
Molecular Theory of Detonation Initiation: Insight from First Principles Modeling of the Decomposition Mechanisms of Organic Nitro Energetic Materials

Abstract
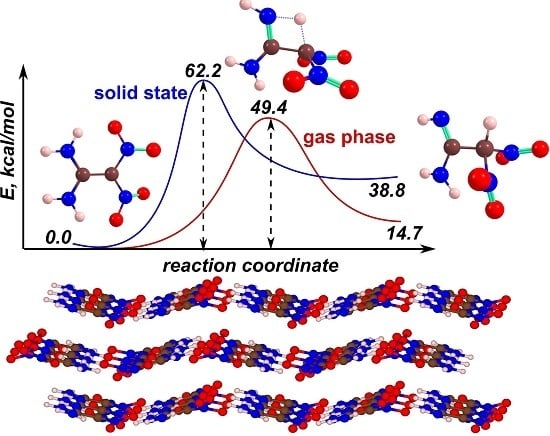
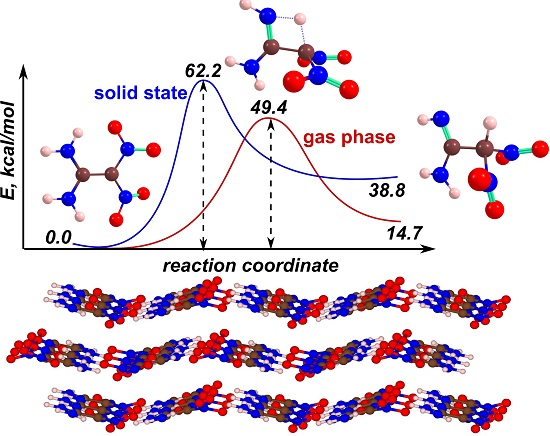
:
1. Introduction
2. Computational Details
2.1. Gas-Phase Calculations
2.2. Periodic Calculations
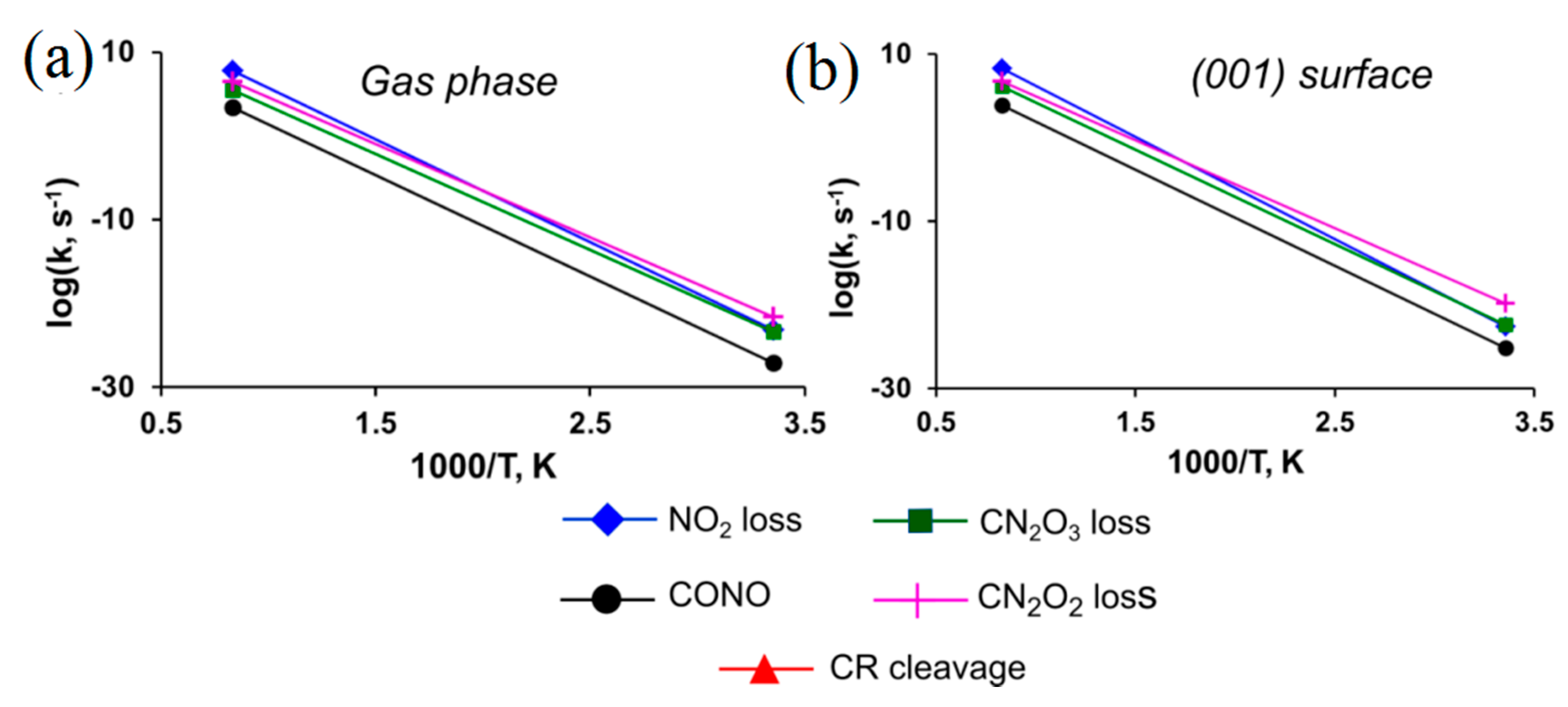
3. Results and Discussion
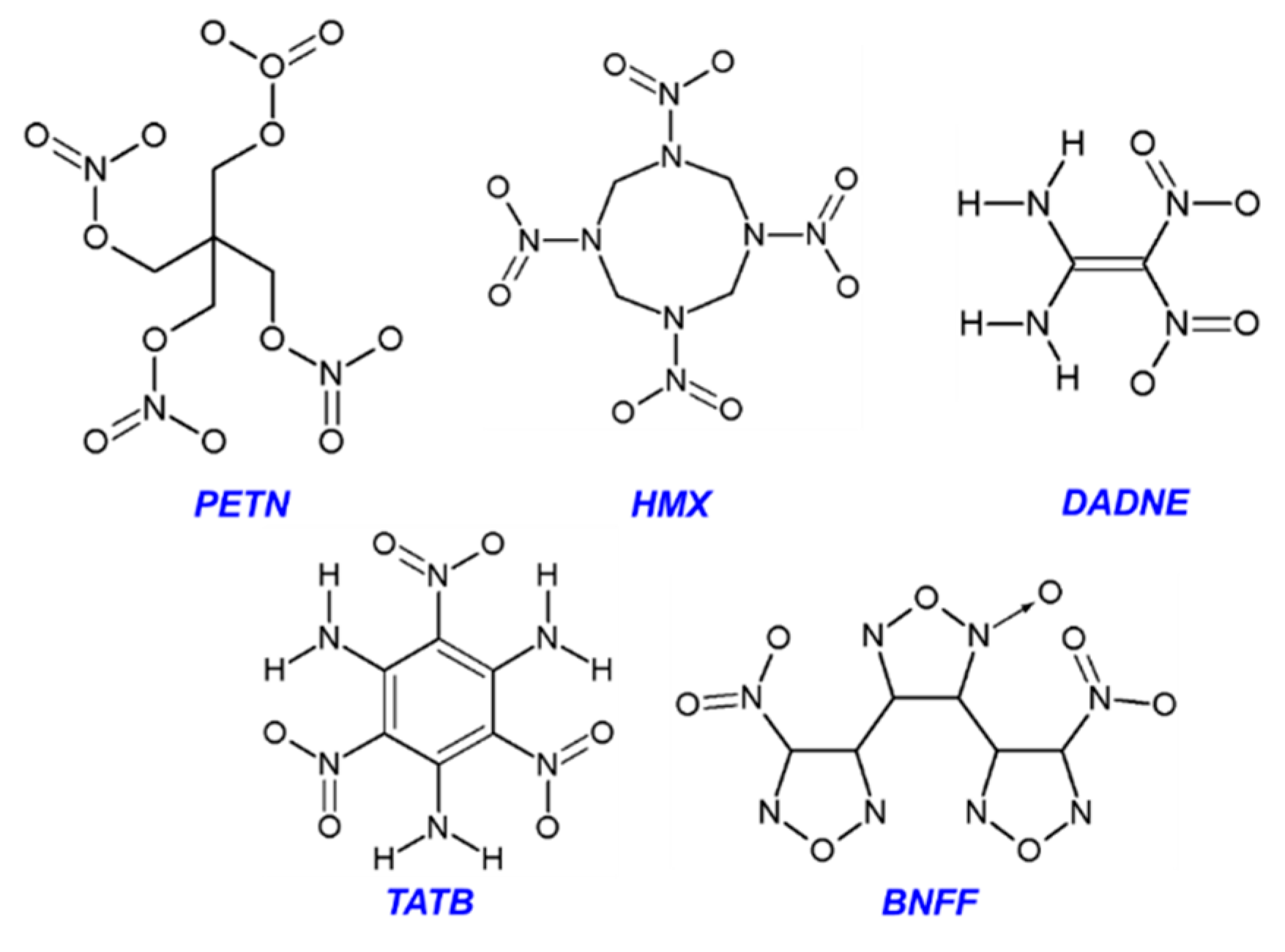
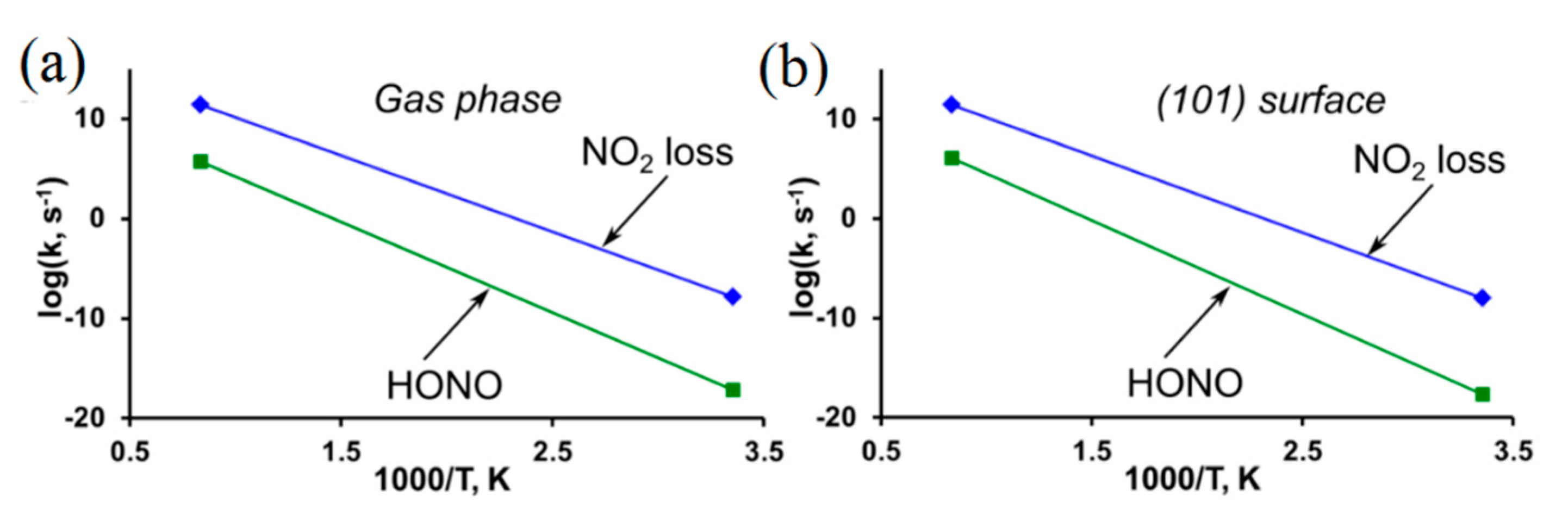
3.1. PETN
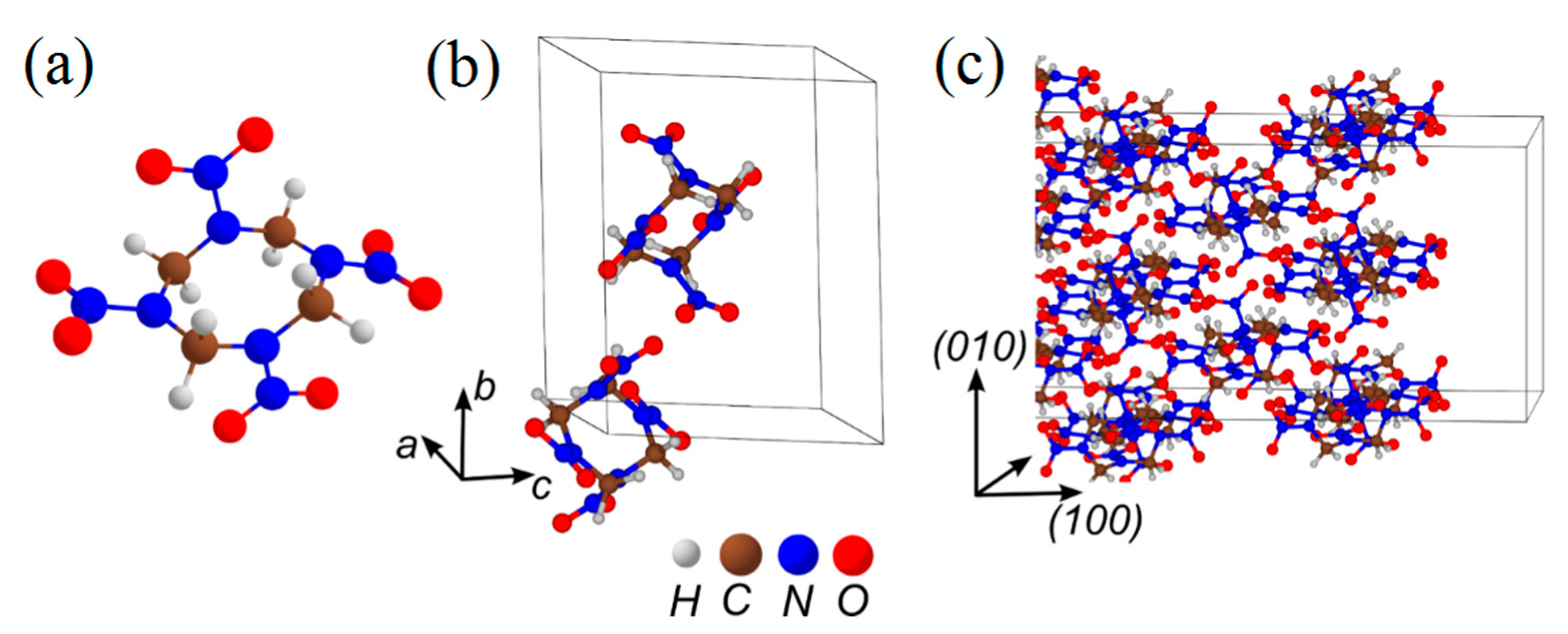
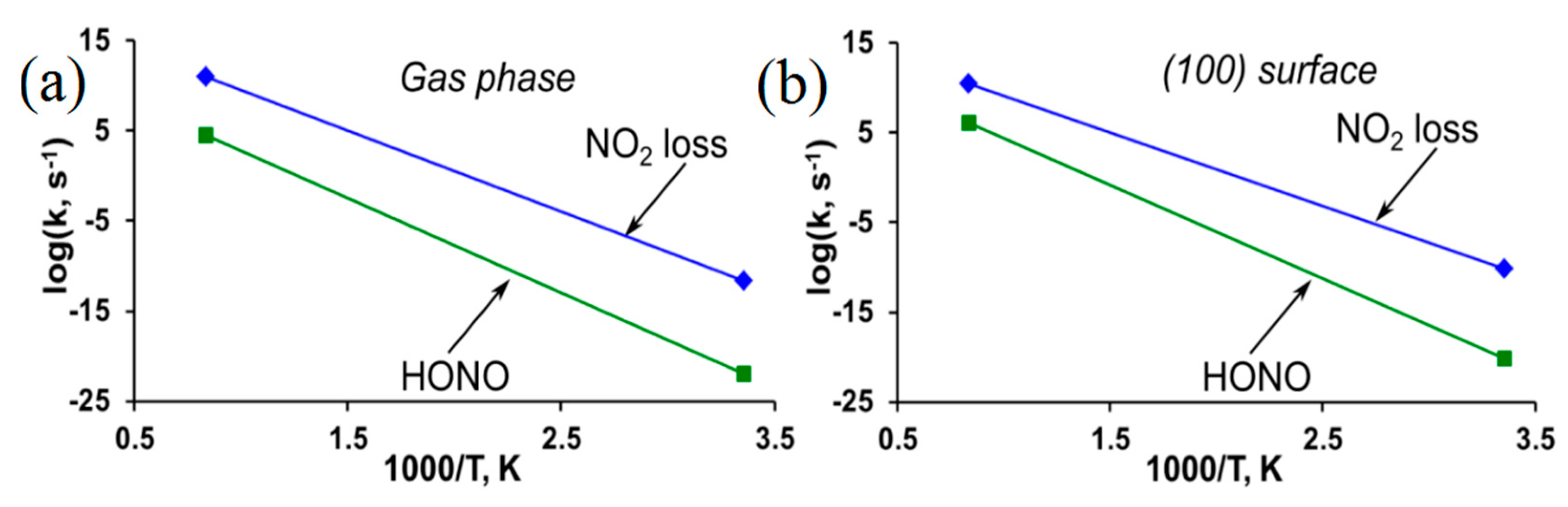
3.2. β-HMX
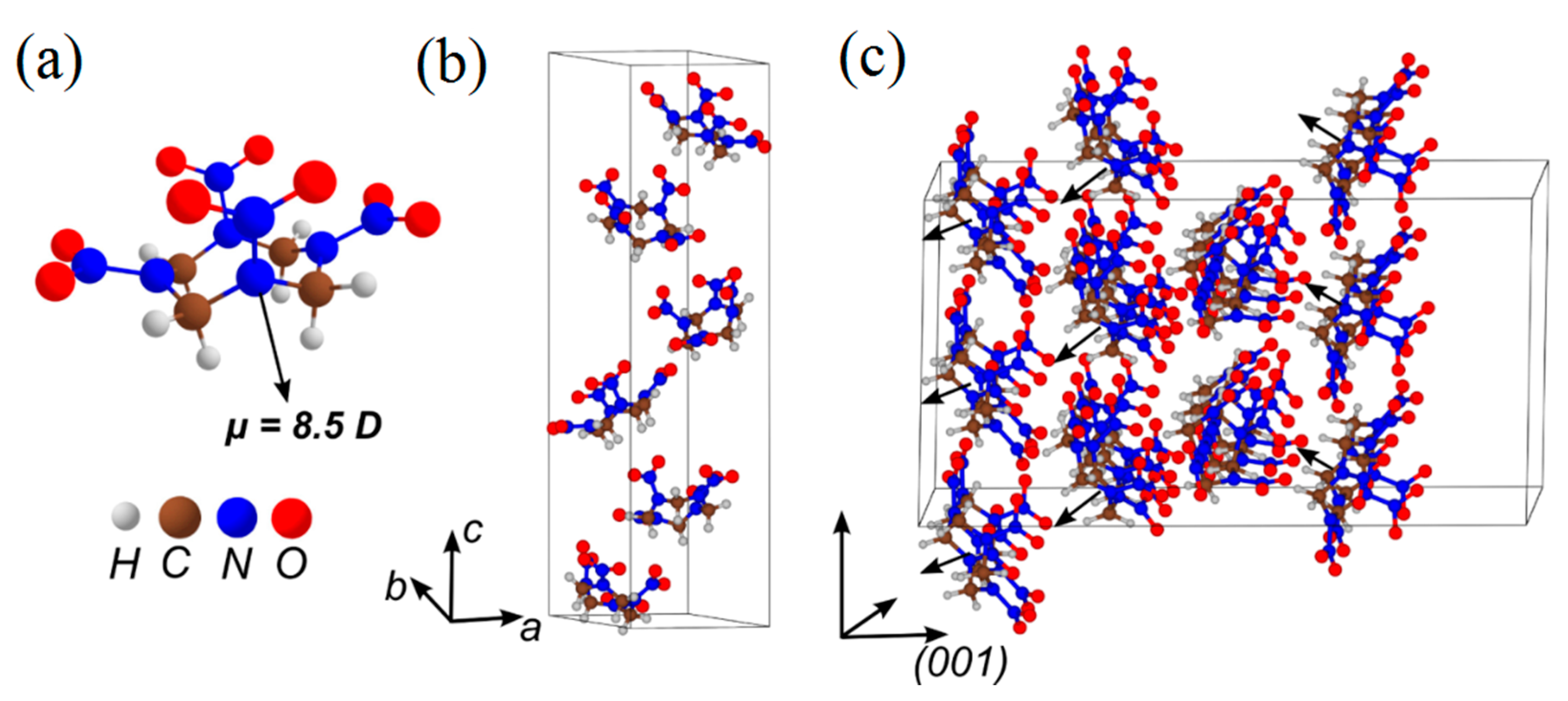
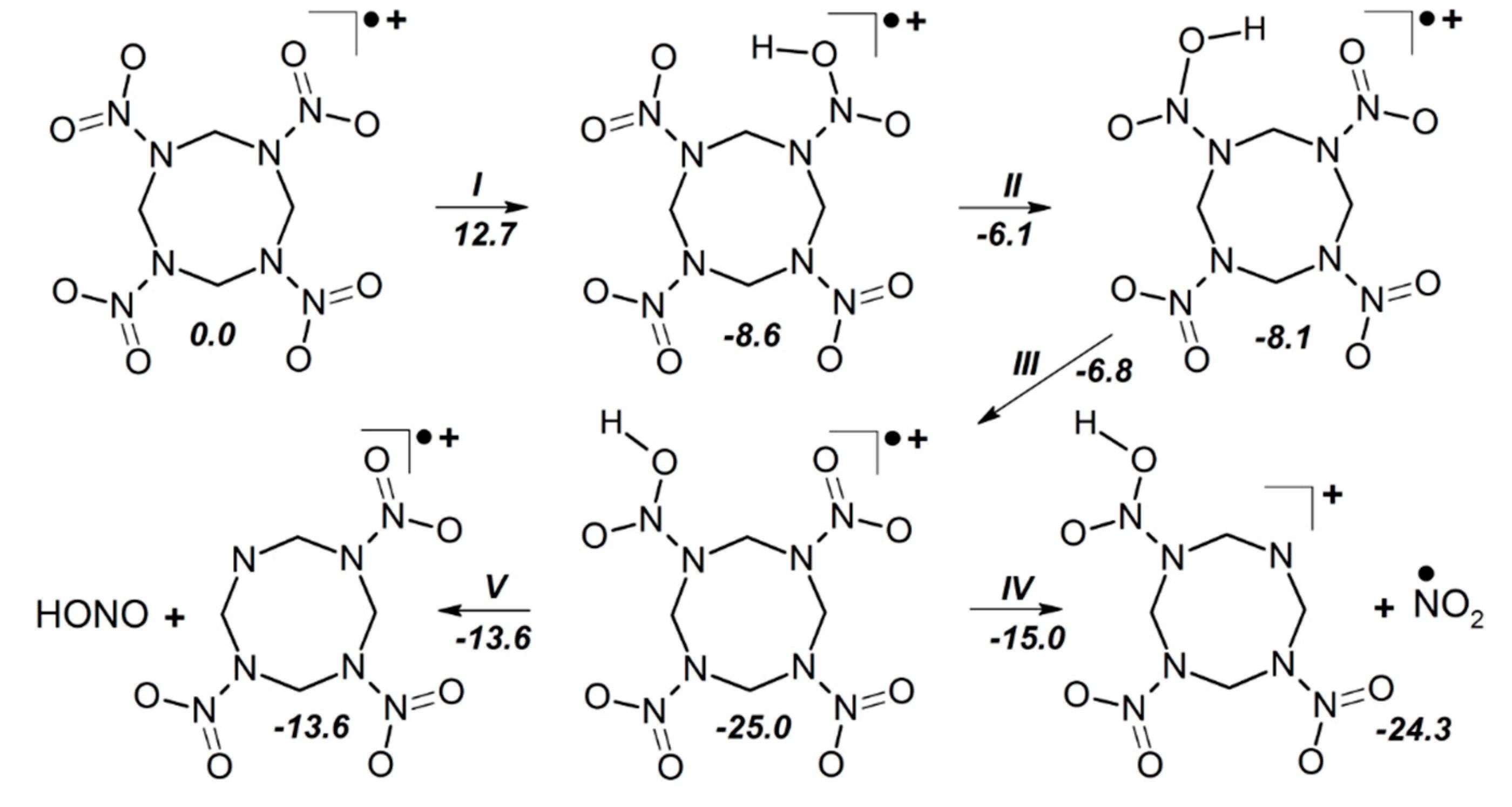
3.3. δ-HMX
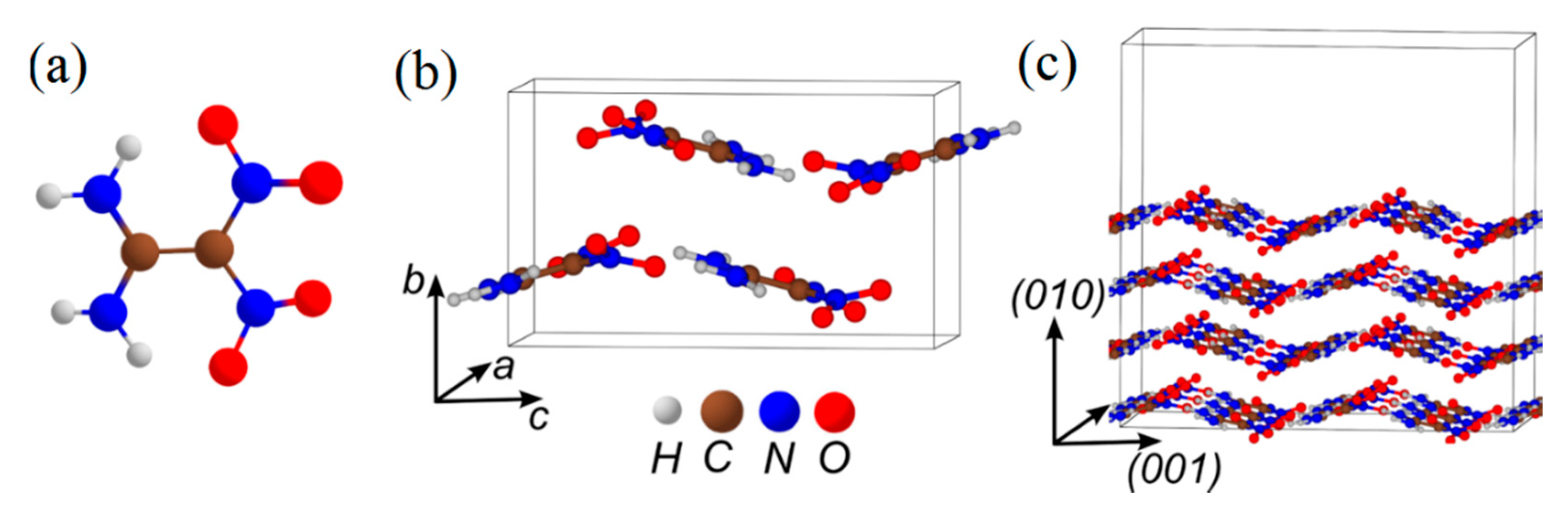
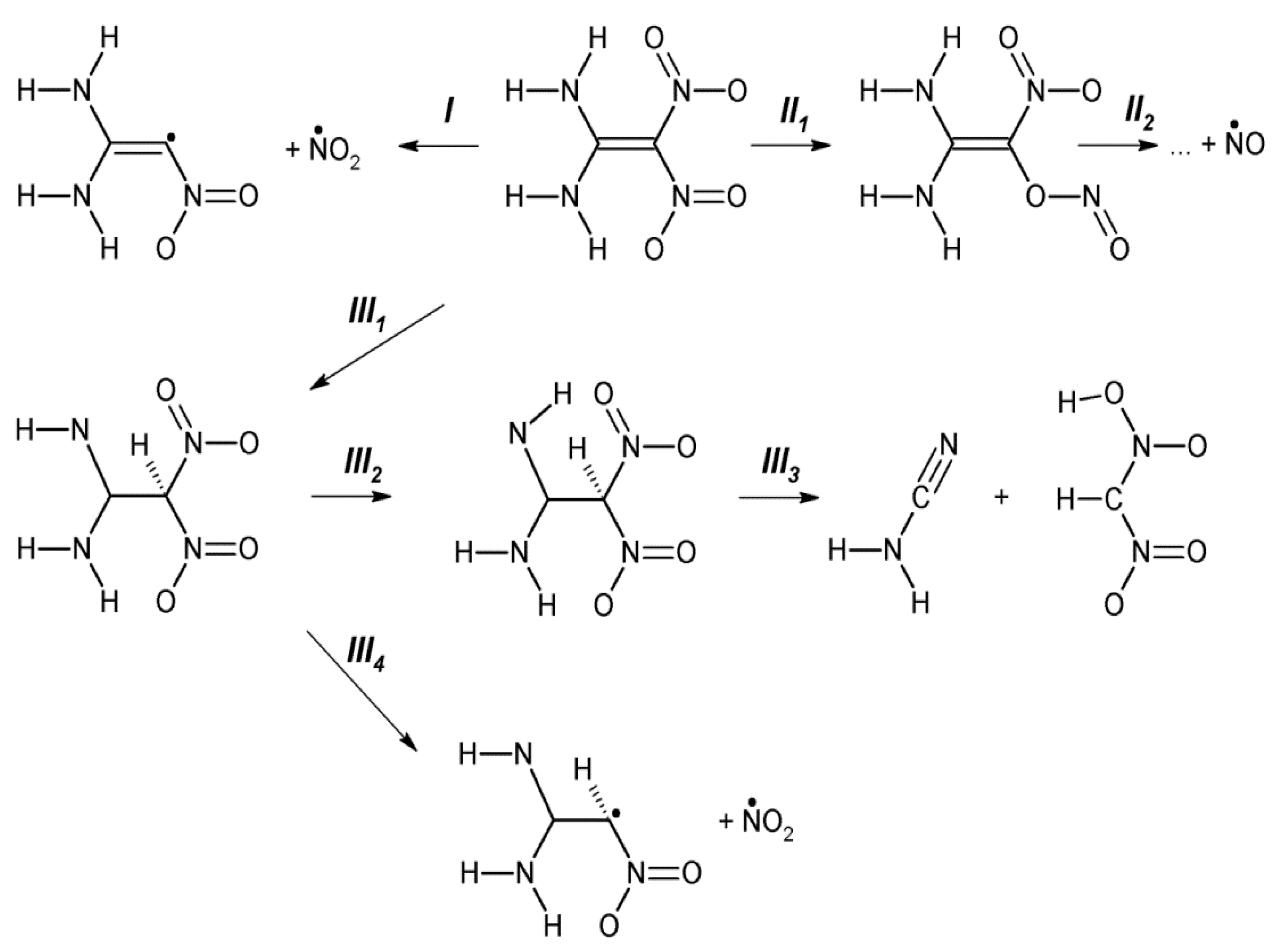
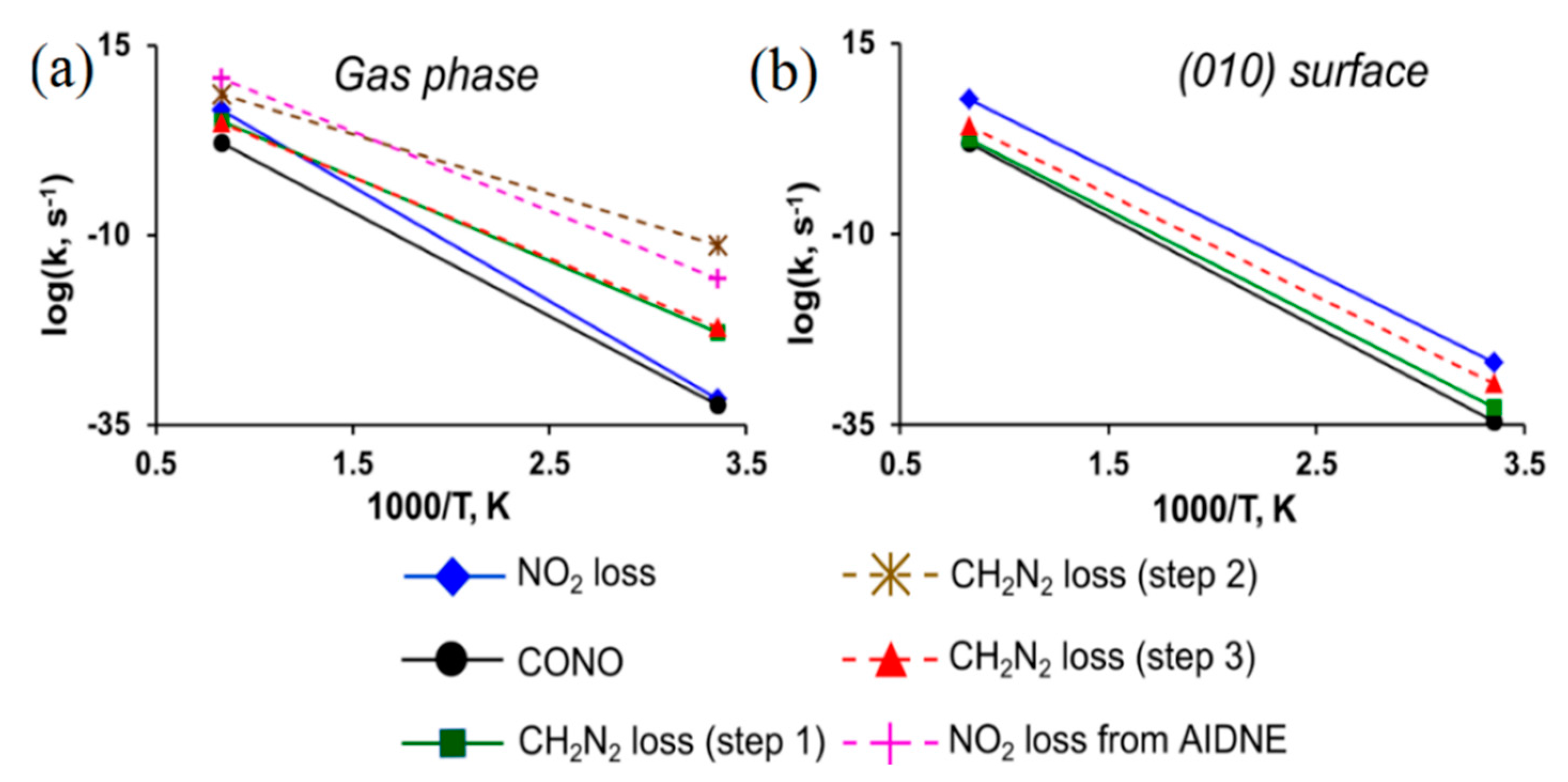
3.4. DADNE
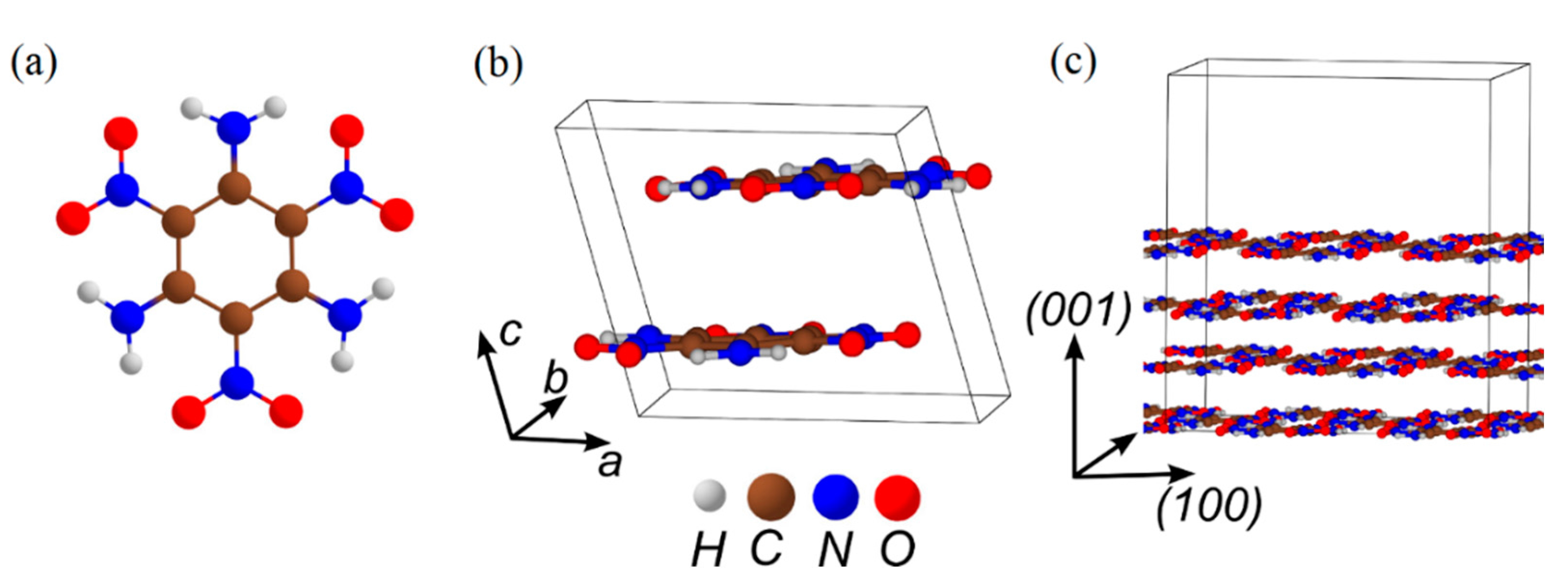
3.5. TATB
3.6. BNFF
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Berthelot, M.; Vieille, P. Sur la vitesse de propagation des phénomènes explosifs dans les gaz. C.R. Acad. Sci. 1881, 93, 18–22. [Google Scholar]
- Mallard, E.; le Chatelier, H. Sur les vitesses de propagation de l’inflammation dans les mélanges gazeux explosifs. C.R. Ac. Sc. 1881, 93, 145–148. [Google Scholar]
- Chapman, D.L. On the rate of explosion in gases. Philos. Mag. 1899, 47, 90–104. [Google Scholar] [CrossRef]
- Jouguet, E. On the propagation of chemical reactions in gases. J. Math. Pures Appl. 1905, 2, 347–425. [Google Scholar]
- Zeldovich, Y.B. On the utilization of detonative combustion energy. Technol. Phys. 1940, 10, 1453–1461. [Google Scholar]
- Döring, W. On detonation processes in gases. Ann. Phys. 1943, 43, 421–436. [Google Scholar] [CrossRef]
- Fickett, W.; Davis, W.C. Detonation: Theory and experiment; Courier Corporation: Mineola, NY, USA, 2012; p. 403. [Google Scholar]
- Fickett, W.; Wood, W.W. Flow calculations for pulsating one-dimensional detonations. Phys. Fluids 1966, 9, 903–916. [Google Scholar] [CrossRef]
- Bridgman, P.W. Water, in the liquid and five solid forms, under pressure. Proc. Am. Acad. Arts Sci. 1912, 47, 441–558. [Google Scholar] [CrossRef]
- Dremin, A.N. Discoveries in detonation of molecular condensed explosives in the 20th century. Combust. Explos. Shock Waves 2000, 36, 704–715. [Google Scholar] [CrossRef]
- Eyring, H.; Daniels, F. The decomposition of nitrogen pentoxide in inert solvents. J. Am. Chem. Soc. 1930, 52, 1472–1486. [Google Scholar] [CrossRef]
- Eyring, H.; Daniels, F. The decomposition of nitrogen pentoxide in chemically active solvents. J. Am. Chem. Soc. 1930, 52, 1486–1492. [Google Scholar] [CrossRef]
- Politzer, P.; Murray, J.S. (Eds.) Energetic materials: Part 1. Decomposition, crystal and molecular properties, and part 2. Detonation, combustion. In Theoretical and Computational Chemistry; Elsevier: Amsterdam, The Netherland, 2003.
- Yan, Q.L.; Zeman, S. Theoretical evaluation of sensitivity and thermal stability for high explosives based on quantum chemistry methods: A brief review. Int. J. Quant. Chem. 2013, 113, 1049–1061. [Google Scholar] [CrossRef]
- Chen, Z.X.; Xiao, H.M. Quantum chemistry derived criteria for impact sensitivity. Propellants Explos. Pyrotech. 2014, 39, 487–495. [Google Scholar] [CrossRef]
- Zhang, L.; Zybin, S.V.; van Duin, A.C.T.; Goddard, W.A., III. Modeling high rate impact sensitivity of perfect RDX and HMX crystals by ReaxFF reactive dynamics. J. Energ. Mater. 2010, 28, 92–127. [Google Scholar] [CrossRef]
- Kunz, A.B. Ab initio investigation of the structure and electronic properties of the energetic solids TATB and RDX. Phys. Rev. B 1996, 53, 9733–9738. [Google Scholar] [CrossRef]
- Kuklja, M.M.; Stefanovich, E.V.; Kunz, A.B. An excitonic mechanism of detonation initiation in explosives. J. Chem. Phys. 2000, 112, 3417–3423. [Google Scholar] [CrossRef]
- Kuklja, M.M.; Aduev, B.P.; Aluker, E.D.; Krasheninin, V.I.; Krechetov, A.G.; Mitrofanov, A.Y. The role of electronic excitations in explosive decomposition of solids. J. Appl. Phys. 2001, 89, 4156–4166. [Google Scholar] [CrossRef]
- Kuklja, M.M.; Rashkeev, S.N. Shear-strain induced structural and electronic modifications of the molecular crystal 1,1-diamino-2,2-dinitroethylene: Slip-plane flow and band gap relaxation. Phys. Rev. B 2007, 75. [Google Scholar] [CrossRef]
- Kuklja, M.M.; Rashkeev, S.N.; Zerilli, F.J. Shear-strain induced decomposition of 1,1-diamino-2,2-dinitroethylene. Appl. Phys. Lett. 2006, 89. [Google Scholar] [CrossRef]
- Sharma, J.; Beard, B.C. Electronic excitations preceding shock initiation in explosives. MRS Proc. 1992, 296. [Google Scholar] [CrossRef]
- Bernstein, E.R. On the release of stored energy from energetic materials. Adv. Quantum Chem. 2014, 69, 31–70. [Google Scholar]
- Chen, S.; Tolbert, W.A.; Dlott, D.D. Direct measurement of ultrafast multiphonon up-pumping in high explosives. J. Phys. Chem. 1994, 98, 7759–7766. [Google Scholar] [CrossRef]
- Coffey, C.S.; Sharma, J. Plastic deformation, energy dissipation, and initiation of crystalline explosives. Phys. Rev. B 1999, 60, 9365–9371. [Google Scholar] [CrossRef]
- Pospíšil, M.; Vávra, P.; Concha, M.C.; Murray, J.S.; Politzer, P. Sensitivity and the available free space per molecule in the unit cell. J. Mol. Model. 2011, 17, 2569–2574. [Google Scholar] [CrossRef] [PubMed]
- Politzer, P.; Murray, J.S. Impact sensitivity and crystal lattice compressibility/free space. J. Mol. Model. 2014, 20. [Google Scholar] [CrossRef] [PubMed]
- Politzer, P.; Murray, J.S. Detonation performance and sensitivity: A quest for balance. Adv. Quantum Chem. 2014, 69, 1–30. [Google Scholar]
- Zhang, C.; Wang, X.; Huang, H. π-Stacked interactions in explosive crystals: Buffers against external mechanical stimuli. J. Am. Chem. Soc. 2008, 130, 8359–8365. [Google Scholar] [CrossRef] [PubMed]
- Brill, T.B.; Reese, C.O. Analysis of intra- and intermolecular interactions relating to the thermophysical behavior of a-, 6-, and b-octahydro-l,3,5,7-tetranltro-1,3,5,7-tetraazocine. J. Phys. Chem. 1980, 84, 1376–1380. [Google Scholar] [CrossRef]
- Taylor, D.E. Prediction of the Impact Sensitivity of Energetic Materials Using Symmetry Adapted Perturbation Theory; Army Research Laboratory Technical Report: Aberdeen, MD, USA, May 2011. [Google Scholar]
- Jones, T. Role of inter- and intramolecular bonding on impact sensitivity. J. Phys. Chem. A 2012, 116, 11008–11014. [Google Scholar] [CrossRef] [PubMed]
- Rice, B.M.; Hare, J.J. A quantum mechanical investigation of the relation between impact sensitivity and the charge distribution in energetic molecules. J. Phys. Chem. A 2002, 106, 1770–1783. [Google Scholar] [CrossRef]
- Murray, J.S.; Lane, P.; Politzer, P.; Bolduc, P.R. A relationship between impact sensitivity and the electrostatic potentials at the midpoints of C-NO2 bonds in nitroaromatics. Chem. Phys. Lett. 1990, 168, 135–139. [Google Scholar] [CrossRef]
- Politzer, P.; Murray, J.S.; Seminario, J.M.; Lane, P.; Grice, M.E.; Concha, M.C. Computational characterization of energetic materials. Comp. Theor. Chem. 2001, 573, 1–10. [Google Scholar] [CrossRef]
- Murray, J.S.; Concha, M.C.; Politzer, P. Links between surface electrostatic potentials of energetic molecules, impact sensitivities and C-NO2/N-NO2 bond dissociation energies. Mol. Phys. 2009, 107, 89–97. [Google Scholar] [CrossRef]
- Mullay, J. A relationship between impact sensitivity and molecular electronegativity. Propellants Explos. Pyrot. 1987, 12, 60–63. [Google Scholar] [CrossRef]
- Badders, N.R.; Wei, C.; Aldeeb, A.A.; Rogers, W.J.; Mannan, M.S. Predicting the impact sensitivities of polynitro compounds using quantum chemical descriptors. J. Energ. Mater. 2006, 24, 17–33. [Google Scholar] [CrossRef]
- Politzer, P.; Murray, J.S.; Lane, P.; Sjoberg, P.; Adolph, H.G. Shock-sensitivity relationships of nitramines and nitroaliphatics. Chem. Phys. Lett. 1991, 181, 78–82. [Google Scholar] [CrossRef]
- Keshavarz, M.H.; Pouretedal, H.R.; Semnani, A. Simple empirical method for prediction of impact sensitivity of selected class of explosives. J. Hazard. Mater. 2005, A124, 27–33. [Google Scholar] [CrossRef] [PubMed]
- Keshavarz, M.H. Prediction of impact sensitivity of nitroaliphatic, nitroaliphatic containing other functional groups and nitrate explosives. J. Hazard. Mater. 2007, 148, 648–652. [Google Scholar] [CrossRef] [PubMed]
- Keshavarz, M.H.; Pouretedal, H.R. Novel correlation for predicting impact sensitivity of nitroheterocyclic energetic molecules. J. Hazard. Mater. 2007, 141, 803–807. [Google Scholar] [CrossRef] [PubMed]
- Owens, F.J. Calculation of energy barriers for bond rupture in some energetic molecules. Theochem. J. Mol. Struct. 1996, 370, 11–16. [Google Scholar] [CrossRef]
- Rice, B.M.; Sahu, S.; Owens, F.J. Density functional calculations of bond dissociation energies for NO2 scission in some nitroaromatic molecules. Theochem. J. Mol. Struct. 2002, 583, 69–72. [Google Scholar] [CrossRef]
- Mathieu, D.; Alaime, T. Predicting impact sensitivities of nitro compounds on the basis of a semi-empirical rate constant. J. Phys. Chem. A 2014, 118, 9720–9726. [Google Scholar] [CrossRef] [PubMed]
- Koch, E.C. Acid-base interactions in energetic materials: I. the hard and soft acids and bases (HSAB) principle—Insights to reactivity and sensitivity of energetic materials. Propellants Explos. Pyrotech. 2005, 30, 5–16. [Google Scholar] [CrossRef]
- Kamlet, M.J.; Adolph, H.G. The relationship of impact sensitivity with structure of organic high explosives. II. Polynitroaromatic explosives. Propellants Explos. 1979, 4, 30–34. [Google Scholar] [CrossRef]
- Zhao, J.; Cheng, X.L.; He, B.; Yang, X.D. Neural networks study on the correlation between impact sensitivity and molecular structures for nitramine explosives. Struct. Chem. 2006, 17, 501–507. [Google Scholar]
- Keshavarz, M.H.; Jaafari, M. Investigation of the various structure parameters for predicting impact sensitivity of energetic molecules via artificial neural network. Propellants Explos. Pyrotech. 2006, 31, 216–225. [Google Scholar] [CrossRef]
- Nefati, H.; Cense, J.M.; Legendre, J.J. Prediction of the impact sensitivity by neural networks. J. Chem. Inf. Comput. Sci. 1996, 36, 804–810. [Google Scholar] [CrossRef]
- Kuklja, M.M. Quantum-chemical modeling of energetic materials: Chemical reactions triggered by defects, deformations, and electronic excitations. Adv. Quantum Chem. 2014, 69, 71–146. [Google Scholar]
- Sharia, O.; Kuklja, M.M. Ab Initio kinetics of gas phase decomposition reactions. J. Phys. Chem. A 2010, 114, 12656–12661. [Google Scholar] [CrossRef] [PubMed]
- Sharia, O.; Kuklja, M.M. Modeling thermal decomposition mechanisms in gaseous and crystalline molecular materials: application to β-HMX. J. Phys. Chem. B 2011, 115, 12677–12686. [Google Scholar] [CrossRef] [PubMed]
- Sharia, O.; Kuklja, M.M. Rapid materials degradation induced by surfaces and voids: Ab Initio modeling of β-octatetramethylene tetranitramine. J. Am. Chem. Soc. 2012, 134, 11815–11820. [Google Scholar] [CrossRef] [PubMed]
- Sharia, O.; Kuklja, M.M. Surface-enhanced decomposition kinetics of molecular materials illustrated with cyclotetramethylene-tetranitramine. J. Phys. Chem. C 2012, 116, 11077–11081. [Google Scholar] [CrossRef]
- Hohenberg, P.; Kohn, W. Inhomogeneous electron gas. Phys. Rev. 1964, 136, B864–B871. [Google Scholar] [CrossRef]
- Kohn, W.; Sham, L.J. Self-consistent equations including exchange and correlation effects. Phys. Rev. A 1965, 140, A1133–A1138. [Google Scholar]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [PubMed]
- Adamo, C.; Barone, V. Toward reliable density functional methods without adjustable parameters: The PBE0 model. J. Chem. Phys. 1999, 110, 6158–6169. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional thermochemistry. III. the role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Lee, C.; Yang, W.; Parr, R.G. Development of the colle-salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef]
- Gaussian 09, Revision B.01 ed; Gaussian, Inc.: Wallingford, CT, USA, 2009.
- Hratchian, H.P.; Schlegel, H.B. Accurate reaction paths using a hessian based predictor-corrector integrator. J. Chem. Phys. 2004, 120, 9918–9924. [Google Scholar] [CrossRef] [PubMed]
- Hratchian, H.P.; Schlegel, H.B. Using hessian updating to increase the efficiency of a hessian based predictor-corrector reaction path following method. J. Chem. Theory Comput. 2005, 1, 61–69. [Google Scholar] [CrossRef] [PubMed]
- Blöchl, P.E. Projector augmented-wave method. Phys. Rev. B 1994, 50, 17953–17979. [Google Scholar] [CrossRef]
- Kresse, G.; Futhmuller, J. Efficiency of Ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comput. Mater. Sci. 1996, 6, 15–50. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmuller, F. Efficient iterative schemes for Ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 1996, 54, 11169–11186. [Google Scholar] [CrossRef]
- Kresse, G.; Hafner, J. Ab Initio molecular dynamics for liquid metals. Phys. Rev. B 1993, 47, 558–561. [Google Scholar] [CrossRef]
- Elban, W.L. Surface energies of high explosives PETN and RDX from contact angle measurements. J. Mater. Sci. 1979, 14, 1008–1011. [Google Scholar]
- Yee, R.Y.; Adicoff, A.; Dibble, E.J. Surface properties of HMX crystal. In Proceedings of the 17th JANNAF Combustion Meeting, Hampton, WV, USA, 22–26 September 1980.
- Henkelman, G.; Uberuaga, B.P.; Jónsson, H. A climbing image nudged elastic band method for finding saddle points and minimum energy paths. J. Chem. Phys. 2000, 113, 9901–9904. [Google Scholar] [CrossRef]
- Hanggi, P.; Talkner, P.; Borkovec, M. Reaction-rate theory: Fifty years after Kramers. Rev. Mod. Phys. 1990, 62, 251–341. [Google Scholar]
- Garrett, B.C.; Truhlar, D.G. Generalized transition state theory. Classical mechanical theory and applications to collinear reactions of hydrogen molecules. J. Phys. Chem. 1979, 83, 1052–1079. [Google Scholar] [CrossRef]
- Sadhukhan, S.; Muñoz, D.; Adamo, C.; Scuseria, G.E. Predicting proton transfer barriers with density functional methods. Chem. Phys. Lett. 1999, 306, 83–87. [Google Scholar] [CrossRef]
- Nachimuthu, S.; Gao, J.; Truhlar, D.G. A benchmark test suite for proton transfer energies and its use to test electronic structure model chemistries. Chem. Phys. 2012, 400, 8–12. [Google Scholar] [CrossRef] [PubMed]
- See for example, Gruzdkov, Y.A.; Gupta, Y.M. Shock wave initiation of pentaerythritol tetranitrate single crystals: Mechanism of anisotropic sensitivity. J. Phys. Chem. A 2000, 104, 11169–11176. [Google Scholar]
- Zhang, G.; Weeks, B.L.; Zhang, X. Crystal growth of organic energetic materials: Pentaerythritol tetranitrate. Cent. Eur. J. Eng. 2012, 2, 336–346. [Google Scholar] [CrossRef]
- Roos, B.D.; Brill, T.B. Thermal decomposition of energetic materials 82. Correlations of gaseous products with the composition of aliphatic nitrate esters. Combust. Flame 2002, 128, 181–190. [Google Scholar] [CrossRef]
- Ng, W.L.; Field, J.E.; Hauser, H.M. Study of the thermal decomposition of pentaerythritol tetranitrate. J. Chem. Soc. Perkin Trans. 1976, 2, 637–639. [Google Scholar] [CrossRef]
- Andreev, K.K.; Kaidymov, B.I. Thermal decomposition of nitrate esters. II. Thermal decomposition of pentaerythritol tetranitrate. Russ. J. Phys. Chem. 1961, 35, 1324–1330. [Google Scholar]
- Chambers, D.M. Perspectives on Pentaerythritol Tetranitrate (PETN) Decomposition; UCRL-JC-148956; Lawrence Livermore National Laboratory: Livermore, CA, USA, July 2002. [Google Scholar]
- Robertson, A.J.B. The thermal decomposition of pentaerythritol tetranitrate, nitroglycerin, ethylenediamine dinitrate and ammonium nitrate. J. Soc. Chem. Ind. 1948, 67, 221–224. [Google Scholar] [CrossRef]
- Rogers, R.N.; Morris, E.D., Jr. On estimating activation energies with a differential scanning calorimeter. Anal. Chem. 1966, 38, 412–414. [Google Scholar] [CrossRef]
- Maycock, J.N.; Pai Verneker, V.R. Characterization of thermal and photosublimation of organic explosives by thermobarogravimetric techniques. Thermochim. Acta 1970, 1, 191–198. [Google Scholar] [CrossRef]
- Volltrauer, H.N. Real time low temperature decomposition of explosives—PETN. J. Hazard. Mater. 1982, 5, 353–357. [Google Scholar] [CrossRef]
- Oxley, J.C.; Smith, J.L.; Brady IV, J.E.; Brown, A.C. Characterization and analysis of tetranitrate esters. Propellants Explos. Pyrotech. 2012, 37, 24–39. [Google Scholar] [CrossRef]
- Tsyshevsky, R.V.; Sharia, O.; Kuklja, M.M. Thermal decomposition mechanisms of nitroesters: Ab Initio modeling of pentaerythritol tetranitrate. J. Phys. Chem. C 2013, 117, 18144–18153. [Google Scholar] [CrossRef]
- Liu, W.-G.; Zybin, S.V.; Dasgupta, S.; Klapötke, T.M.; Goddard, W.A., III. Explanation of the colossal detonation sensitivity of silicon pentaerythritol tetranitrate (Si-PETN) explosive. J. Am. Chem. Soc. 2009, 131, 7490–7491. [Google Scholar] [CrossRef] [PubMed]
- Hiskey, M.A.; Brower, K.R.; Oxley, J.C. Thermal decomposition of nitrate esters. J. Phys. Chem. 1991, 95, 3955–3960. [Google Scholar] [CrossRef]
- Choi, C.S.; Boutin, H.P. A study of the crystal structure of β-cyclotetramethylene tetranitramine by neutron diffraction. Acta. Crystallogr. B 1970, 26, 1235–1240. [Google Scholar] [CrossRef]
- Oyumi, Y.; Brill, T.B. Thermal decomposition of energetic materials 3. A high-rate, in situ, FTIR study of the thermolysis of RDX and HMX with pressure and heating rate as variables. Combust. Flame 1985, 62, 213–224. [Google Scholar] [CrossRef]
- Glascoe, E.A.; Hsu, P.C.; Springer, H.K.; de Haven, M.R.; Tan, N.; Turner, H.C. The response of the HMX-based material PBXN-9 to thermal insults: Thermal decomposition kinetics and morphological changes. Thermochim. Acta. 2011, 515, 58–66. [Google Scholar] [CrossRef]
- Brill, T.B.; James, K.J. Kinetics and mechanisms of thermal decomposition of nitroaromatic explosives. Chem. Rev. 1993, 93, 2667–2692. [Google Scholar] [CrossRef]
- Brill, T.B.; Gongwer, P.E.; Williams, G.K. Thermal decomposition of energetic materials. 66. Kinetic compensation effects in HMX, RDX, and NTO. J. Phys. Chem. 1994, 98, 12242–12247. [Google Scholar] [CrossRef]
- Sharia, O.; Tsyshevsky, R.; Kuklja, M.M. Surface-accelerated decomposition of δ-HMX. J. Phys. Chem. Lett. 2013, 4, 730–734. [Google Scholar] [CrossRef] [PubMed]
- Kuklja, M.M.; Tsyshevsky, R.V.; Sharia, O. Effect of polar surfaces on decomposition of molecular materials. J. Am. Chem. Soc. 2014, 136, 13289–13302. [Google Scholar] [CrossRef] [PubMed]
- Chakraborty, D.; Muller, R.P.; Dasgupta, S.; Goddard, W.A., III. Mechanism for unimolecular decomposition of HMX (1,3,5,7-tetranitro-1,3,5,7-tetrazocine), an Ab Initio study. J. Phys. Chem. A 2001, 105, 1302–1314. [Google Scholar] [CrossRef]
- Maksimov, Y.Y.; Apol’kova, V.N.; Braverman, O.V.; Solov’ev, A.I. Kinetics of the thermal decomposition of cyclotrimethylenetrinitramine and cyclotetramethylenetetranitramine in the gas phase. Russ. J. Phys. Chem. 1985, 59, 201–204. [Google Scholar]
- Burov, Y.M.; Nazin, G.M. Influence of structure on rate of decomposition of secondary nitramines in the gas phase. Kinet. Catal. 1982, 23, 5–10. [Google Scholar]
- Shaw, R.; Walker, F.E. Estimated kinetics and thermochemistry of some initial unimolecular reactions in the thermal decomposition of 1,3,5,7-tetranitro-1,3,5,7-tetraazacyclooctane in the gas phase. J. Phys. Chem. 1977, 81, 2572–2576. [Google Scholar] [CrossRef]
- Cobbledick, R.E.; Small, R.W.H. The crystal structure of the δ-form of 1,3,5,7-tetranitro-1,3,5,7-tetraazacyclooctane (δ-HMX). Acta. Crystallogr. B 1974, 30, 1918–1922. [Google Scholar] [CrossRef]
- Herrmann, M.; Engel, W.; Eisenreich, N. Thermal expansion, transitions, sensitivities and burning rates of HMX. Propellants Explos. Pyrotech. 1992, 17, 190–195. [Google Scholar] [CrossRef]
- Henson, B.F.; Asay, B.W.; Sander, R.K.; Son, S.F.; Robinson, J.M.; Dickson, P.M. Dynamic measurement of the HMX β–δ phase transition by second harmonic generation. Phys. Rev. Lett. 1999, 82, 1213–1216. [Google Scholar] [CrossRef]
- Henson, B.F.; Smilowitz, L.; Asay, B.W.; Dickson, P.M. The β–δ phase transition in the energetic nitramine octahydro-1,3,5,7-tetranitro-1,3,5,7-tetrazocine: Thermodynamics. J. Chem. Phys. 2002, 117, 3780–3788. [Google Scholar] [CrossRef]
- Henson, B.F.; Smilowitz, L.; Asay, B.W.; Dickson, P.M. The β–δ phase transition in the energetic nitramine-octahydro-1,3,5,7-tetranitro-1,3,5,7-tetrazocine: Kinetics. J. Chem. Phys. 2002, 117, 3789–3798. [Google Scholar] [CrossRef]
- Urtiew, P.A.; Forbes, J.W.; Tarver, C.M.; Vandersall, K.S.; Garcia, F.; Greenwood, D.W.; Hsu, P.C.; Maienschein, J.L. Shock sensitivity of LX-04 containing delta phase HMX at elevated temperatures. AIP Conf. Proc. 2004, 706, 1053–1056. [Google Scholar]
- Asay, B.W.; Henson, B.F.; Smilowitz, L.B.; Dickson, P.M. On the difference in impact sensitivity of β and δ HMX. J. Energ. Mater. 2003, 21, 223–235. [Google Scholar] [CrossRef]
- Willey, T.M.; Lauderbach, L.; Gagliardi, F.; van Buuren, T.; Glascoe, E.A.; Tringe, J.W.; Lee, J.R.I.; Springer, H.K.; Ilavsky, J. Mesoscale evolution of voids and microstructural changes in HMX-based explosives during heating through the β-δ phase transition. J. Appl. Phys. 2015, 118. [Google Scholar] [CrossRef]
- Tarver, C.M.; Tran, T.D. Thermal decomposition models for HMX-based plastic bonded explosives. Combust. Flame 2004, 137, 50–62. [Google Scholar] [CrossRef]
- Brill, T.B.; Karpowicz, R.J. Solid phase transition kinetics. The role of intermolecular forces in the condensed-phase decomposition of octahydro-1,3,5,7-tetranitro-1,3,5,7-tetrazocine. J. Phys. Chem. 1982, 86, 4260–4265. [Google Scholar] [CrossRef]
- Karpowicz, R.J.; Brill, T.B. Kinetic data for solid phase transitions by Fourier transform infrared spectroscopy. Appl. Spectrosc. 1983, 37, 79–81. [Google Scholar] [CrossRef]
- Whelan, D.J.; Fitzgerald, M.R. A Reassessment of the Kinetics of the Thermal Decomposition of the High Explosive, Delta-HMX, in the Range 508 K to 524 K, as Studied by Isothermal Gravimetry; DSTO-TR-0635; DSTO Aeronautical and Maritime Research Laboratory: Melbourne, Australia, 1998. [Google Scholar]
- Bulusu, S.; Weinstein, D.I.; Autera, J.R.; Velicky, R.W. Deuterium kinetic isotope effect in the thermal decomposition of 1,3,5-trinitro-1,3,5-triazacyclohexane and 1,3,5,7-tetranitro-1,3,5,7-tetraazacyclooctane: its use as an experimental probe for their shock-induced chemistry. J. Phys. Chem. 1986, 90, 4121–4126. [Google Scholar] [CrossRef]
- Fox, P.G.; Hutchinson, R.W. Slow thermal decomposition. In Energetic Materials, Physics and Chemistry of Inorganic Azides; Fair, H.D., Walker, R.F., Eds.; Plenum Press: New York, NY, USA, 1977; p. 255. [Google Scholar]
- Latypov, N.V.; Bergman, J.; Langlet, A.; Wellmar, U.; Bemm, U. Synthesis and reactions of 1,1-diamino-2,2-dinitroethylene. Tetrahedron 1998, 54, 11525–11536. [Google Scholar] [CrossRef]
- Bemm, U.; Östmark, H. 1,1-Diamino-2,2-dinitroethylene: A novel energetic material with infinite layers in two dimensions. Acta. Crystallogr. 1998, C54, 1997–1999. [Google Scholar] [CrossRef]
- Östmark, H.; Langlet, A.; Bergman, H.; Wingborg, U.; Wellmar, U.; Bemm, U. FOX-7—A new explosive with low sensitivity and high performance. In Proceedings of 11th International Symposium on Detonation, Snowmass, CO, USA, 1998.
- Lochert, I.J. FOX-7—A New Insensitive Explosive; ADA399359; DSTO Aeronautical and Maritime Research Laboratory: Fishermans Bend, Australia, November 2001. [Google Scholar]
- Burnham, A.K.; Weese, R.K.; Wang, R.; Kwok, Q.S.M.; Jones, D.E.G. Thermal properties of FOX-7. In Proceedings of the 36th International Annual Conference of ICT & 32nd International Pyrotechnics Seminar, Karlsruhe, Germany, 28 June–1 July 2005.
- De Klerk, W.; Popescu, C.; van der Heijden, A. Study on the decomposition kinetics of FOX-7 and HNF. J. Therm. Anal. Cal. 2003, 72, 955–966. [Google Scholar] [CrossRef]
- Politzer, P.; Concha, M.C.; Grice, M.E.; Murray, J.S.; Lane, P.; Habibollazadeh, D. Computational investigation of the structures and relative stabilities of amino/nitro derivatives of ethylene. Theochem. J. Mol. Struct. 1998, 452, 75–83. [Google Scholar] [CrossRef]
- Kimmel, A.V.; Sushko, P.V.; Shluger, A.L.; Kuklja, M.M. Effect of charged and excited states on the decomposition of 1,1-diamino-2,2-dinitroethylene molecules. J. Chem. Phys. 2007, 126, 234711–234721. [Google Scholar] [CrossRef] [PubMed]
- Gindulyte, A.; Massa, L.; Huang, L.; Karle, J. Proposed mechanism of 1,1-diamino-dinitroethylene decomposition: a density functional theory study. J. Phys. Chem. A 1999, 103, 11045–11051. [Google Scholar] [CrossRef]
- Kuklja, M.M.; Rashkeev, S.N. Self-accelerated mechanochemistry in nitroarenes. J. Phys. Chem. Lett. 2010, 1, 363–367. [Google Scholar] [CrossRef]
- Booth, R.S.; Butler, L.J. Thermal decomposition pathways for 1,1-diamino-2,2-dinitroethene (FOX-7). J. Chem. Phys. 2014, 141. [Google Scholar] [CrossRef] [PubMed]
- Kiselev, V.G.; Gritsan, N.P. Unexpected primary reactions for thermolysis of 1,1-diamino-2,2-dinitroethylene (FOX-7) revealed by Ab Initio calculations. J. Phys. Chem. A 2014, 118, 8002–8008. [Google Scholar] [CrossRef] [PubMed]
- Gibbs, T.R.; Popolato, A. (Eds.) LASL Explosive Property Data; University of California Press: Oakland, CA, USA, 1980.
- Rogers, R.N.; Janney, J.L.; Ebinger, M.H. Kinetic-isotope effects in thermal explosions. Thermochim. Acta 1982, 59, 287–297. [Google Scholar] [CrossRef]
- Stolovy, A.; Jones, E.C., Jr.; Aviles, J.B., Jr.; Namenson, A.I.; Fraser, W.A. Exothermic reactions in TATB initiated by an electron beam. J. Chem. Phys. 1983, 78, 229–235. [Google Scholar] [CrossRef]
- Wu, C.J.; Fried, L.E. Ring closure mediated by intramolecular hydrogen transfer in the decomposition of a push-pull nitroaromatic: TATB. J. Phys. Chem. A 2000, 104, 6447–6452. [Google Scholar] [CrossRef]
- Shao, J.; Cheng, X.; Yang, X. The C−NO2 bond dissociation energies of some nitroaromatic compounds: DFT study. Struct. Chem. 2006, 17, 547–550. [Google Scholar] [CrossRef]
- Manaa, M.R.; Fried, L.E. The reactivity of energetic materials under high pressure and temperature. In Energetic Materials; Sabin, J.R., Ed.; Academic Press: Cambridge, MA, USA, 2014. [Google Scholar]
- Kimmel, A.V.; Sushko, P.V.; Shluger, A.L.; Kuklja, M.M. Effect of molecular and lattice structure on hydrogen transfer in molecular crystals of diamino-dinitroethylene and triamino-trinitrobenzene. J. Phys. Chem. A 2008, 112, 4496–4500. [Google Scholar] [CrossRef] [PubMed]
- Tsyshevsky, R.; Pagoria, P.; Zhang, M.; Racoveanu, A.; de Hope, A.; Parrish, D.; Kuklja, M.M. Searching for low-sensitivity cast-melt high-energy-density materials: Synthesis, characterization, and decomposition kinetics of 3,4-bis(4-nitro-1,2,5-oxadiazol-3-yl)-1,2,5-oxadiazole-2-oxide. J. Phys. Chem. C 2015, 119, 3509–3521. [Google Scholar] [CrossRef]
- Zhao, F.Q.; Chen, P.; Hu, R.Z.; Luo, Y.; Zhang, Z.Z.; Zhou, Y.S.; Yang, X.W.; Gao, Y.; Gao, S.L.; Shi, Q.Z. Thermochemical properties and non-isothermal decomposition reaction kinetics of 3,4-dinitrofurazanfuroxan (DNTF). J. Hazard. Mater. 2004, 113, 67–71. [Google Scholar]
- Zeldovich, Y.B. On The Theory of the Propagation of Detonations on Gaseous System; NACA-TM-1261; National Advisory Committee for Aeronautics: Washington, DC, USA, 1950. (In Russian) [Google Scholar]
- Von Neumann, J. Theory of Detonation Waves; ADB967734; Institute for Advanced Study: Princeton, NJ, USA, 1942. [Google Scholar]
- Lee, E.L.; Tarver, C.M. Phenomenological model of shock initiation in heterogeneous explosives. Phys. Fluids 1980, 23. [Google Scholar] [CrossRef]
- McGuire, R.R.; Tarver, C.M. Chemical-Decomposition Models for the Thermal Explosion of Confined HMX, TATB, RDX, and TNT Explosives. In Proceedings of the 7th Symposium (International) on Detonation, Annapolis, MD, USA, 16–19 June 1981.
- Tarver, C.M.; Forbes, J.W.; Urtiew, P.A. Nonequilibrium Zeldovich-von Neumann-Doring theory and reactive flow modeling of detonation. Russ. J. Phys. Chem. B 2007, 1, 39–45. [Google Scholar] [CrossRef]
- Kuklja, M.M.; Rashkeev, S.N. Interplay of decomposition mechanisms at shear-strain interface. J. Phys. Chem. C 2009, 113, 17–20. [Google Scholar] [CrossRef]
- Tsyshevsky, R.V.; Rashkeev, S.N.; Kuklja, M.M. Defect states at organic-inorganic interfaces: Insight from first principles calculations for pentaerythritol tetranitrate on MgO surface. Surf. Sci. 2015, 19, 637–638. [Google Scholar] [CrossRef]
- Tsyshevsky, R.V.; Zverev, A.; Mitrofanov, A.; Rashkeev, S.N.; Kuklja, M.M. Photochemistry of the α-Al2O3-PETN interface. Molecules 2016. submitted for publication. [Google Scholar]
- Aluker, E.D.; Krechetov, A.G.; Mitrofanov, A.Y.; Nurmukhametov, D.R.; Kuklja, M.M. Laser initiation of energetic materials: Selective photoinitiation regime in pentaerythritol tetranitrate. J. Phys. Chem. C 2011, 115, 6893–6901. [Google Scholar] [CrossRef]
- Aluker, E.D.; Krechetov, A.G.; Mitrofanov, A.Y.; Zverev, A.S.; Kuklja, M.M. Understanding limits of the thermal mechanism of laser initiation of energetic materials. J. Phys. Chem. C 2012, 116, 24482–24486. [Google Scholar] [CrossRef]
- Aluker, E.D.; Krechetov, A.G.; Mitrofanov, A.Y.; Zverev, A.S.; Kuklja, M.M. Topography of photochemical initiation in molecular materials. Molecules 2013, 18, 14148–14160. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.A.; Carter, E.A. Orbital-free kinetic-energy density functional theory. In Theoretical Methods in Condensed Phase Chemistry; Schwartz, S.D., Ed.; Springer: Dordrecht, The Netherlands, 2002; pp. 117–184. [Google Scholar]
- Vikram, G.; Bhattacharya, K.; Ortiz, M. Quasi-continuum orbital-free density-functional theory: A route to multi-million atom non-periodic DFT calculation. J. Mech. Phys. Solids 2007, 55, 697–718. [Google Scholar]
- Dreizler, R.M.; Gross, E.K.U. Density Functional Theory: An Approach to the Quantum Many-Body Problem; Springer-Verlag: Berlin, Germany, 2012; p. 304. [Google Scholar]
- Taylor, D.E.; Rice, B.M. Quantum-informed multiscale M & S for energetic materials. In Energetic Materials; Sabin, J.R., Ed.; Academic Press: Cambridge, MA, USA, 2014. [Google Scholar]

















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
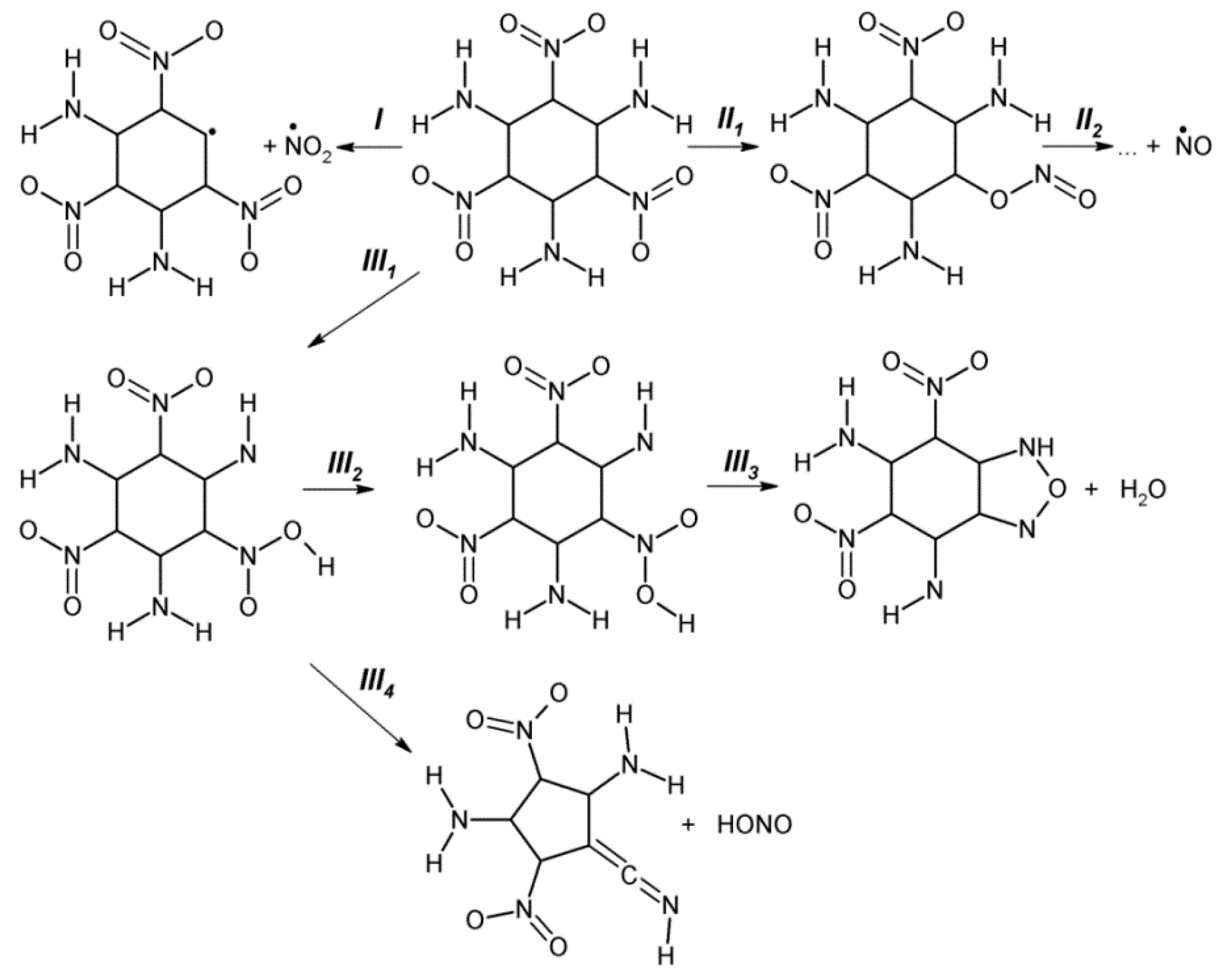{kind=link}
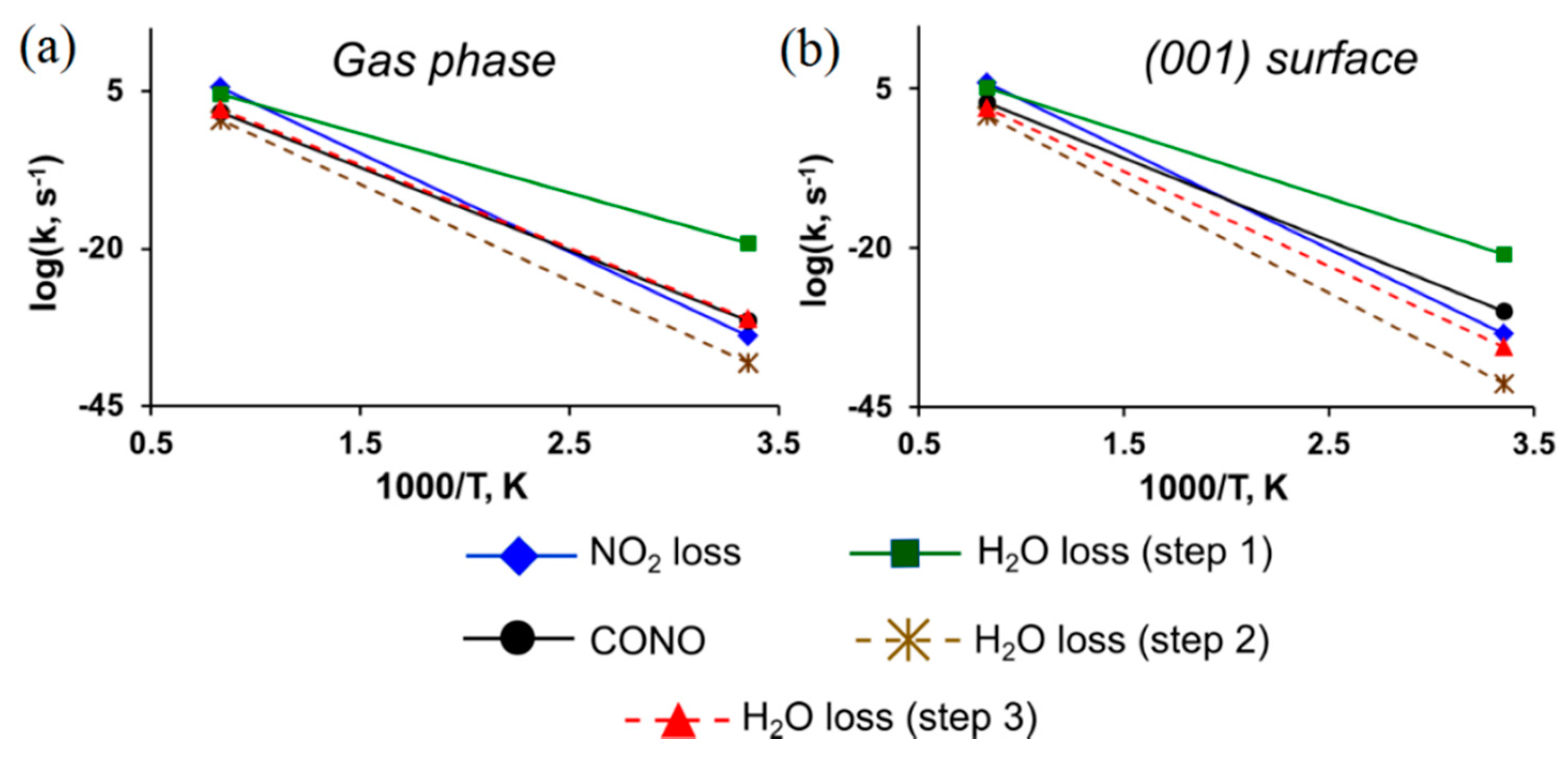{kind=link}
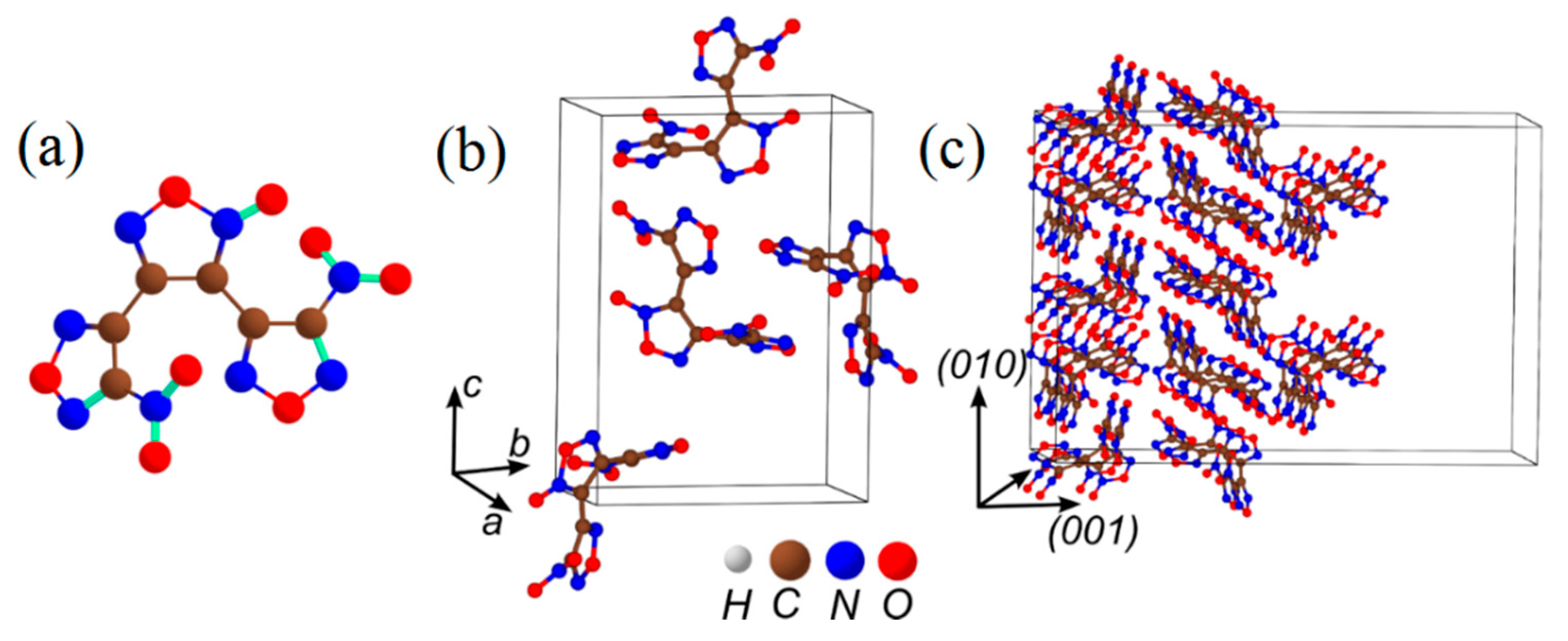{kind=link}
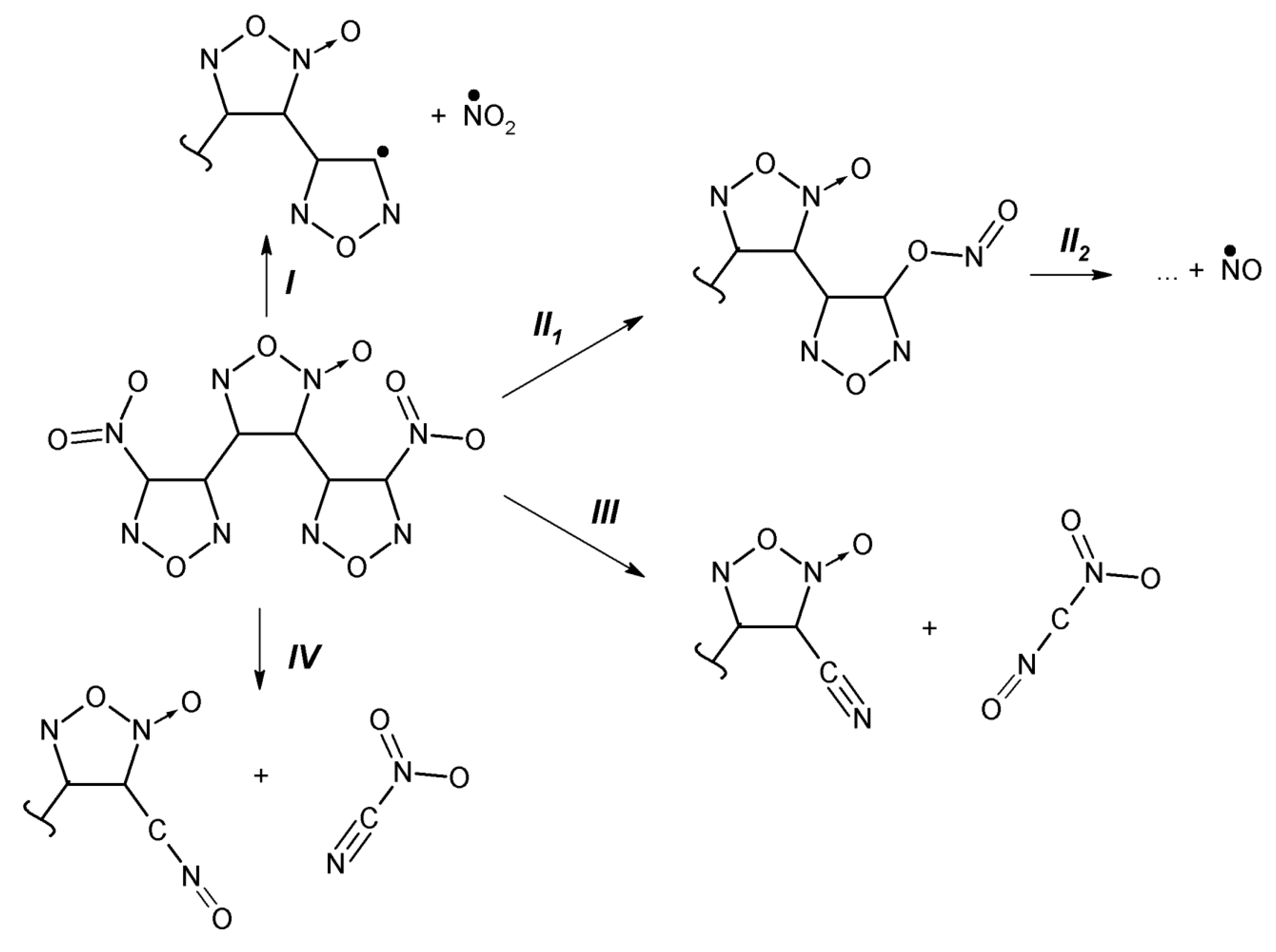{kind=link}
{kind=link}
| Reaction | Isolated Molecule | (mnl) Crystal Surface | ||||||
|---|---|---|---|---|---|---|---|---|
| E | EZPE | log A | E | EZPE | log A | |||
| PETN (C5H8N4O12) | (101) | |||||||
| I | NO2• loss | 41.8 (41.8) a | 36.6 [34.9] b | 17.5 | 40.0 (40.0) | 35.3 | 17.8 | |
| II | HONO | 33.6 (−18.6) | 28.8 [40.5] | 13.4 | 34.6 (−10.4) | 29.5 [40.9] | 14.2 | |
| β-HMX (C4H8N8O8) | (100) | |||||||
| I | NO2• loss | 44.8 (44.8) | 40.2 [40.9] | 18.3 | 40.1 (40.1) | 37.4 | 17.3 | |
| II | HONO | 40.4 (−2.4) | 35.8 [47.1] | 13.4 | 41.0 (−3.2) | 38.1 [47.7] | 14.8 | |
| δ-HMX (C4H8N8O8) | Polar (001) | |||||||
| I | NO2• loss | charge state [0] b | 45.1 (45.1) | 40.3 [41.5] | 18.4 | - | ||
| [+] c | 12.7 (−24.3) | - [10.1] | 13.4 | ~20.7 (−6.0) | ||||
| [−] c | 17.0 (17.0) | - [15.4] | - | ~20.1 (20.1) | ||||
| II | HONO | [0] b | 38.0 (0.7) | 33.3 [44.1] | 13.3 | - | ||
| [+] c | 12.7 (−13.6) | - [10.1] | 13.4 | ~20.7 (12.9) | ||||
| DADNE (C2H4N4O4) | (010) | |||||||
| I | NO2• loss | 69.1 (69.1) | 65.4 [68.6] | 19.0 | 66.4 (66.4) | 62.3 | 19.0 | |
| II | CONO | 54.3 (−1.0) | 52.1 [61.5] | 13.5 | 55.3 (0.1) | 52.6 [64.6] | 13.8 | |
| III | 1 | CH2N2 loss (step1) | 49.0 (14.7) | 45.5 [49.4] | 14.3 | 57.3 (35.2) | 53.3 [62.2] | 14.0 |
| 2 | CH2N2 loss (step2) | 34.0 (14.4) | 32.1 [34.4] | 14.9 | 56.5 (38.8) | 53.4 [60.7] | 15.4 | |
| 3 | CH2N2 loss (step3) | 44.9 (34.6) | 41.0 [47.9] | 13.7 | 52.9 (44.5) | 48.6 [58.8] | 15.2 | |
| 4 | NO2• loss from AIDNE | 50.7 (50.7) | 46.2 [47.8] | 19.1 | 67.1 (67.1) | 61.9 | 19.3 | |
| TATB (C6H6N6O6) | (001) | |||||||
| I | NO2• loss | 74.9 (74.9) | 70.7 [65.8] | 18 | 75.0 (75.0) | 71.1 | 19 | |
| II | CONO | 54.9 (4.3) | 52.5 [58.6] | 13.1 | 54.0 (5.6) | 51.5 [58.0] | 13.8 | |
| III | 1 | H2O loss (step1) | 44.5 (37.5) | 43.2 [42.1] | 13.0 | 49.0 (43.0) | 47.3 [46.1] | 13.8 |
| 2 | H2O loss (step2) | 65.3 (40.9) | 62.6 [68.6] | 13.4 | 74.6 (44.5) | 71.3 [74.3] | 14.7 | |
| 3 | H2O loss (step3) | 52.8 (41.7) | 47.5 [58.8] | 13.3 | 59.1 (49.5) | 53.2 [65.5] | 14.3 | |
| 4 | HONO loss | 95.4 (42.5) | 91.3 [94.1] | 13.4 | - | - | - | |
| BNFF (C6N8O8) | (001) | |||||||
| I | NO2• loss | 63.2 (63.2) | 59.3 [56.2] | 17.9 | 60.1 (60.1) | 56.2 | 18.4 | |
| II | CONO | 49.5 (−9.5) | 47.2 [54.3] | 13.4 | 49.1 (−11.0) | 46.8 [51.4] | 13.3 | |
| III | RC CN2O3 | 48.7 (36.4) | 45.7 [50.5] | 15.2 | 48.0 (31.7) | 45.1 [49.4] | 15.5 | |
| IV | RC CN2O2 | 47.1 (36.6) | 44.0 [48.9] | 16.3 | 44.2 (31.2) | 41.4 [45.8] | 15.4 | |
© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tsyshevsky, R.V.; Sharia, O.; Kuklja, M.M. Molecular Theory of Detonation Initiation: Insight from First Principles Modeling of the Decomposition Mechanisms of Organic Nitro Energetic Materials. Molecules 2016, 21, 236. https://doi.org/10.3390/molecules21020236
Tsyshevsky RV, Sharia O, Kuklja MM. Molecular Theory of Detonation Initiation: Insight from First Principles Modeling of the Decomposition Mechanisms of Organic Nitro Energetic Materials. Molecules. 2016; 21(2):236. https://doi.org/10.3390/molecules21020236
Chicago/Turabian StyleTsyshevsky, Roman V., Onise Sharia, and Maija M. Kuklja. 2016. "Molecular Theory of Detonation Initiation: Insight from First Principles Modeling of the Decomposition Mechanisms of Organic Nitro Energetic Materials" Molecules 21, no. 2: 236. https://doi.org/10.3390/molecules21020236