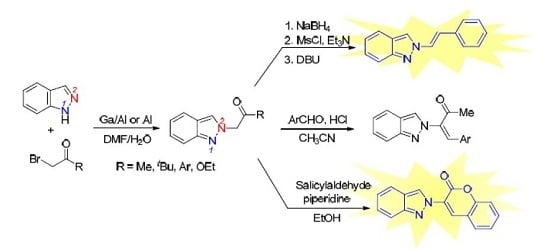
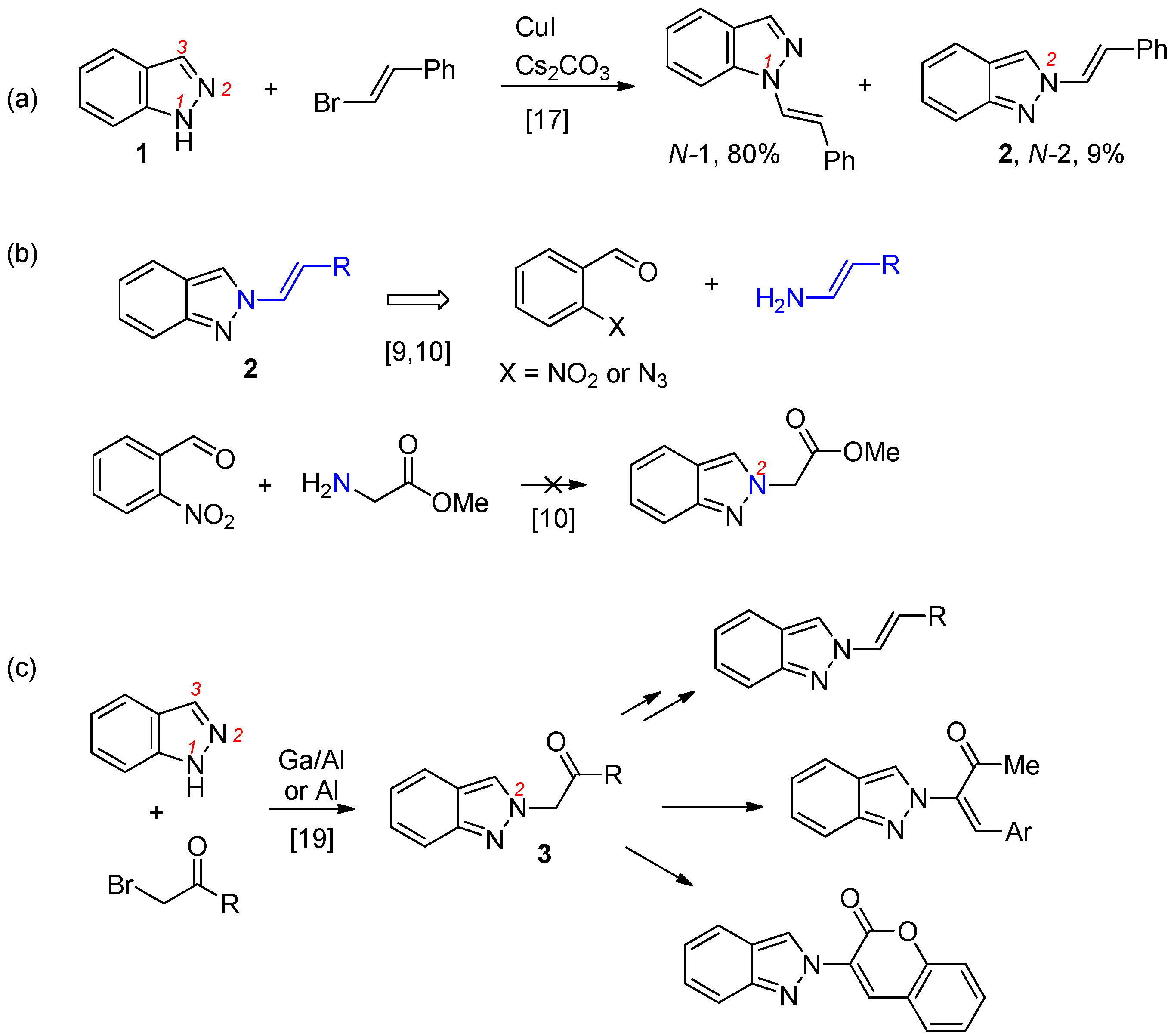
3.2.2. General Procedure for the Synthesis of 2-Alkenyl-2H-indazoles 2 (Route B)
To a cold solution (ice bath 0–5 °C) of ketone 3 (0.5 mmol) in MeOH/CH2Cl2 (1/1, 10 mL) was added NaBH4 (0.5 mmol, 1.0 equiv.) slowly. The resulting mixture was warmed to ambient temperature (ice bath removal). Reaction was monitored by TLC until no starting material was observed and normally the reaction was stirred at ambient temperature for 2 h. The reaction was then cooled to 0–5 °C and quenched with 2 N HCl (10 mL). CH2Cl2 (10 mL) was added and the mixture was transferred to a separatory funnel. The aqueous layer was back extracted with CH2Cl2 (10 mL × 2). The combined organic layers were dried over Na2SO4, filtered, and concentrated in a rotary evaporator. The residue was dissolved in CH2Cl2 (5 mL) and the mixture was cooled 0–5 °C (ice bath) under N2. Et3N (0.25 mL, 1.8 mmol) was added followed by MsCl (0.07 mL, 0.9 mmol). The resulting mixture was warmed to ambient temperature (ice bath removal). Reaction was monitored by TLC until no starting material was observed and normally the reaction was stirred at ambient temperature for 2 h. The resulting mixture was cooled to 0–5 °C and sat. NaHCO3 (5 mL) was added slowly. The mixture was transferred to a separatory funnel. The aqueous layer was back extracted with CH2Cl2 (10 mL × 2). The combined organic layers were dried over Na2SO4, filtered and concentrated in a rotary evaporator. The residue was dissolved in DBU (0.6 mL) and the mixture was heated to 90 °C under N2 overnight. The mixture was cooled to ambient temperature. EtOAc (10 mL) and H2O (10 mL) were added and the mixture was transferred to a separatory funnel. The aqueous layer was back extracted with EtOAc (10 mL × 2). The combined organic layers were dried over Na2SO4, filtered, and concentrated in a rotary evaporator. The residue was purified by silica gel chromatography using EtOAc/hexanes (1/9) as eluent to give the product 2.
(E)-2-Styryl-2H-indazole (2a): Following the general procedure, the title compound was obtained (Route A: 72 mg, 65%, Route B: 99 mg, 90%). A yellow solid, mp 131–133 °C; TLC (EtOAc/hexanes (1:4)) Rf = 0.56; 1H-NMR (CDCl3) δ 7.07 (t, J = 7.8 Hz, 1 H), 7.25–7.37 (m, 4H), 7.43–7.48 (m, 3H), 7.58–7.65 (m, 2H), 7.75 (d, J = 7.8 Hz, 1H), 7.97 (s, 1H); 13C-NMR (CDCl3) δ 117.2 (CH), 120.1 (CH), 120.9 (CH), 121.8 (CH), 122.0 (C), 122.2 (CH), 126.2 (CH), 126.4 (CH × 2), 127.0 (CH), 128.0 (CH), 128.7 (CH × 2), 134.1 (C), 149.4 (C); IR (neat) 3131, 2950, 1645 cm−1; EI-MS m/z (rel intensity) 220 ([M]+, 56), 219 (100), 129 (97), 77 (22); HRMS [M]+ calcd for C15H12N2: 220.1000, found 220.1004.
2-(4-Bromostyryl)-2H-indazole (2b): Following the general procedure, the title compound was obtained (Route A: 78 mg, 52%, Route B: 147 mg, 98%). A yellow solid, mp 154–156 °C; TLC (EtOAc/hexanes (1:4)) Rf = 0.50; 1H-NMR (CDCl3) δ 7.08 (dd, J = 7.8, 6.6 Hz, 1H), 7.28–7.41 (m, 3H), 7.46–7.51 (m, 3H), 7.62–7.75 (m, 3H), 8.10 (s, 0.8H), 8.13 (s, 0.1H); 13C-NMR (CDCl3) δ 117.3, 119.8, 120.1, 121.1, 121.9, 122.0, 122.1, 122.4, 122.5, 126.5, 126.7, 127.1, 127.3, 127.9, 128.1, 128.8, 131.9, 133.2, 134.2, 149.6; IR (neat) 3062, 2950, 1653 cm−1; EI-MS m/z (rel intensity) 300 ([M + 2]+, 71), 299 (100), 298 ([M]+, 73), 118 (97); HRMS [M]+ calcd for C15H11BrN2: 298.0106, found 298.0101.
(E)-2-(4-Chlorostyryl)-2H-indazole (2c): Following the general procedure, the title compound was obtained (Route A: 61 mg, 48%, Route B: 96 mg, 75%). A yellow solid, mp 150–151 °C; TLC (EtOAc/hexanes (1:4)) Rf = 0.50; 1H-NMR (CDCl3) δ 7.06 (dd, J = 8.1, 6.9 Hz, 1H), 7.26–7.40 (m, 6H), 7.55–7.59 (m, 2H), 7.71 (d, J = 9.0 Hz, 1H), 7.98 (s, 1H); 13C-NMR (CDCl3) δ 117.3 (CH), 119.7 (CH), 120.1 (CH), 122.0 (CH), 122.1 (C), 122.4 (CH), 126.6 (CH), 127.3 (CH), 127.5 (CH × 2), 128.9 (CH × 2), 132.7 (C), 133.7 (C), 149.6 (C); IR (neat) 3031, 2950, 1393 cm−1; EI-MS m/z (rel intensity) 256 ([M + 2]+, 25), 254 ([M]+, 81), 253 (100), 118 (80); HRMS [M]+ calcd for C15H11ClN2: 254.0611, found 254.0617.
(E)-2-(4-Methylstyryl)-2H-indazole (2d): Following the general procedure, the title compound was obtained (Route A: 74 mg, 63%, Route B: 93 mg, 79%). A white solid, mp 149–151 °C; TLC (EtOAc/hexanes (1:4)) Rf = 0.60; 1H-NMR (CDCl3) δ 2.33 (s, 3H), 7.06 (t, J = 7.8 Hz, 1H), 7.15 (d, J = 7.8 Hz, 2H), 7.23–7.45 (m, 4H), 7.58–7.63 (m, 2H), 7.74 (d, J = 8.7 Hz, 1H), 7.98 (s, 1H); 13C-NMR (CDCl3) δ 21.1 (CH3), 117.2 (CH), 120.1 (CH), 121.0 (CH), 121.6 (CH), 122.1 (C), 122.2 (CH), 125.6 (CH), 126.3 (CH × 2), 126.9 (CH), 129.4 (CH × 2), 131.3 (C), 138.1 (C), 149.4 (C); IR (neat) 3121, 1602, 1367 cm−1; EI-MS m/z (rel intensity) 234 ([M]+, 71), 233 (100), 218 (14), 118 (27); HRMS [M]+ calcd for C16H14N2: 234.1157, found 234.1159.
(E)-2-(4-Methoxystyryl)-2H-indazole (2e): Following the general procedure, the title compound was obtained (Route A: 101 mg, 81%, Route B: 109 mg, 87%). A white solid, mp 137–139 °C; TLC (EtOAc/hexanes (1:4)) Rf = 0.45; 1H-NMR (CDCl3) δ 3.78 (s, 3H), 6.88 (d, J = 8.7 Hz, 2H), 7.06 (t, J = 8.7 Hz, 1H), 7.26 (t, J = 8.7 Hz, 1H), 7.31–7.43 (m, 3H), 7.55 (d, J = 14.7 Hz, 1H), 7.60 (d, J = 8.7 Hz, 1H), 7.72 (d, J = 8.7 Hz, 1H), 8.00 (s, 1H); 13C-NMR (CDCl3) δ 55.1 (CH3), 114.2 (CH × 2), 117.2 (CH), 120.0 (CH), 120.8 (CH), 121.5 (CH), 122.1 (C), 122.2 (CH), 124.6 (CH), 126.7 (C), 126.9 (CH), 127.8 (CH × 2), 149.3 (C), 159.6 (C); IR (neat) 3120, 1609, 1517 cm−1; EI-MS m/z (rel intensity) 250 ([M]+, 100), 249 (58), 132 (93), 118 (19); HRMS [M]+ calcd for C16H14N2O: 250.1106, found 250.1104.
(E)-2-(2-(Naphthalen-2-yl)vinyl)-2H-indazole (2f): Following the general procedure, the title compound was obtained (Route A: 85 mg, 63%, Route B: 115 mg, 85%). A yellow solid, mp 195–196 °C; TLC (EtOAc/hexanes (1:4)) Rf = 0.52; 1H-NMR (DMSO-d6) δ 7.08 (t, J = 7.8 Hz, 1H), 7.31 (t, J = 7.8 Hz, 1H), 7.49–7.55 (m, 2H), 7.67–7.77 (m, 3H), 7.88–7.96 (m, 4H), 8.08 (s, 1H), 8.41 (d, J = 14.1 Hz, 1 H), 8.68 (s, 1H); 13C-NMR (DMSO-d6) δ 117.0 (CH), 120.3 (CH), 120.9 (CH), 121.9 (CH × 2), 123.4 (CH), 123.7 (CH), 126.2 (CH), 126.6 (CH), 126.8 (CH), 127.0 (CH), 127.6 (CH), 127.7 (C), 127.8 (CH), 128.4 (CH), 132.2 (C), 132.6 (C), 133.2 (C) 148.9 (C); IR (neat) 3062, 2950, 1626 cm−1; EI-MS m/z (rel intensity) 270 ([M]+, 93), 269 (100), 152 (90), 118 (11); HRMS [M]+ calcd for C19H14N2: 270.1157, found 270.1150.
2-(Prop-1-en-1-yl)-2H-indazole (2g): Following the general procedure, the title compound was obtained (Route A: 57 mg, 72%, Route B: 66 mg, 84%). A yellow oil; TLC (EtOAc/hexanes (1:2)) Rf = 0.58; 1H-NMR (CDCl3) δ 1.85 (dd, J = 6.9, 1.8 Hz, 2.60H), 2.02 (dd, J = 6.9, 1.5 Hz, 0.40H), 5.60–5.62 (m, 0.13H), 6.39–6.99 (m, 0.87H), 7.00–7.07 (m, 2H), 7.21–7.28 (m, 1H), 7.55–7.72 (m, 2H), 7.89 (s, 0.87H), 7.96 (s, 0.13H), 8.65 (s, 0.87H), 8.70 (s, 0.13H); 13C-NMR (CDCl3) δ 12.9, 14.8, 117.3, 117.7, 120.0, 120.5, 121.8, 121.9, 126.5, 128.3, 149.0; IR (neat) 3113, 2933, 1624 cm−1; EI-MS m/z (rel intensity) 158 ([M]+, 46), 157 (28), 131 (100), 118 (12); HRMS [M]+ calcd for C10H10N2: 158.0844, found 158.0843.
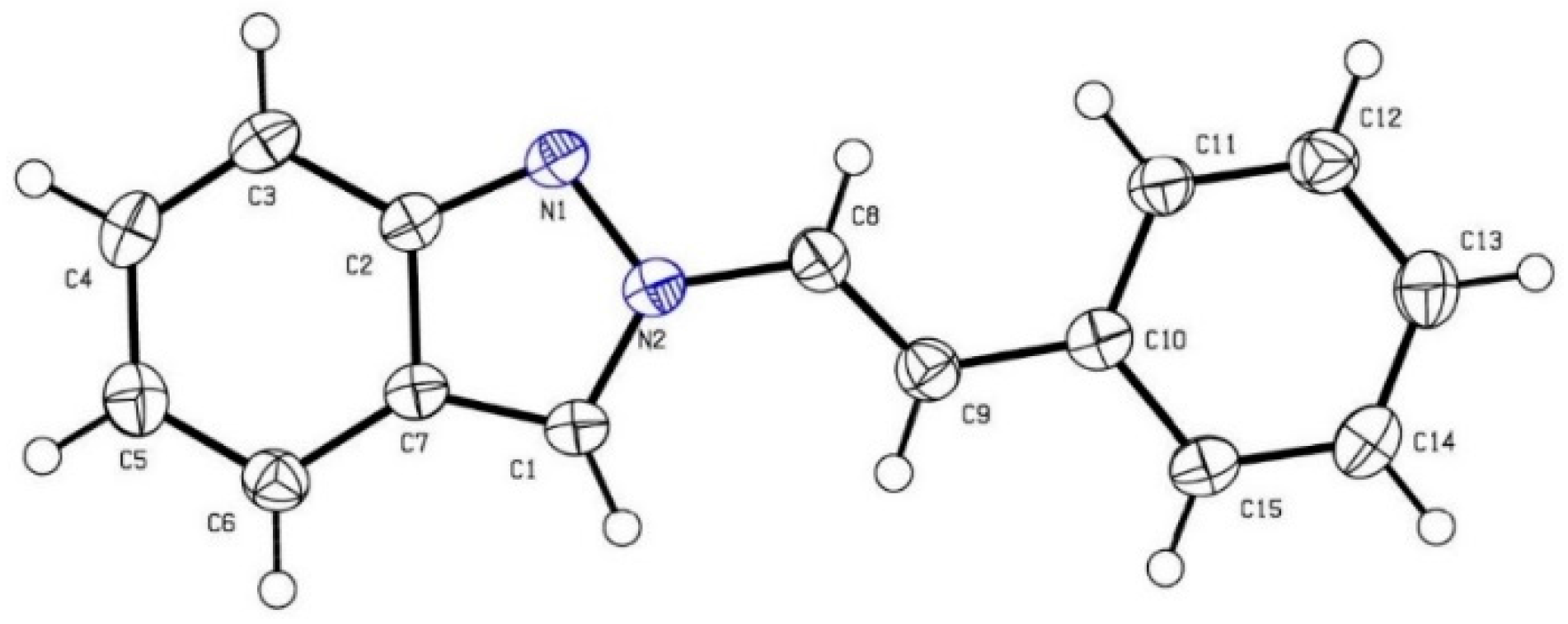
(E)-2-(3,3-Dimethylbut-1-en-1-yl)-2H-indazole (2h): Following the general procedure, the title compound was obtained (Route A: 24 mg, 24%, Route B: 70 mg, 70%). An oil; TLC (EtOAc/hexanes (1:2)) Rf = 0.70; 1H-NMR (CDCl3) δ 1.18 (s, 9H), 6.59 (d, J = 14.4 Hz, 1H), 7.01 (d, J = 14.4 Hz, 1H), 7.05 (d, J = 8.7 Hz, 1H), 7.27 (d, J = 8.7 Hz, 1H), 7.61 (d, J = 8.7 Hz, 1H), 7.68 (d, J = 8.7 Hz, 1H), 8.00 (s, 1H); 13C-NMR (CDCl3) δ 25.6 (CH3 × 3), 32.3 (C), 117.3 (CH), 120.0 (CH), 121.0 (CH), 121.8 (C), 122.0 (CH), 124.5 (CH), 126.5 (CH), 133.4 (CH), 149.1 (C); IR (neat) 3062, 2950, 1633 cm−1; EI-MS m/z (rel intensity) 200 ([M]+, 74), 185 (100), 131 (57), 118 (58); HRMS [M]+ calcd for C13H16N2: 200.1313, found 200.1315.
6-Nitro-2-(prop-1-en-1-yl)-2H-indazole (2i): Following the general procedure, the title compound was obtained (Route B: 81 mg, 80%). A green solid, mp 103–105 °C; TLC (EtOAc/hexanes (1:4)) Rf = 0.20; 1H-NMR (acetone-d6) δ 1.92 (d, J = 6.9 Hz, 2.85H), 2.15 (d, J = 6.9 Hz, 0.15H), 5.78–5.88 (m, 0.05H), 6.68–6.80 (m, 0.95H), 7.33–7.40 (m, 1H), 7.78–7.91 (m, 2H), 8.54 (s, 1.9H), 8.55 (s, 0.1H); 13C-NMR (acetone-d6) δ 15.0 (CH3), 115.7 (CH), 116.2 (CH), 121.1 (CH), 123.2 (CH), 123.8 (CH), 125.3 (C), 129.5 (CH), 147.7 (C), 147.8 (C); IR (neat) 3051, 1514, 1342 cm−1; EI-MS m/z (rel intensity) 203 ([M]+, 100), 176 (95), 156 (27), 130 (47); HRMS [M]+ calcd for C10H9N3O2: 203.0695, found 203.0699.
(E)-6-Bromo-2-styryl-2H-indazole (2j): Following the general procedure, the title compound was obtained (Route A: 112 mg, 75%, Route B: 120 mg, 80%). A yellow solid, mp 118–120 °C; TLC (EtOAc/hexanes (1:4)) Rf = 0.50; 1H-NMR (CDCl3) δ 7.15 (dd, J = 9.0, 1.5 Hz, 1H), 7.28–7.40 (m, 3H), 7.44–7.52 (m, 4H), 7.67 (d, J = 14.4 Hz, 1H), 7.89 (s, 1H), 8.09 (s, 1H); 13C-NMR (CDCl3) δ 119.7 (CH), 120.6 (C), 121.1 (C), 121.5 (CH), 121.7 (CH), 122.2 (CH), 126.0 (CH), 126.1 (CH × 2), 126.5 (CH), 128.3 (CH), 128.8 (CH × 2), 133.9 (C), 150.0 (C); IR (neat) 3062, 2950, 1625 cm−1; EI-MS m/z (rel intensity) 300 ([M + 2]+, 56), 299 (100), 298 ([M]+, 61), 77 (32); HRMS [M]+ calcd for C15H11BrN2: 298.0106, found 298.0109.
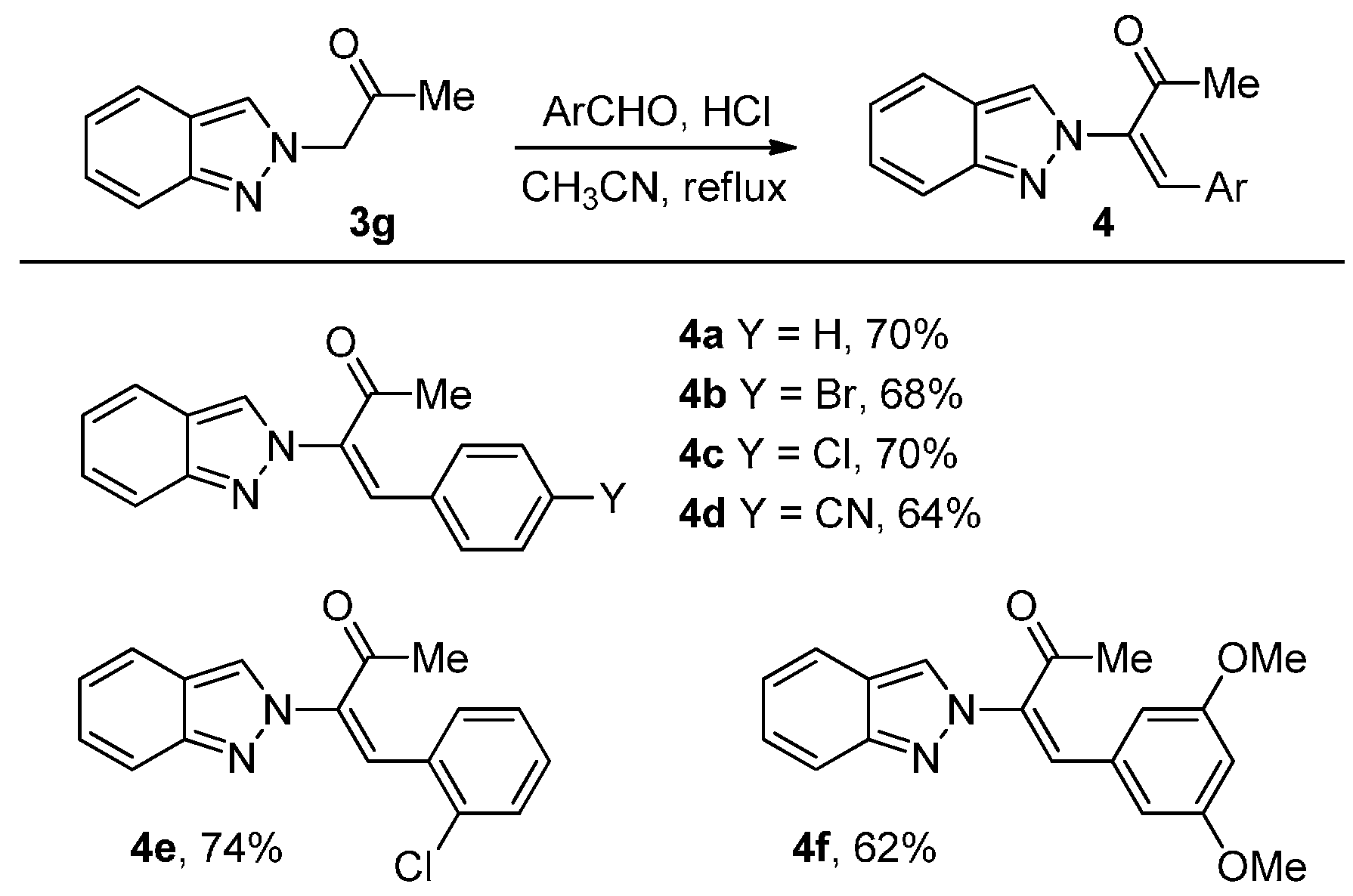
3.2.3. General procedure for the synthesis of 2-alkenyl-2H-indazoles 4:
A mixture of ketone 3g (1.0 mmol), aldehyde (1.5 mmol), and conc. HCl (3 drops) in CH3CN (1 mL) was heated to reflux under nitrogen. Reaction was monitored by TLC until no starting material was observed and normally the reaction was stirred under reflux for 4 h. The reaction was then cooled to ambient temperature and quenched with sat. NaHCO3 until pH was 7. CH2Cl2 (10 mL) was added and the mixture was transferred to a separatory funnel. The aqueous layer was back extracted with CH2Cl2 (10 mL × 2). The combined organic layers were dried over MgSO4, filtered, and concentrated in a rotary evaporator. The residue was purified by silica gel chromatography using EtOAc/hexanes (1/10) as eluent to give the product 4.
(E)-3-(2H-Indazol-2-yl)-4-phenylbut-3-en-2-one (4a): Following the general procedure, the title compound was obtained (184 mg, 70%). A yellow oil; TLC (EtOAc/hexanes (1:2)) Rf = 0.35; 1H-NMR (acetone-d6) δ 2.20 (s, 3H), 6.83 (d, J = 7.5 Hz, 2H), 7.06–7.16 (m, 3H), 7.25–7.33 (m, 2H), 7.69–7.73 (m, 2H), 7.95 (s, 1H), 8.22 (s, 1H); 13C-NMR (acetone-d6) δ 26.3 (CH3), 119.2 (CH), 122.2 (CH), 123.3 (CH), 123.9 (C), 126.9 (CH), 127.8 (CH), 130.0 (CH × 2), 131.7 (CH × 2), 132.2 (CH), 133.1 (C), 138.2 (C), 138.3 (CH), 151.0 (C), 194.8 (C); IR (neat) 3062, 1683, 1610 cm−1; EI-MS m/z (rel intensity) 262 ([M]+, 46), 261 (100), 219 (34), 118 (41); HRMS [M]+ calcd for C17H14N2O: 262.1106, found 262.1099.
(E)-4-(4-Bromophenyl)-3-(2H-indazol-2-yl)but-3-en-2-one (4b): Following the general procedure, the title compound was obtained (232 mg, 68%). A yellow oil; TLC (EtOAc/hexanes (1:2)) Rf = 0.38; 1H-NMR (CDCl3) δ 2.12 (s, 3H), 6.55 (d, J = 8.7 Hz, 2H), 7.09–7.14 (m, 1H), 7.24 (d, J = 8.7 Hz, 2H), 7.30–7.36 (m, 1H), 7.64 (d, J = 8.7 Hz, 1H), 7.74–7.80 (m, 2H), 7.90 (s, 1H); 13C-NMR (CDCl3) δ 25.6 (CH3), 118.0 (CH), 120.4 (CH), 122.4 (C), 122.6 (CH), 124.6 (CH), 125.7 (C), 127.0 (CH), 129.9 (C), 131.6 (CH × 2), 132.0 (CH × 2), 135.7 (CH), 136.4 (C), 149.8 (C), 193.9 (C); IR (neat) 3062, 1674, 1619 cm−1; EI-MS m/z (rel intensity) 342 ([M + 2]+, 49), 341 (100), 340 ([M]+, 49), 118 (88); HRMS [M]+ calcd for C17H13BrN2O: 340.0211, found 340.0214.
(E)-4-(4-Chlorophenyl)-3-(2H-indazol-2-yl)but-3-en-2-one (4c): Following the general procedure, the title compound was obtained (208 mg, 70%). A yellow oil; TLC (EtOAc/hexanes (1:2)) Rf = 0.38; 1H-NMR (CDCl3) δ 2.12 (s, 3 H), 6.62 (d, J = 8.7 Hz, 2H), 7.05–7.14 (m, 3H), 7.30–7.36 (m, 2H), 7.64 (d, J = 8.7 Hz, 1H), 7.75–7.80 (m, 2H), 7.90 (s, 1H); 13C-NMR (CDCl3) δ 25.6 (CH3), 118.1 (CH), 120.4 (CH), 122.4 (C), 122.6 (CH), 124.6 (CH), 127.0 (CH), 129.1 (CH × 2), 129.5 (C), 131.5 (CH × 2), 135.7 (CH), 136.4 (C), 137.2 (C), 149.9 (C), 194.0 (C); IR (neat) 3062, 1683, 1491 cm−1; EI-MS m/z (rel intensity) 298 ([M + 2]+, 18), 296 ([M]+, 51), 295 (100), 118 (64); HRMS [M]+ calcd for C17H13ClN2O: 296.0716, found 296.0708.
(E)-4-(2-(2H-Indazol-2-yl)-3-oxobut-1-en-1-yl)benzonitrile (4d): Following the general procedure, the title compound was obtained (184 mg, 64%). A yellow oil; TLC (EtOAc/hexanes (1:2)) Rf = 0.20; 1H-NMR (CDCl3) δ 2.16 (s, 3H), 6.68 (d, J = 8.7 Hz, 2H), 7.13 (t, J = 8.1 Hz, 1H), 7.32–7.41 (m, 3H), 7.63 (d, J = 8.1 Hz, 1H), 7.74 (d, J = 8.1 Hz, 1H), 7.78 (s, 1H), 7.88 (s, 1H); 13C-NMR (CDCl3) δ 25.8 (CH3), 113.8 (C), 117.8 (C), 118.0 (CH), 120.4 (CH), 122.4 (C), 122.9 (CH), 124.7 (CH), 127.3 (CH), 130.4 (CH × 2), 132.3 (CH × 2), 134.3 (CH), 135.4 (C), 138.1 (C), 149.9 (C), 193.9 (C); IR (neat) 3064, 1677, 1631 cm−1; EI-MS m/z (rel intensity) 287 ([M]+, 55), 286 (100), 244 (25), 118 (30); HRMS [M]+ calcd for C18H13N3O: 287.1059, found 287.1049.
(E)-4-(2-Chlorophenyl)-3-(2H-indazol-2-yl)but-3-en-2-one (4e): Following the general procedure, the title compound was obtained (220 mg, 74%). A yellow oil; TLC (EtOAc/hexanes (1:2)) Rf = 0.50; 1H-NMR (CDCl3) δ 2.21 (s, 3H), 6.30 (d, J = 8.1 Hz, 1H), 6.78 (d, J = 8.1 Hz, 1H), 7.03–7.16 (m, 2H), 7.26–7.37 (m, 2H), 7.55 (d, J = 8.1 Hz, 1H), 7.75 (d, J = 8.1 Hz, 1H), 7.79 (s, 1H), 8.10 (s, 1H); 13C-NMR (CDCl3) δ 25.9 (CH3), 118.0 (CH), 120.4 (CH), 122.1 (C), 122.4 (CH), 125.1 (CH), 126.9 (CH), 127.0 (CH), 129.4 (CH), 129.6 (CH), 129.9 (C), 131.4 (CH), 132.5 (CH), 135.7 (C), 137.9 (C), 149.7 (C), 193.8 (C); IR (neat) 3053, 1693, 1624 cm−1; EI-MS m/z (rel intensity) 296 ([M]+, 3), 262 (18), 261 (100), 118 (9); HRMS [M]+ calcd for C17H13ClN2O: 296.0716, found 296.0718.
(E)-4-(3,5-Dimethoxyphenyl)-3-(2H-indazol-2-yl)but-3-en-2-one (4f): Following the general procedure, the title compound was obtained (200 mg, 62%). A brown solid, mp 94–96 °C; TLC (EtOAc/hexanes (1:2)) Rf = 0.35; 1H-NMR (CDCl3) δ 2.19 (s, 3H), 3.22 (s, 6H), 5.72 (d, J = 2.4 Hz, 2H), 6.30 (t, J = 2.4 Hz, 1H), 7.04–7.09 (m, 1H), 7.25–7.30 (m, 1H), 7.60 (d, J = 8.1 Hz, 1H), 7.72–7.75 (m, 2 H), 7.90 (s, 1H); 13C- NMR (CDCl3) δ 25.7 (CH3), 54.6 (CH3 × 2), 104.4 (CH), 107.4 (CH × 2), 117.7 (CH), 120.3 (CH), 122.4 (C), 122.5 (CH), 125.1 (CH), 126.9 (CH), 132.3 (C), 136.2 (C), 137.9 (CH), 149.7 (C), 160.4 (C × 2), 194.0 (C); IR (neat) 3120, 1697, 1583 cm−1; EI-MS m/z (rel intensity) 322 ([M]+, 78), 321 (100), 279 (21), 189 (32); HRMS [M]+ calcd for C19H18N2O3: 322.1317, found 322.1318.
2-(1-Methyl-1H-inden-2-yl)-2H-indazole (5). To a solution of ketone 4a (184 mg, 0.7 mmol) in MeOH (7 mL) was added CeCl3–7H2O (135 mg, 0.36 mmol) followed by NaBH4 (28 mg, 0.7 mmol, 1.0 equiv.) at ambient temperature. The reaction was stirred at ambient temperature for 2 h and then quenched with H2O (10 mL). CH2Cl2 (10 mL) was added and the mixture was transferred to a separatory funnel. The aqueous layer was back extracted with CH2Cl2 (10 mL × 2). The combined organic layers were dried over MgSO4, filtered, and concentrated in a rotary evaporator. The residue (174 mg) was dissolved in DCE (2.4 mL) and PPA (0.66 mL) was added. The mixture was heated to 90 °C under N2 for 2 h. The resulting mixture was cooled to 0–5 °C and sat. NaHCO3 was added slowly until pH was 7. CH2Cl2 (10 mL) was added and the mixture was transferred to a separatory funnel. The aqueous layer was back extracted with CH2Cl2 (10 mL × 2). The combined organic layers were dried over MgSO4, filtered, and concentrated in a rotary evaporator. The residue was purified by silica gel chromatography using EtOAc/hexanes (1/200) as eluent to give the product 5. (121 mg, 70%). A yellow solid, mp 74–75 °C; TLC (EtOAc/hexanes (1:2)) Rf = 0.63; 1H-NMR (CDCl3) δ 1.52 (d, J = 7.5 Hz, 3H), 4.23 (q, J = 7.5 Hz, 1H), 7.10 (t, J = 7.5 Hz, 2H), 7.23–7.45 (m, 5H), 7.66 (d, J = 8.7 Hz, 1H), 7.80 (d, J = 8.7 Hz, 1H), 8.20 (s, 1H); 13C-NMR (CDCl3) δ 16.9 (CH3), 43.2 (CH), 117.4 (CH), 117.6 (CH), 120.1 (CH), 121.0 (CH), 121.5 (CH), 122.2 (C), 122.4 (CH), 122.7 (CH), 125.4 (CH), 126.9 (CH), 127.0 (CH), 141.0 (C), 145.9 (C), 149.6 (C), 149.7 (C); IR (neat) 3062, 1634, 1486 cm−1; EI-MS m/z (rel intensity) 246 ([M]+, 100), 245 (92), 231 (34), 128 (45); HRMS [M]+ calcd for C17H14N2: 246.1157, found 246.1155.
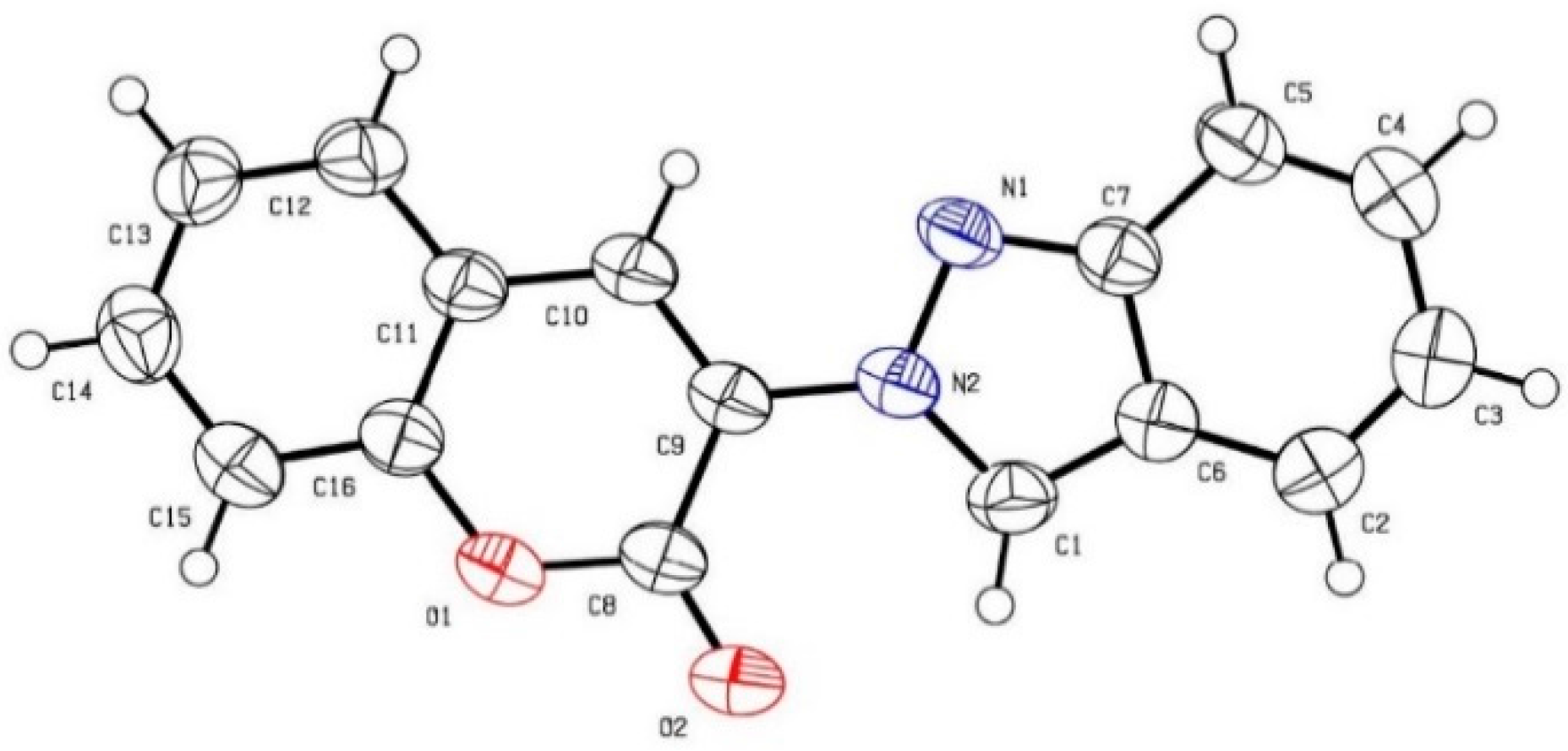
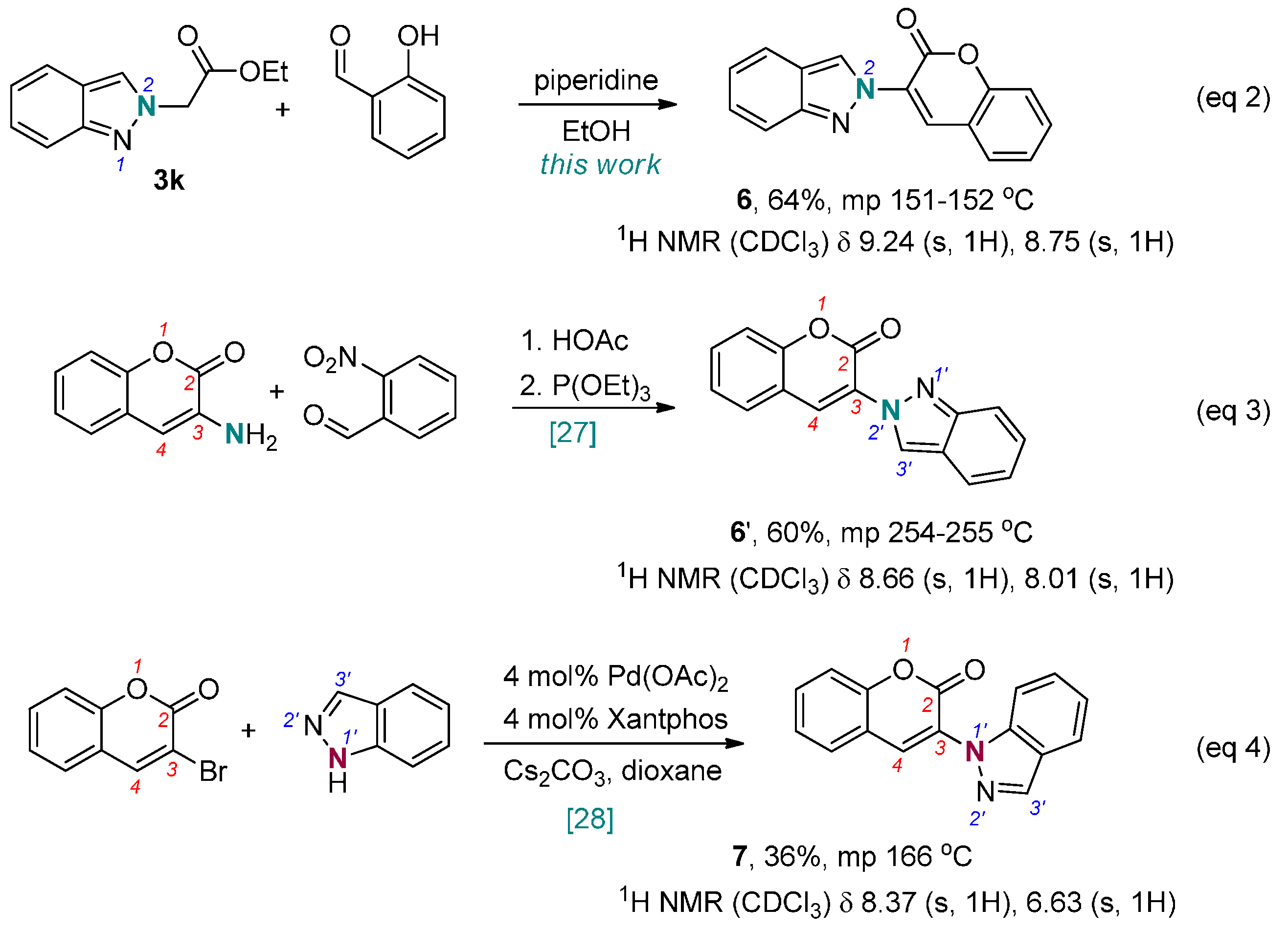
3-(2H-Indazol-2-yl)-2H-chromen-2-one (6). A mixture of ethyl 2-(2H-indazol-2-yl)acetate (204 mg, 1.0 mmol), 2-hydroxybenzaldehyde (122 mg, 1.0 mmol), and piperidine (43 mg, 0.5 mmol) in EtOH (1 mL) was heated to reflux. After 15 h, the reaction was then cooled to ambient temperature and quenched with sat. NH4Cl until pH was 7. CH2Cl2 (10 mL) was added and the mixture was transferred to a separatory funnel. The aqueous layer was back extracted with CH2Cl2 (10 mL × 2). The combined organic layers were dried over MgSO4, filtered, and concentrated in a rotary evaporator. The residue was purified by silica gel chromatography using EtOAc/hexanes (1/10) as eluent to give the product 6 (167 mg, 64%). A yellow solid, mp 151–152 °C; TLC (EtOAc/hexanes (1:4)) Rf = 0.45; 1H-NMR (DMSO-d6) δ 7.11 (d, J = 7.8 Hz, 1H), 7.35 (t, J = 7.8 Hz, 1H), 7.46 (t, J = 7.8 Hz, 1H), 7.54 (d, J = 7.8 Hz, 1H), 7.67–7.73 (m, 2H), 7.82 (d, J = 7.8 Hz, 1H), 8.05 (d, J = 7.8 Hz, 1H), 8.87 (s, 1H), 9.17 (s, 1H); 13C-NMR (DMSO-d6) δ 116.1 (CH), 117.0 (CH), 118.6 (C), 121.5 (CH), 122.0 (C), 122.4 (CH), 125.4 (CH), 125.5 (CH), 125.8 (C), 127.9 (CH), 129.5 (CH), 132.5 (CH), 134.1 (CH), 148.6 (C), 152.0 (C), 156.2 (C); IR (neat) 3154, 1722, 1600 cm−1; EI-MS m/z (rel intensity) 262 ([M]+, 79), 236 (77), 127 (64), 105 (100); HRMS [M]+ calcd for C16H10N2O2: 262.0742, found 262.0734.











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
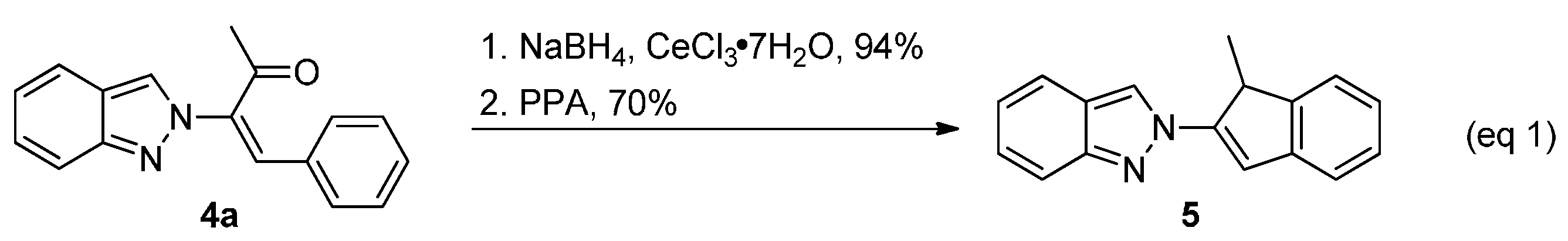{kind=link}
{kind=link}









