydrous ether was stirred for 4 h at room temperature. The suspension was filtered to remove the extra periodic acid. The filtrate was washed with saturated NaHCO3 solution to neutral and then washed with brine. The organic phase was dried over Na2SO4, filtered and concentrated. The residue was purified by column chromatography on silica gel (ethyl acetate/petroleum ether = 1:10) to give compound 12c as a colorless oil (3.36 g, 78%). n-Butyl lithium (10.5 mL, 1.6 M) was added dropwise to a solution of (Z)-hex-3-ene triphenylphosphonium iodine (6.61 g, 14 mmol) in 80 mL of THF at −30 °C. After 0.5 h, a solution of compound 12c (1.1 g, 4 mmol) in 5 mL of THF was added to the mixture, which was then allowed to warm to 0 °C. The reaction was continued for another 6 h. The resulting mixture was filtered. The filtrate was dried over Na2SO4, filtered and concentrated. The residue was purified by column chromatography on silica gel (ethyl acetate/petroleum ether = 1:20) to give compound 13c as a colorless oil (0.48 g, 36%). Compound 13c (0.47 g, 1.4 mmol) was added to a solution of concentrated HCl (6 mL) in water/methanol (100 mL, v/v = 1:9). The mixture was stirred for 3 h at room temperature and then neutralized with NaOH solution. The resulting mixture was concentrated to remove methanol. The residue was diluted with EtOAc and washed with water. The organic phase was dried over Na2SO4, filtered and concentrated. The residue was purified by column chromatography on silica gel (ethyl acetate/petroleum ether = 1:2) to give compound 3 as a colorless oil (0.39 g, 75%). 1H-NMR (400 MHz, CDCl3) δ: 0.88 (t, J = 6.8 Hz, 3H, CH3), 0.96(t, J = 7.5 Hz, 3H, CH3), 1.26–1.72(m, 14H, CH2), 2.02–2.06(m, 2H, CH2), 2.20 (brs, 2H, 2OH), 2.60(brs, 1H, OH), 2.77–2.86 (m, 2H, CH2), 3.44–3.48 (m, 1H, CH), 3.65(s, 1H, CH), 4.23 (s, 1H, CH), 5.30–5.37 (m, 1H, =CH), 5.43–5.49(m, 1H, =CH), 5.58–5.64 (m, 1H, =CH), 5.77–5.84(m, 1H, =CH); HRMS (ESI) m/z 321.2405 (calcd. for C18H34O3Na [M + Na]+, 321.2400).
(5S,6R,7R,Z)-Octadeca-1,3-diene-5,6,7-triol (4). n-Butyl lithium (8.5 mL, 1.6 M) was added dropwise to a solution of allyl triphenylphosphonium bromide (4.02 g, 10.5 mmol) in 60 mL of THF at −10 °C. After 0.5 h, a solution of compound 12a (0.94 g, 3.0 mmol) in 5 mL of THF was added to the mixture, which was then allowed to warm to 20 °C. The reaction was continued for another 6 h. The resulting mixture was filtered. The filtrate was dried over Na2SO4, filtered and concentrated. The residue was purified by column chromatography on silica gel (ethyl acetate/petroleum ether = 1:20) to give compound 14a as a colorless oil (0.39 g, 40%). Compound 14a (0.37 g, 1.1 mmol) was added to a solution of concentrated HCl (10 mL) in water/methanol (70 mL, v/v = 1:9). The mixture was stirred for 3 h at room temperature and then neutralized with NaOH solution. The resulting mixture was concentrated to remove methanol. The residue was diluted with EtOAc and washed with water. The organic phase was dried over Na2SO4, filtered and concentrated. The residue was purified by column chromatography on silica gel (ethyl acetate/petroleum ether = 1:3) to give compound 4 as a colorless oil (0.24 g, 74%). 1H-NMR (400 MHz, CDCl3) δ: 0.88 (t, J = 6.8 Hz, 3H, CH3), 1.01–1.73 (m, 20H, 10CH2), 3.34–3.37 (m, 1H, CH), 3.54–3.97 (m, 2H, 2CH), 5.14–5.34 (m, 2H, =CH2), 5.57–5.82 (m, 1H, =CH), 6.22–6.72 (m, 2H, 2CH=); HRMS (ESI) m/z 321.2404 (calcd. for C18H34O3Na [M + Na]+, 321.2400).
(5S,6R,7R,Z)-Heptadeca-1,3-diene-5,6,7-triol (5). n-Butyl lithium (11.8 mL, 1.6 M) was added dropwise to a solution of allyl triphenylphosphonium bromide (6.77 g, 15.75 mmol) in 80 mL of THF at −10 °C. After 0.5 h, a solution of compound 12b (1.35 g, 4.5 mmol) in 5 mL of THF was added to the mixture, which was then allowed to warm to 20 °C. The reaction was continued for another 6 h. The resulting mixture was filtered. The filtrate was dried over Na2SO4, filtered and concentrated. The residue was purified by column chromatography on silica gel (ethyl acetate/petroleum ether = 1:20) to give compound 14b as a colorless oil (0.65 g, 45%). Compound 14b (0.65 g, 2 mmol) was added to a solution of concentrated HCl (8 mL) in water/methanol (50 mL, v/v = 1:9). The mixture was stirred for 3 h at room temperature and then neutralized with NaOH solution. The resulting mixture was concentrated to remove methanol. The residue was diluted with EtOAc and washed with water. The organic phase was dried over Na2SO4, filtered and concentrated. The residue was purified by column chromatography on silica gel (ethyl acetate/petroleum ether = 1:3) to give compound 5 as a colorless oil (0.38 g, 68%). 1H-NMR (400 MHz, CDCl3) δ: 0.87 (t, J = 6.8 Hz, 3H), 1.25–1.62 (m, 18H), 2.27–2.85 (brs, 1H, OH), 3.04–3.18 (brs, 1H, OH), 3.83–3.89 (m, 2H, 2CH), 3.99–4.00 (m, 1H, CH), 4.04 (s, 1H, OH), 4.74–4.89 (m, 2H, =CH2), 5.22–5.55 (m, 2H, 2=CH), 6.15–6.66 (m, 1H, =CH); HRMS (ESI) m/z 307.2250 (calcd. for C17H32O3Na [M + Na]+, 307.2244).
(5S,6R,7R,Z)-Pentadeca-1,3-diene-5,6,7-triol (6). n-Butyl lithium (10.5 mL, 1.6 M) was added dropwise to a solution of allyl triphenylphosphonium bromide (6.02 g, 14 mmol) in 80 mL of THF at −10 °C. After 0.5 h, a solution of compound 12c (1.1 g, 4 mmol) in 5 mL of THF was added to the mixture, which was then allowed to warm to 20 °C. The reaction was continued for another 6 h. The resulting mixture was filtered. The filtrate was dried over Na2SO4, filtered and concentrated. The residue was purified by column chromatography on silica gel (ethyl acetate/petroleum ether = 1:20) to give compound 14c as a colorless oil (0.49 g, 41%). Compound 14c (0.48 g, 1.6 mmol) was added to a solution of concentrated HCl (6.7 mL) in water/methanol (40 mL, v/v = 1:9). The mixture was stirred for 3 h at room temperature and then neutralized with NaOH solution. The resulting mixture was concentrated to remove methanol. The residue was diluted with EtOAc and washed with water. The organic phase was dried over Na2SO4, filtered and concentrated. The residue was purified by column chromatography on silica gel (ethyl acetate/petroleum ether = 1:3) to give compound 6 as a colorless oil (0.31 g, 76%). 1H NMR (400 MHz, CDCl3) δ: 0.89 (t, J = 6.4 Hz, 3H, CH3), 1.25–1.59 (m, 14H, 7CH2), 2.98 (brs, 3H, 3OH), 3.33–3.36 (m, 0.5H, CH), 3.41–3.42 (m, 0.5H, CH), 3.50–3.53 (m, 0.5H, CH), 3.62–3.65 (m, 0.5H, CH), 3.77–3.86 (m, 0.5H, CH), 3.94–4.03 (m, 0.5H, CH), 4.31–4.78 (m, 1H, =CH2), 5.12–5.33 (m, 1.5H, =CH2, =CH), 5.49–5.58 (m, 0.5H, =CH), 5.71–5.85 (m, 0.5H, =CH), 6.13–6.24 (m, 0.5H, =CH), 6.29–6.37 (m, 0.5H, =CH), 6.56–6.70 (m, 0.5H, =CH); HRMS (ESI) m/z 279.1937 (calcd. for C15H28O3Na [M + Na]+, 279.1931).
(3R,4S)-Methyl 3,4-dihydroxy-5-undecyl-tetrahydrofuran-2-acetate (7). n-Butyl lithium (5.5 mL, 1.6 M) was added dropwise to a solution of triethyl phosphonoacetate (1.93 g, 8.6 mmol) in 70 mL of THF at −30 °C. After 0.5 h, a solution of compound 12a (1.89 g, 6.0 mmol) in 5 mL of THF was added to the mixture, which was then allowed to warm to room temperature. The reaction mixture was stirred overnight and then filtered. The filtrate was dried over Na2SO4, filtered and concentrated. The residue was purified by column chromatography on silica gel (ethyl acetate/petroleum ether = 1:20) to give compound 15a as a colorless oil (1.02 g, 44%). Compound 15a (1.0 g, 2.6 mmol) was added to a solution of concentrated HCl (20 mL) in water/methanol (150 mL, v/v = 1:9). The mixture was stirred for 3 h at room temperature and then neutralized with NaOH solution. The resulting mixture was concentrated to remove methanol. The residue was diluted with EtOAc and washed with water. The organic phase was dried over Na2SO4, filtered and concentrated. The residue was purified by column chromatography on silica gel (ethyl acetate/petroleum ether = 1:3) to give compound 7 as a colorless oil (0.51 g, 69%). 1H-NMR (400 MHz, CDCl3) δ: 0.86 (t, J = 6.8 Hz, 3H, CH3), 1.24–1.72 (m, 20H, 10CH2), 2.58–2.86 (m, 2H, CH2), 3.60–3.90 (m containing s, 5H, CH, OCH3), 3.96–4.02 (m, 1H, CH), 4.79–4.98 (m, 1H, CH); HRMS (ESI) m/z 353.2287 (calcd. for C18H34O5Na [M + Na]+, 353.2298).
(3R,4S)-Methyl 5-decyl-3,4-dihydroxytetrahydrofuran-2-acetate (8). n-Butyl lithium (4.1 mL, 1.6 M) was added dropwise to a solution of triethyl phosphonoacetate (1.45 g, 6.45 mmol) in 70 mL of THF at −30 °C. After 0.5 h, a solution of compound 12b (1.3 g, 4.3 mmol) in 5 mL of THF was added to the mixture, which was then allowed to warm to room temperature. The reaction mixture was stirred overnight and then filtered. The filtrate was dried over Na2SO4, filtered and concentrated. The residue was purified by column chromatography on silica gel (ethyl acetate/petroleum ether = 1:20) to give compound 15b as a colorless oil (0.76 g, 48%). Compound 15b (0.74 g, 2 mmol) was added to a solution of concentrated HCl (8 mL) in water/methanol (50 mL, v/v = 1:9). The mixture was stirred for 3 h at room temperature and then neutralized with NaOH solution. The resulting mixture was concentrated to remove methanol. The residue was diluted with EtOAc and washed with water. The organic phase was dried over Na2SO4, filtered and concentrated. The residue was purified by column chromatography on silica gel (ethyl acetate/petroleum ether = 1:3) to give compound 8 as a colorless oil (0.47 g, 75%). 1H-NMR (400 MHz, CDCl3) δ: 0.86 (t, J = 0.90 Hz, 3H, CH3), 1.23–1.55 (m, 18H, 9CH2), 2.61 (dd, J = 16.2, 7.4 Hz, 1H, CH2), 2.75 (dd, J = 16.2, 5.9 Hz, 1H, CH2), 3.78 (s, 3H, OCH3), 3.71–3.75 (m, 1H, CH), 3.78–3.85 (m, 2H, 2CH), 3.95–4.00 (m, 1H, CH); 13C NMR (100 MHz, CDCl3) δ: 14.1 (CH3), 22.6 (CH2), 25.5 (CH2), 29.3 (CH2), 29.5 (2CH2), 29.6 (3CH2), 31.9 (CH2), 38.3 (CH2), 52.0 (OCH3), 74.8 (CH), 74.9 (CH), 78.4 (CH), 84.3 (CH),172.5 (C=O); HRMS (ESI) m/z 339.2144 (calcd. for C17H32O5Na [M + Na]+, 339.2142).
(3R,4S)-Methyl 3,4-dihydroxy-5-octyltetrahydrofuran-2-acetate (9). n-Butyl lithium (4.3 mL, 1.6 M) was added dropwise to a solution of triethyl phosphonoacetate (1.28 g, 5.7 mmol) in 80 mL of THF at −10 °C. After 0.5 h, a solution of compound 12c (1.04 g, 3.8 mmol) in 5 mL of THF was added to the mixture, which was then allowed to warm to 20 °C. The reaction was continued for another 6 h. The resulting mixture was filtered. The filtrate was dried over Na2SO4, filtered and concentrated. The residue was purified by column chromatography on silica gel (ethyl acetate/petroleum ether = 1:20) to give compound 15c as a colorless oil (0.53 g, 41%). Compound 15c (0.52 g, 1.5 mmol) was added to a solution of concentrated HCl (6 mL) in water/methanol (40 mL, v/v = 1:9). The mixture was stirred for 3 h at room temperature and then neutralized with NaOH solution. The resulting mixture was concentrated to remove methanol. The residue was diluted with EtOAc and washed with water. The organic phase was dried over Na2SO4, filtered and concentrated. The residue was purified by column chromatography on silica gel (ethyl acetate/petroleum ether = 1:3) to give compound 9 as a colorless oil (0.32 g, 74%). 1H-NMR (400 MHz, CDCl3) δ: 0.85 (t, J = 7.2 Hz, 3H, CH3), 1.23–1.73 (m, 14H, 7 CH2), 2.58–2.85 (m, 2H, CH2), 3.57–4.01 (m containing s, 6H, CH3, 3CH), 4.55–4.99 (m, 1H, CH); HRMS (ESI) m/z 289.2007 (calcd. for C15H29O5 [M + H]+, 289.2010).







{kind=link}
{kind=link}
{kind=link}