
Focus on Chirality of HIV-1 Non-Nucleoside Reverse Transcriptase Inhibitors


Abstract
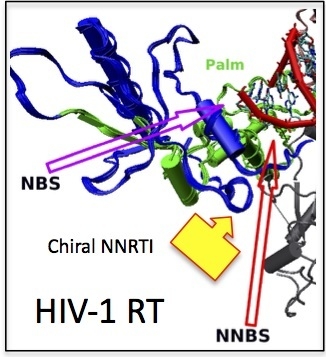
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
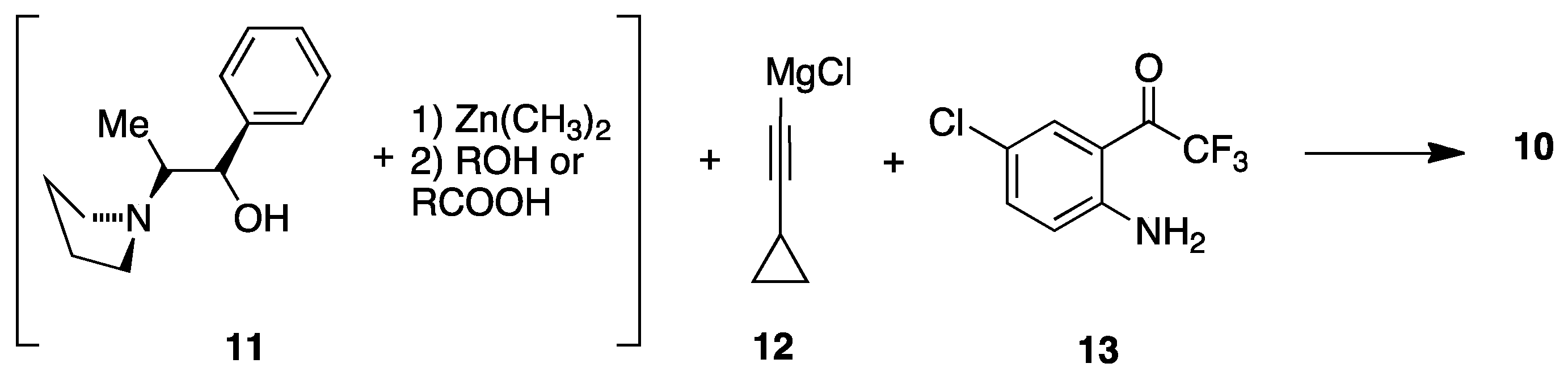{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
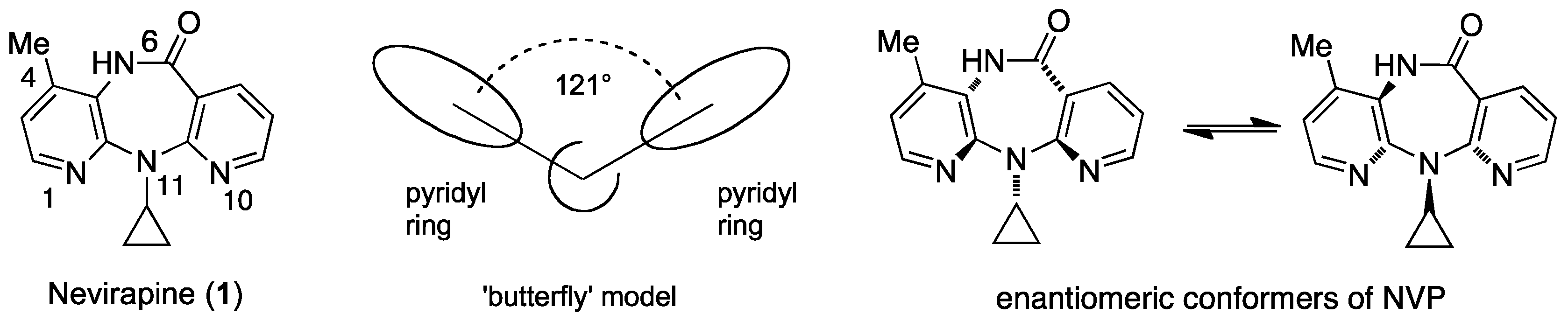
2. Nevirapine
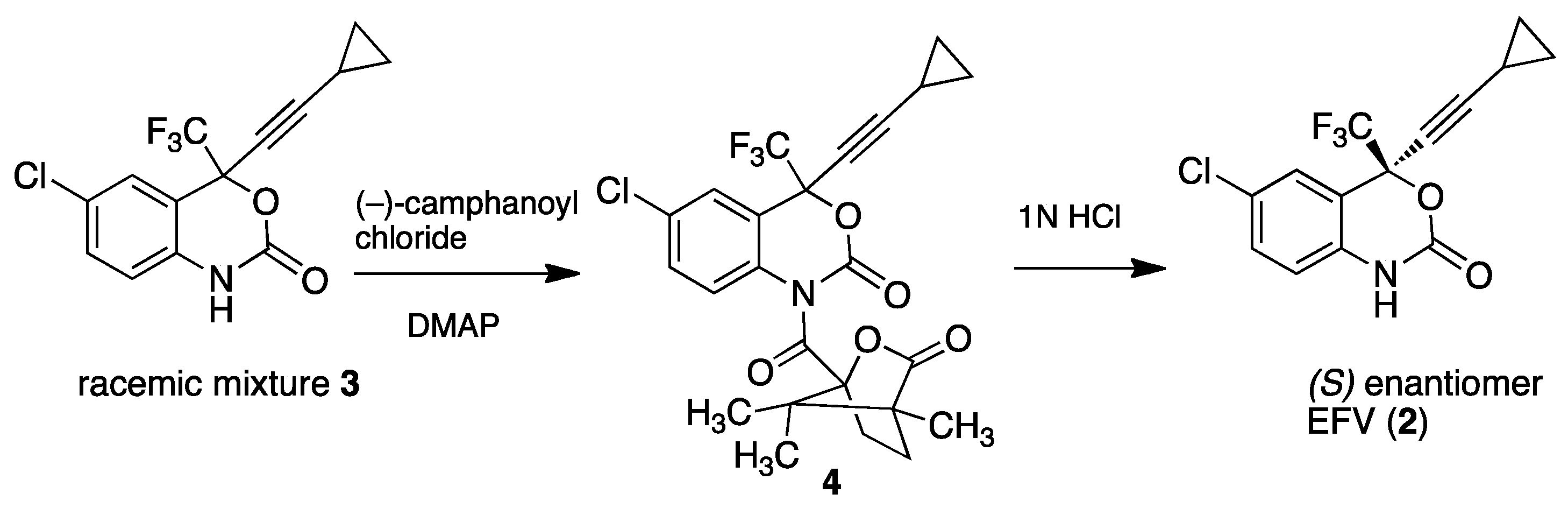
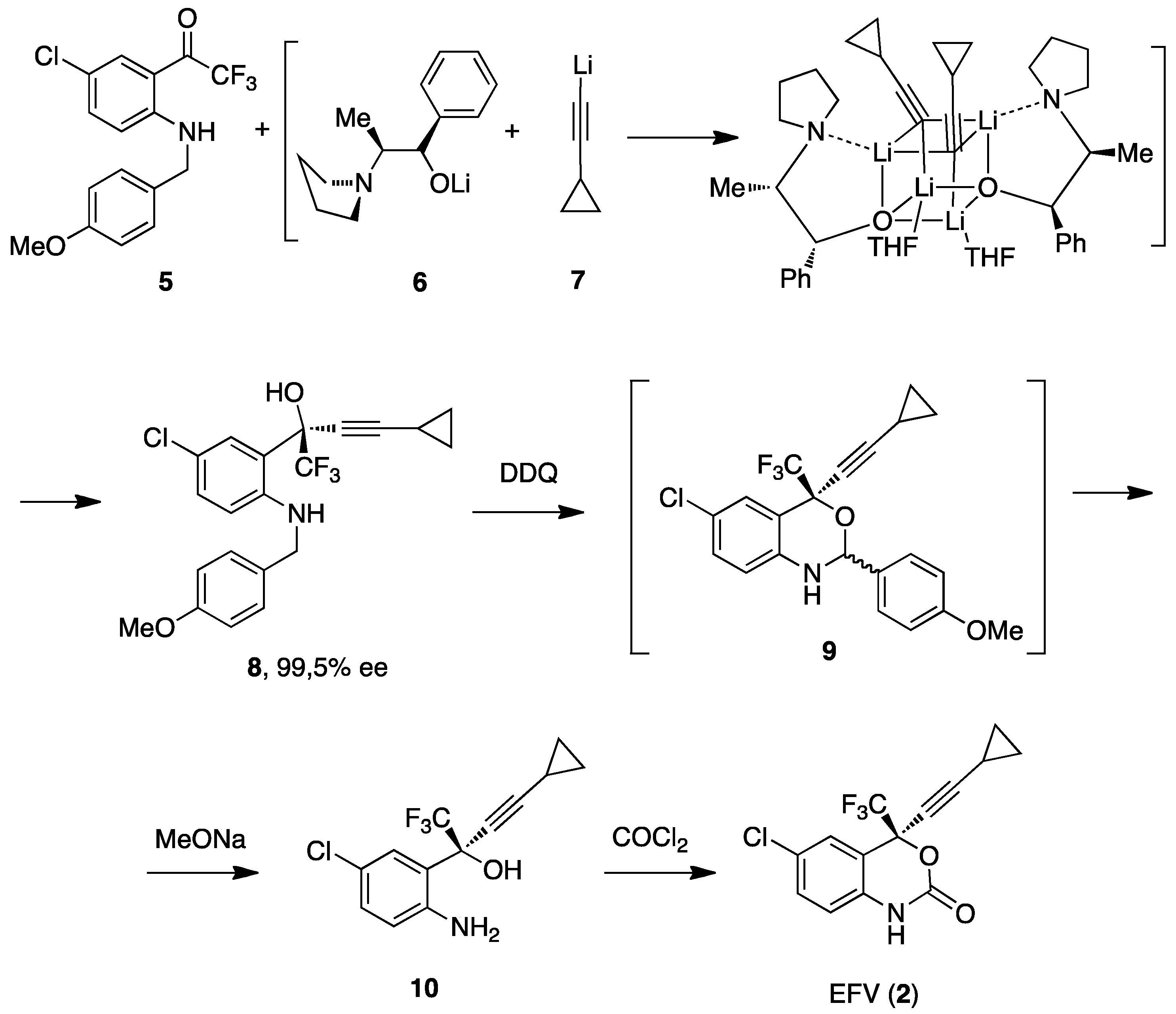
3. Efavirenz
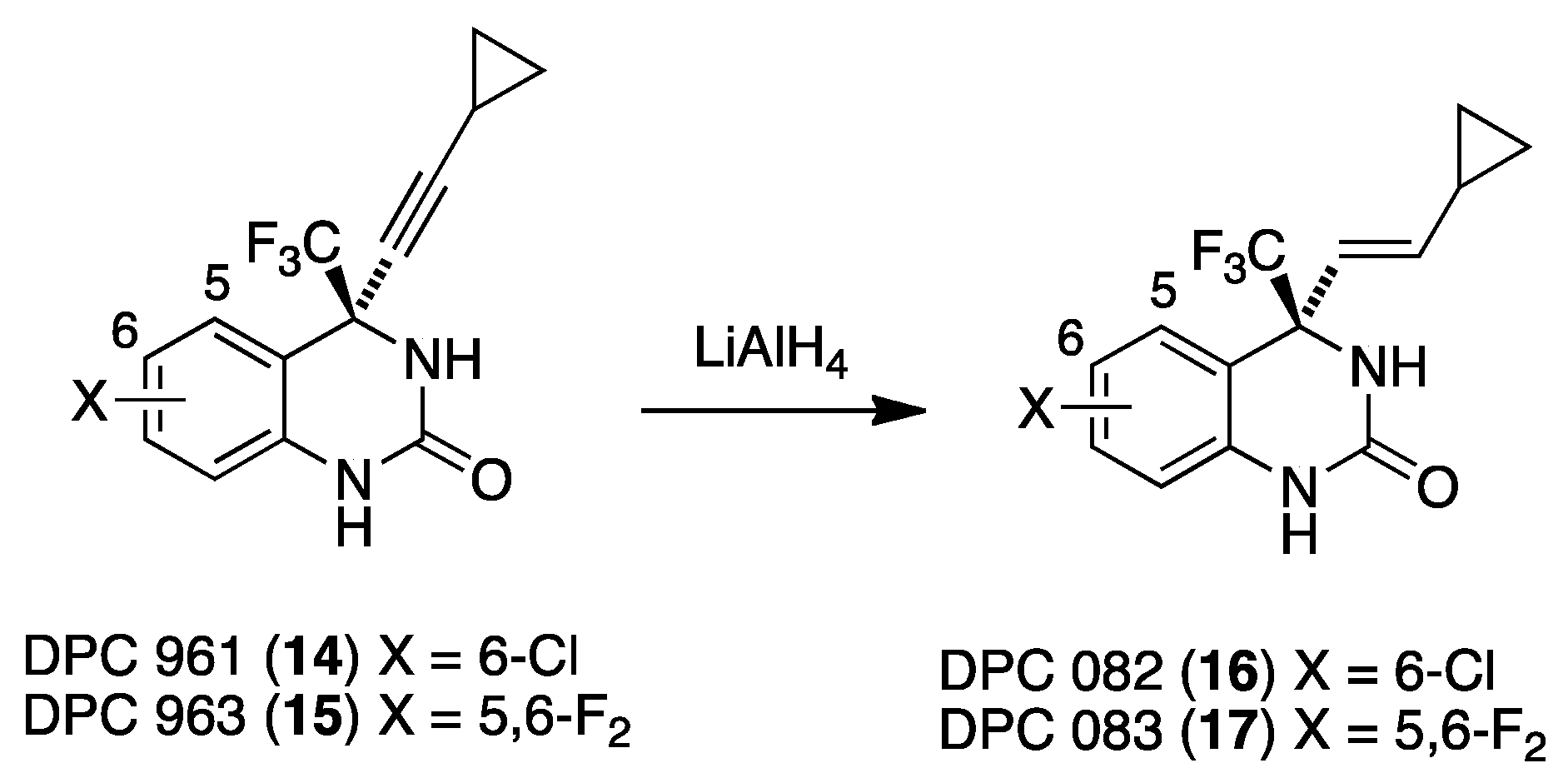
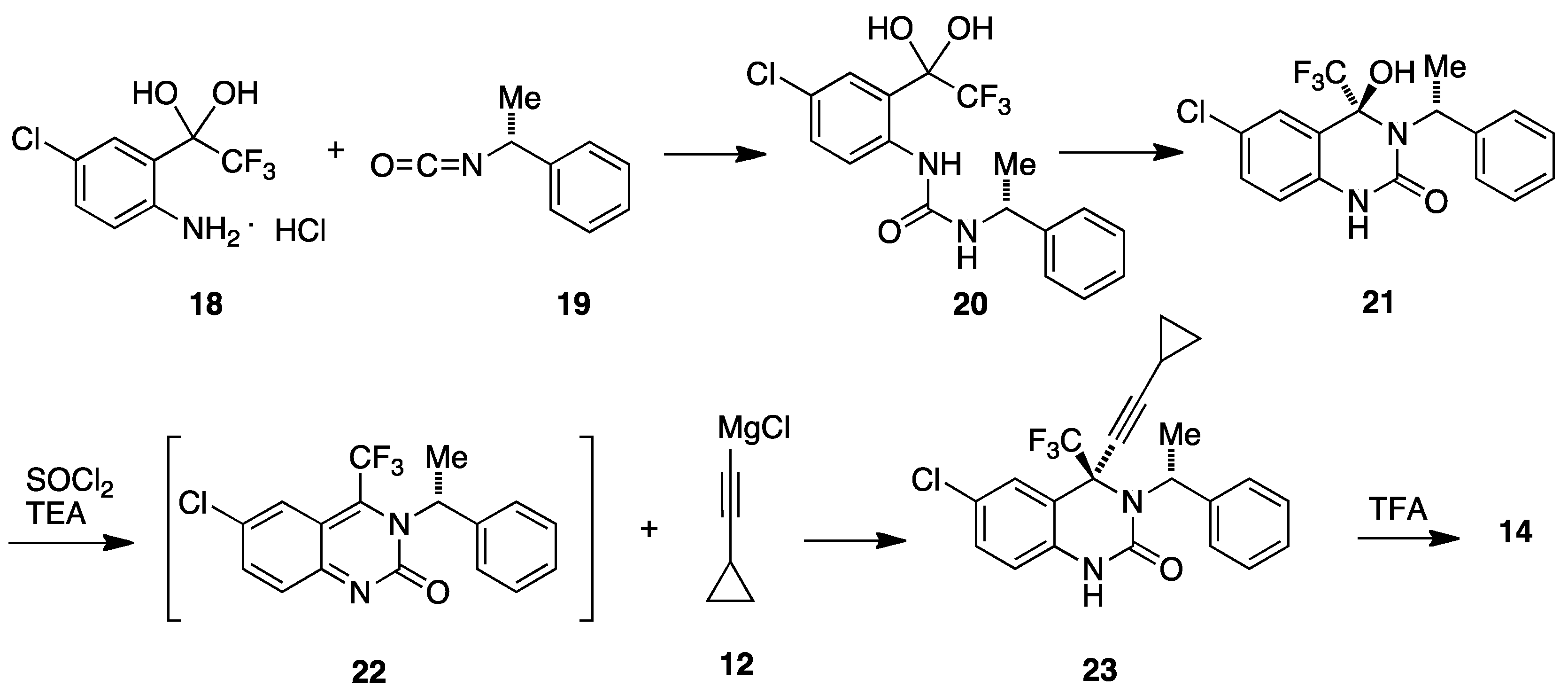
4. DuPont Compounds (DPC)
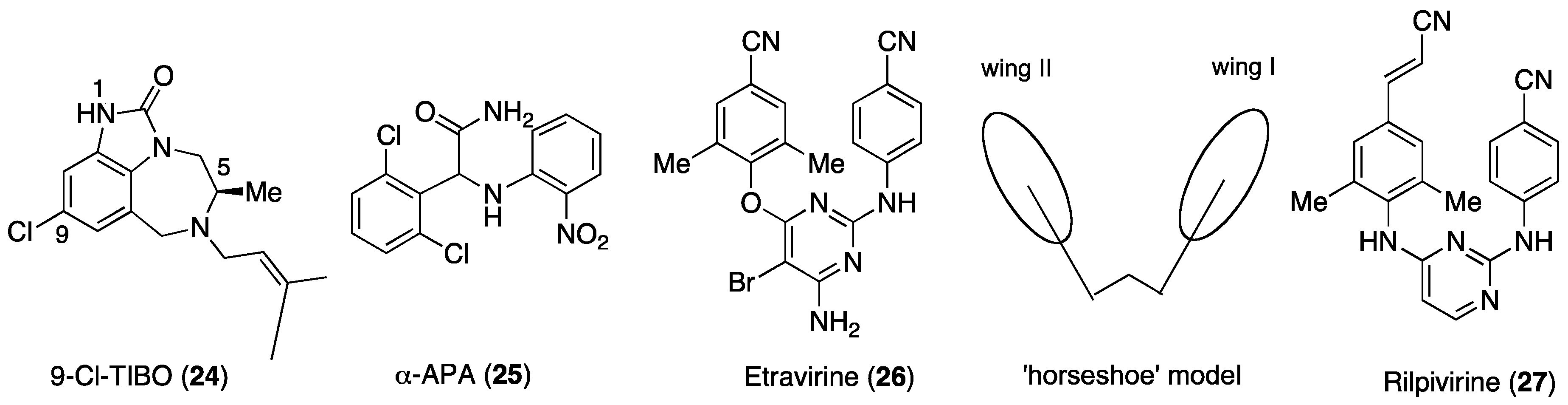
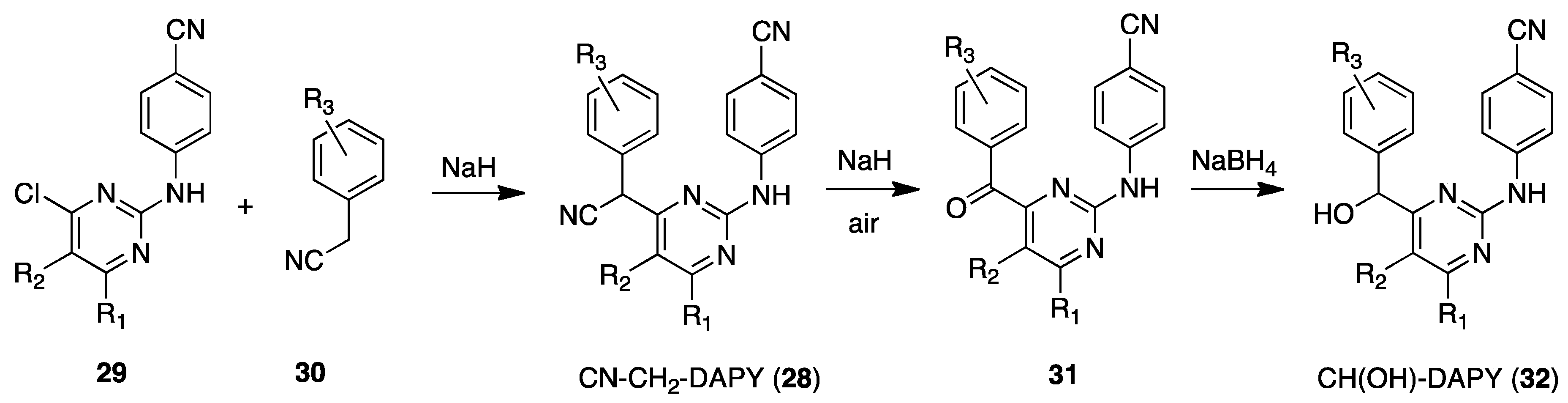
5. DAPY
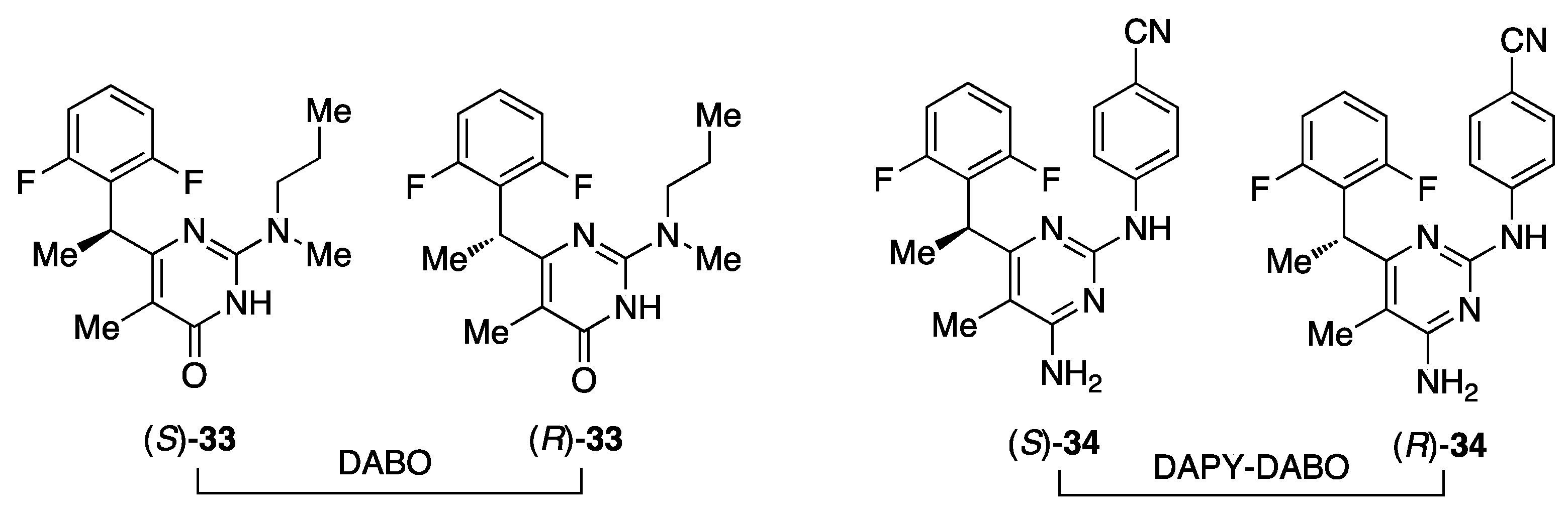
6. DABO and DAPY-DABO Hybrids
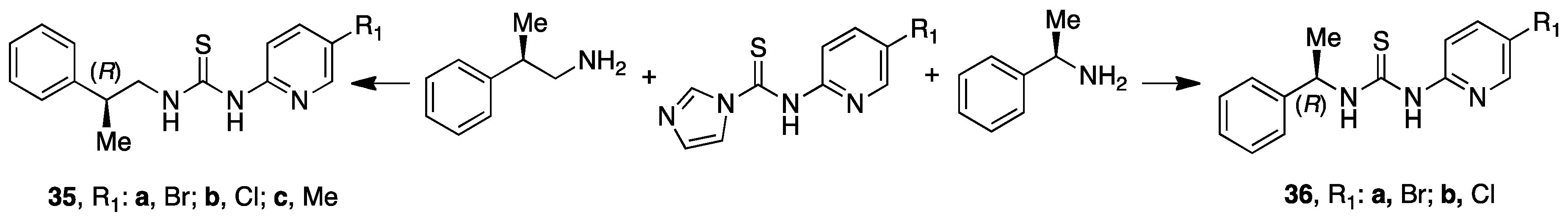
7. PETT
8. IAS
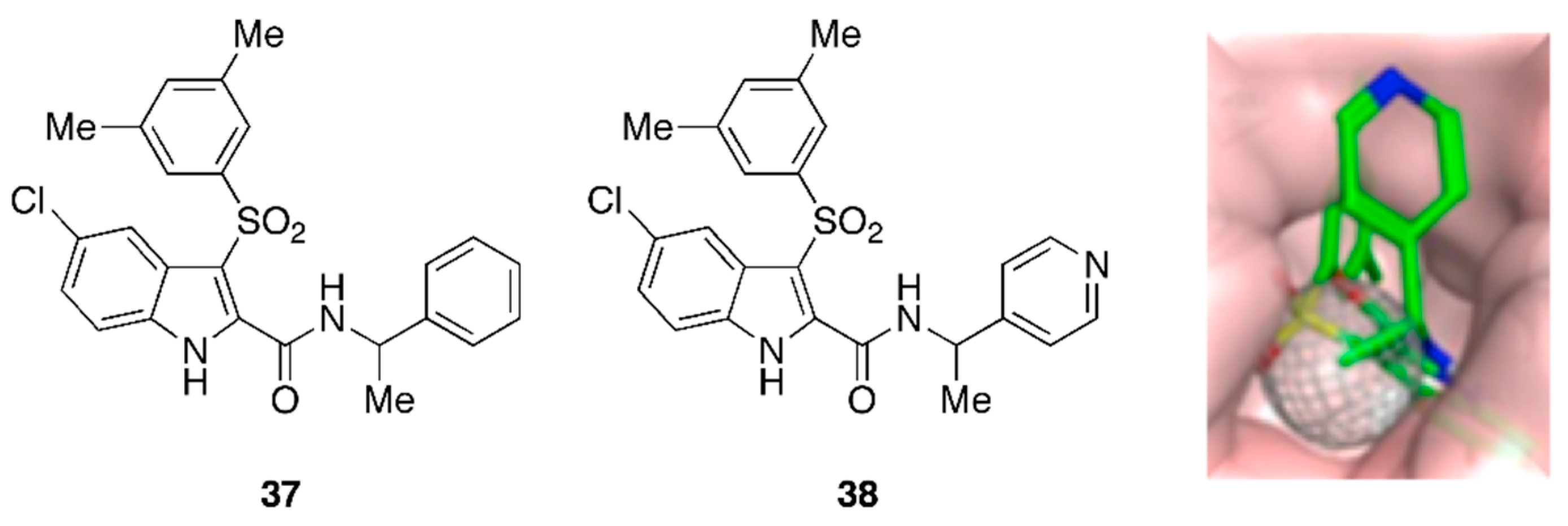
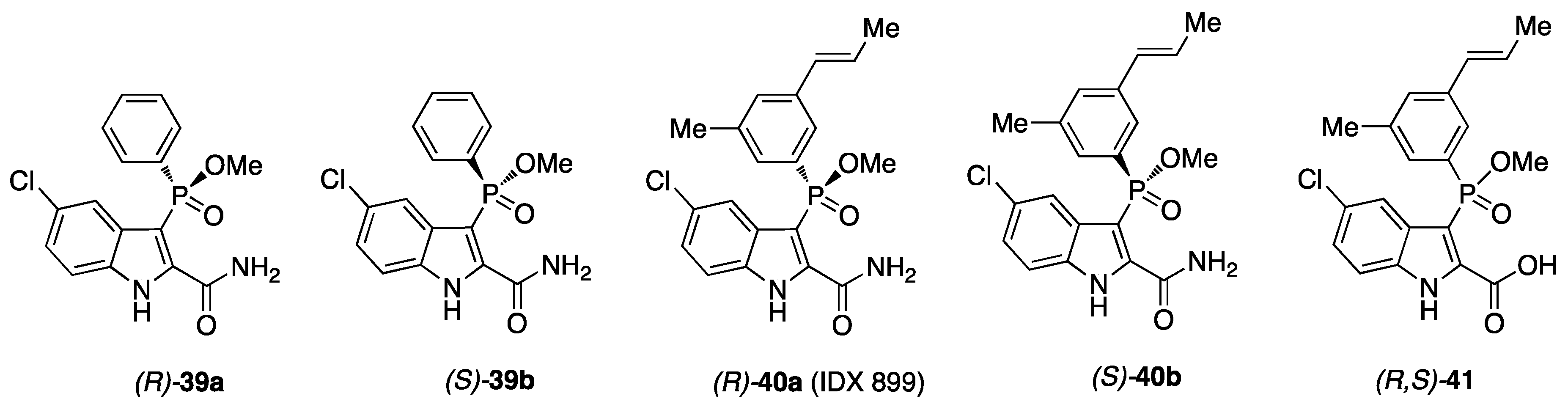
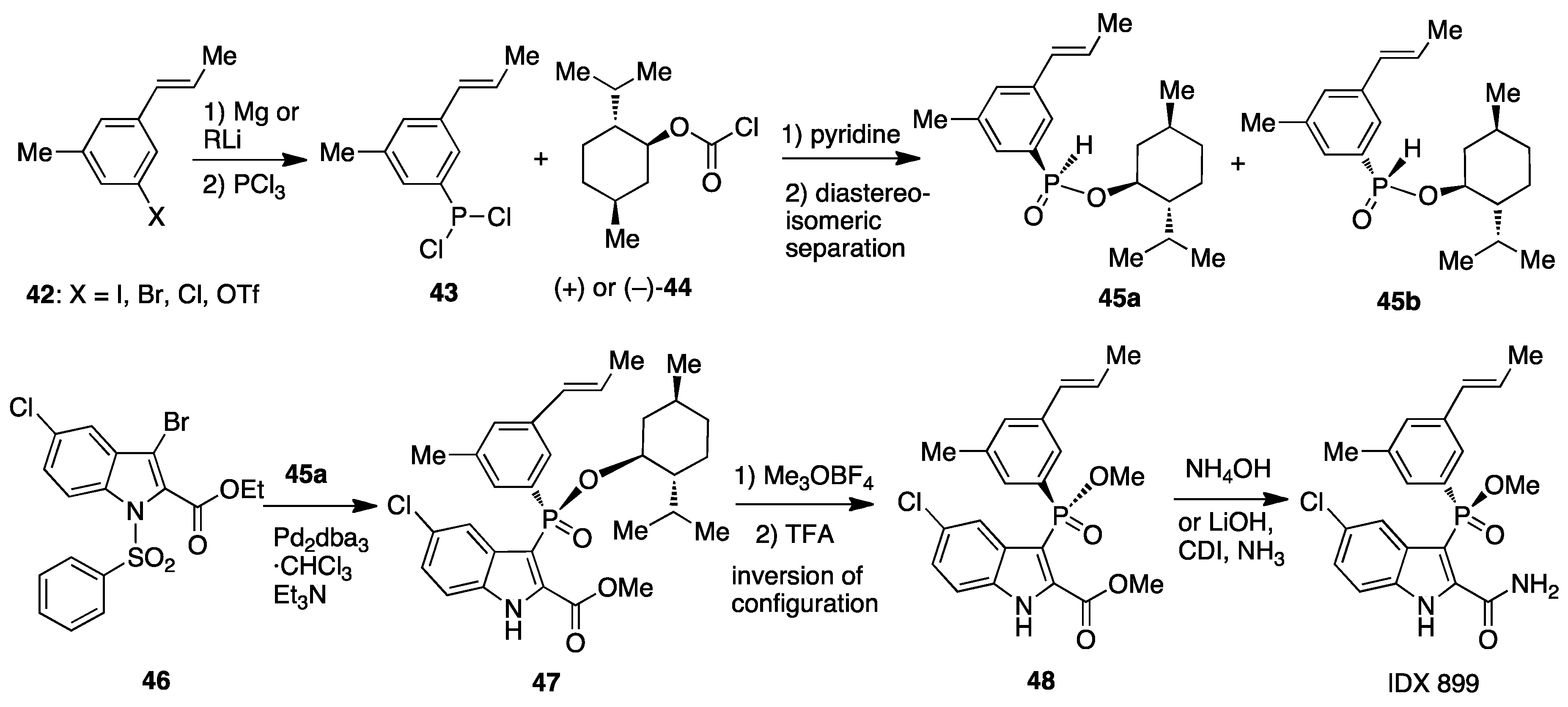
9. API
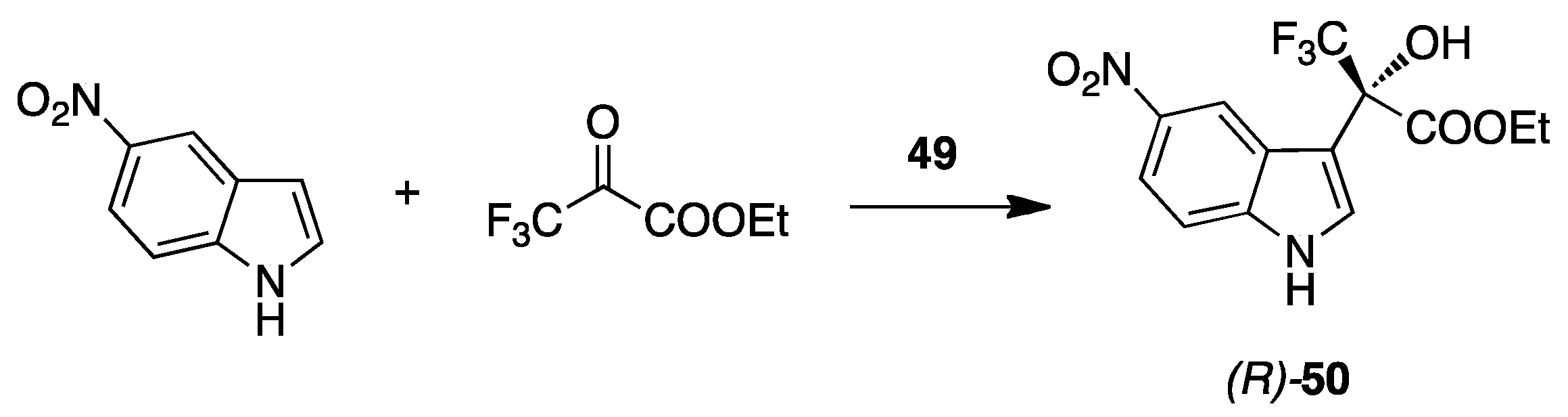
10. TFMI
11. Conclusions
Acknowledgments
Conflicts of Interest
References
- World Health Organization. HIV/AIDS, Fact Sheet n. 360, Updated November 2015. Available online: http://www.who.int/mediacentre/factsheets/fs360/en/ (accessed on 7 December 2015).
- Cohen, M.S.; Chen, Y.Q.; McCauley, M.; Gamble, T.; Hosseinipour, M.C.; Kumarasamy, N.; Hakim, J.G.; Kumwenda, J.; Grinsztejn, B.; Pilotto, J.H.; et al. Prevention of HIV-1 infection with early antiretroviral therapy. N. Engl. J. Med. 2011, 365, 493–505. [Google Scholar] [CrossRef] [PubMed]
- Veillette, M.; Richard, J.; Pazgler, M.; Lewis, G.K.; Parsons, M.S.; Finzi, A. Role of HIV-1 envelope glycoproteins conformation and accessory proteins on ADCC responses. Curr. HIV Res. 2016, 14, 9–23. [Google Scholar] [CrossRef] [PubMed]
- Landais, E.; Huang, X.; Havenar-Daughton, C.; Murrell, B.; Price, M.A.; Wickramasinghe, L.; Ramos, A.; Bian, C.B.; Simek, M.; Allen, S.; et al. Broadly neutralizing antibody responses in a large longitudinal sub-saharan HIV primary infection cohort. PLoS Pathog. 2016, 12, e1005369. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grand, R.M.; Lama, J.R.; Anderson, P.L.; McMahan, V.; Liu, A.Y.; Vargas, L.; Goicochea, P.; Casapía, M.; Guanira-Carranza, J.V.; Ramirez-Cardich, M.E.; et al. Preexposure chemoprophylaxis for HIV prevention in men who have sex with men. New Engl. J. Med. 2010, 363, 2587–2599. [Google Scholar]
- Esposito, F.; Corona, A.; Tramontano, E. HIV-1 reverse transcriptase still remains a new drug target: Structure, function, classical inhibitors, and new inhibitors with innovative mechanisms of actions. Mol. Biol. Int. 2012, 2012. [Google Scholar] [CrossRef] [PubMed]
- Menéndez-Arias, L. Molecular basis of human immunodeficiency virus type 1 drug resistance: Overview and recent developments. Antivir. Res. 2013, 98, 93–120. [Google Scholar] [CrossRef] [PubMed]
- U.S. FDA. Antiretroviral Drugs Used in the Treatment of HIV Infection; U.S. FDA: Silver Spring, MD, USA, 2015. [Google Scholar]
- Cortez, K.J.; Maldarelli, F. Clinical management of HIV drug resistance. Viruses 2011, 3, 347–378. [Google Scholar] [CrossRef] [PubMed]
- Hawkins, T. Understanding and managing the adverse effects of antiretroviral therapy. Antivir. Res. 2010, 85, 201–209. [Google Scholar] [CrossRef] [PubMed]
- De Clercq, E. Antiretroviral drugs. Curr. Opin. Pharmacol. 2010, 10, 507–515. [Google Scholar] [CrossRef] [PubMed]
- De Clercq, E. The nucleoside reverse transcriptase inhibitors, nonnucleoside reverse transcriptase inhibitors, and protease inhibitors in the treatment of HIV infections (AIDS). Adv. Pharmacol. 2013, 67, 317–358. [Google Scholar] [PubMed]
- De Clercq, E. Dancing with chemical formulae of antivirals: A personal account. Biochem. Pharmacol. 2013, 86, 711–725. [Google Scholar] [CrossRef] [PubMed]
- De Clercq, E. Dancing with chemical formulae of antivirals: A panoramic view. Biochem. Pharmacol. 2013, 86, 1397–1410. [Google Scholar] [CrossRef] [PubMed]
- AIDSinfo. Side Effects of HIV Medicines. Available online: https://aidsinfo.nih.gov (accessed on 7 January 2016).
- Mehellou, Y.; de Clercq, E. Twenty-six years of anti-HIV drug discovery: Where do we stand and where do we go? J. Med. Chem. 2010, 53, 521–538. [Google Scholar] [CrossRef] [PubMed]
- Vere Hodge, R.A. Meeting report: 28th International conference on antiviral research in Rome, Italy. Antivir. Res. 2015, 123, 172–187. [Google Scholar] [CrossRef] [PubMed]
- Zhan, P.; Pannecouque, C.; de Clercq, E.; Liu, X. Anti-HIV drug discovery and development: Current innovations and future trends. J. Med. Chem. 2015. [Google Scholar] [CrossRef] [PubMed]
- Maga, G.; Spadari, S. Combinations against combinations: Associations of anti-HIV 1 reverse transcriptase drugs challenged by constellations of drug resistance mutations. Curr. Drug Metab. 2002, 3, 73–95. [Google Scholar] [CrossRef] [PubMed]
- Pauwels, R.; Andries, K.; Desmyter, J.; Schols, D.; Kukla, M.J.; Breslin, H.J.; Raeymaeckers, A.; van Gelder, J.; Woestenborghs, R.; Heykants, J. Potent and selective inhibition of HIV-1 replication in vitro by a novel series of TIBO derivatives. Nature 1990, 343, 470–474. [Google Scholar] [CrossRef] [PubMed]
- Jacobo-Molina, A.; Clarck, A.D., Jr.; Williams, R.L.; Nanni, R.L.; Clarck, P.; Ferris, A.L.; Highes, S.H.; Arnold, E. Crystals of a ternary complex of human immunodeficiency virus type 1 reverse transcriptase with a monoclonal antibody Fab fragment and double-stranded DNA diffract x-rays to 3.5 Å resolution. Proc. Natl. Acad. Sci. USA 1991, 88, 10895–10899. [Google Scholar] [CrossRef] [PubMed]
- Tantillo, C.; Ding, J.; Jacobo-Molina, A.; Nanni, R.G.; Boyer, P.L.; Hughes, S.H.; Pauwels, R.; Andries, K.; Janssen, P.A.J.; Arnold, E. Locations of anti-AIDS drug binding sites and resistant mutations in the three dimensional structure of HIV-1 reverse trancriptase. J. Mol. Biol. 1994, 243, 369–387. [Google Scholar] [CrossRef] [PubMed]
- Kohlstaedt, L.A.; Wang, J.; Friedman, J.M.; Rice, P.A.; Steitz, T.A. Crystal structure at 3.5 Å resolution of HIV-1 reverse transcriptase complexed with an inhibitor. Science 1992, 256, 1783–1790. [Google Scholar] [CrossRef] [PubMed]
- Ren, J.; Esnouf, R.; Garman, E.; Somers, D.; Ross, C.; Kirby, I.; Keeling, J.; Darby, G.; Jones, Y.; Stuart, D.; et al. High resolution structures of HIV-1 RT from four RT-inhibitor complexes. Nat. Struct. Biol. 1995, 2, 293–302. [Google Scholar] [CrossRef] [PubMed]
- Das, K.; Ding, J.; Hsiou, Y.; Clark, A.D., Jr.; Moereels, H.; Koymans, L.; Andries, K.; Pauwels, R.; Janssen, P.A.; Boyer, P.L.; et al. Crystal structures of 8-Cl and 9-Cl TIBO complexed with wild-type HIV-1 RT and 8-Cl TIBO complexed with the Tyr181Cys HIV-1 RT drug-resistant mutant. J. Mol. Biol. 1996, 264, 1085–1100. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Abbondanzieri, E.A.; Rausch, J.W.; le Grice, S.F.J.; Zhuang, X. Slide into action: Dynamic shuttling of HIV reverse transcriptase on nucleic acid substrates. Science 2008, 322, 1092–1097. [Google Scholar] [CrossRef] [PubMed]
- Esnouf, R.J.; Ren, C.; Ross, C.; Jones, Y.; Stammers, D.; Stuart, D. Mechanism of inhibition of HIV-1 reverse transcriptase by non-nucleoside inhibitors. Nat. Struct. Biol. 1995, 2, 303–308. [Google Scholar] [CrossRef] [PubMed]
- AIDSinfo. Guidelines for the Use of Antiretroviral Agents in HIV-1-Infected Adults and Adolescents. Available online: http://aidsinfo.nih.gov/guidelines (accessed on 17 December 2015).
- Hui, D.Y. Effects of HIV protease inhibitor therapy on lipid metabolism. Prog. Lipid Res. 2003, 42, 81–92. [Google Scholar] [CrossRef]
- Delaugerre, C.; Rohban, R.; Simon, A.; Mouroux, M.; Tricot, C.; Agher, R.; Huraux, J.M.; Katlama, C.; Calvez, V. Resistance profile and cross-resistance of HIV-1 among patients failing a non-nucleoside reverse transcriptase inhibitor-containing regimen. J. Virol. Med. 2001, 65, 445–448. [Google Scholar] [CrossRef] [PubMed]
- Brown, C. Chirality in Drug Design and Synthesis; Academic Press: London, UK, 1990. [Google Scholar]
- Federsel, H.J. Chiral drug discovery and development from concept stage to market launch. In Compregensive Medicinal Chemistry II; Elsevier: Amsterdam, The Netherlands, 2007; Volume 2. [Google Scholar]
- Soudijn, W. Advantages and disadvantages in the application of biological racemates of specific isomers in drugs. In Stereochemistry and Biological Activity of Drugs; Blackwell Scientific Publications: Oxford, UK, 1983. [Google Scholar]
- Borman, S. FDA issues flexible policy on chiral drugs. Chem. Eng. News 1992, 70, 5. [Google Scholar] [CrossRef]
- Stinson, S.C. In wake of new FDA guidelines, most drug firms are developing single enantiomers, spawning a “chirotechnology” industry. Chem. Eng. News 1992, 70, 46–79. [Google Scholar] [CrossRef]
- Yamaguchi, T.; Iwanami, N.; Shudo, K.; Saneyoshi, M. Chiral discrimination of enantiomeric 2′-deoxythymidine 5′-triphosphate by HIV-1 reverse transcriptase and eukaryotic DNA polymerases. Biochem. Biophys. Commun. 1994, 200, 1023–1027. [Google Scholar] [CrossRef] [PubMed]
- Focher, F.; Maga, G.; Bendiscioli, A.; Capobianco, M.; Colonna, F.; Garbesi, A.; Spadari, S. Stereospecificity of human DNA polymerases alpha, beta, gamma, delta and epsilon, HIV-reverse transcriptase, HSV-1 DNA polymerase, calf thymus terminal transferase and Escherichia coli DNA polymerase I in recognizing d- and l-thymidine 5′-triphosphate as substrate. Nucleic Acid Res. 1995, 23, 2480–2847. [Google Scholar]
- Maga, G.; Amacker, M.; Hübscher, U.; Gosselin, G.; Imbach, J.L.; Mathé, C.; Faraj, A.; Sommadossi, J.P.; Spadari, S. Molecular basis for the enantioselectivity of HIV-1 reverse transcriptase: Role of the 3′-hydroxyl group of the l-(β)-ribose in chiral discrimination between d- and l-enantiomers of deoxy- and dideoxy-nucleoside triphosphate analogs. Nucleic Acid Res. 1999, 27, 972–978. [Google Scholar] [CrossRef] [PubMed]
- Gosselin, G.; Schinazi, R.F.; Sommadossi, J.P.; Mathi, C.; Bergogne, M.C.; Aubertin, A.M.; Kirn, A.; Imbach, J.L. Anti-human immunodeficiency virus activities of the 1-L enantiomer of 2′,3′-dideoxycytidine and its 5-fluoro derivative in vitro in vitro. Antimicrob. Agents Chemother. 1994, 38, 1292–1297. [Google Scholar] [CrossRef] [PubMed]
- Schinazi, R.F.; McMillan, A.; Cannon, D.; Mathis, R.; Lloyd, R.M.; Peck, A.; Sommadossi, J.-P.; St. Clair, M.; Wilson, J.; Furman, P.A.; et al. Selective inhibition of human immunodeficiency viruses by racemates and enantiomers of cis-5-fluoro-1-[2-(hydroxymethyl)-1,3-oxathiolan-5-yl]cytosine. Antimicrob. Agents Chemother. 1992, 36, 2423–2431. [Google Scholar] [CrossRef] [PubMed]
- Gao, Q.; Gu, Z.; Parniak, M.A.; Cameron, J.; Cammack, N.; Boucher, C.; Wainberg, M.A. The same mutation that encodes low-level human immunodeficiency virus type 1 resistance to 2′,3′-dideoxyinosine and 2′,3′-dideoxycytidine confers high level resistance to the (−) enantiomer of 2′,3′-dideoxy-3′-thiacytidine. Antimicrob. Agents Chemother. 1993, 37, 1390–1392. [Google Scholar] [CrossRef] [PubMed]
- Merluzzi, V.J.; Hargrave, K.D.; Labadia, M.; Grozinger, K.; Skoog, M.; Wu, J.C.; Shih, C.K.; Eckner, K.; Hattox, H.; Adams, J.; et al. Inhibition of HIV-1 replication by a nonnucleoside reverse transcriptase inhibitor. Science 1990, 250, 1411–1413. [Google Scholar] [CrossRef] [PubMed]
- Adams, J.; Merluzzi, V.J. Discovery of nevirapine, a nonnucleoside inhibitor of HIV-1 reverse rranscriptase. In The Search for Antiviral Drugs; Springer Verlag: New York, NY, USA, 1993; Chapter 3; pp. 5–70. [Google Scholar]
- Pedersen, O.S.; Pedersen, E.B. Non-nucleoside reverse transcriptase inhibitors: The NNRTI boom. Antivir. Chem. Chemother. 1999, 10, 285–314. [Google Scholar] [CrossRef] [PubMed]
- Santoro, M.M.; Sabin, C.; Forbici, F.; Bansi, L.; Dunn, D.; Fearnhill, E.; Boumis, E.; Nicastri, E.; Antinori, A.; Palamara, G.; et al. Drug-resistance development differs between HIV-1-infected patients failing first-line antiretroviral therapy containing nonnucleoside reverse transcriptase inhibitors with and without thymidine analogues. HIV Med. 2013, 14, 571–577. [Google Scholar] [CrossRef] [PubMed]
- Ibe, S.; Sugiura, W. Clinical significance of HIV reverse-transcriptase inhibitor-resistance mutations. Future Microbiol. 2011, 6, 295–315. [Google Scholar] [CrossRef] [PubMed]
- Mui, P.W.; Jacober, S.P.; Hargrave, K.D.; Adams, D. Crystal structure of nevirapine, a non-nucleoside inhibitor of HIV-1 reverse transcriptase, and computational alignment with a structurally diverse inhibitor. J. Med. Chem. 1992, 35, 201–202. [Google Scholar] [CrossRef] [PubMed]
- Burke, E.W.D.; Morris, G.A.; Vincent, M.A.; Hillier, I.H.; Clayden, J. Is nevirapine atropisomeric? Experimental and computational evidence for rapid conformational inversion. Org. Biomol. Chem. 2012, 10, 716–719. [Google Scholar] [CrossRef] [PubMed]
- Ayala, A.P.; Siesler, H.W.; Wardwll, S.M.M.S.V.; Boechat, N.; Dabbene, V.; Cuffini, S.L. Vibrational spectra and quantum mechanical calculations of antiretroviral drugs: Nevirapine. J. Mol. Struct. 2007, 828, 201–210. [Google Scholar] [CrossRef]
- Carlier, P.R.; Zhao, H.; DeGuzman, J.; Lam, P.C.H. Enantioselective synthesis of “quaternary” 1,4-benzodiazepin-2-one scaffolds via memory of chirality. J. Am. Chem. Soc. 2003, 125, 11482–11483. [Google Scholar] [CrossRef] [PubMed]
- Chattopadhyay, S.K.; Chatterjee, A.; Tandon, S.; Maulik, P.R.; Kant, R. Isolation of optically active nevirapine, a dipyridodiazepinone metabolite from the seeds of Cleome viscosa. Tetrahedron 2011, 67, 452–454. [Google Scholar] [CrossRef]
- De Clercq, E. Anti-HIV drugs: 25 compounds approved within 25 years after the discovery of HIV. Int. J. Antimicrob. Agents 2009, 33, 307–320. [Google Scholar] [CrossRef] [PubMed]
- Ogunjale, A.O.; Okwundu, C.I. Rilpivirine versus efavirenz for initial therapy in treatment-naive, HIV-1-infected patients. Cochrane Database Syst. Rev. 2014. [Google Scholar] [CrossRef]
- Young, S.D.; Britcher, S.F.; Tran, L.O.; Payne, L.S.; Lumma, W.C.; Lyle, T.A.; Huff, J.R.; Anderson, P.S.; Olsen, D.B.; Carroll, S.S.; et al. L-743,726 (DMP-266): A novel, highly potent nonnucleoside inhibitor of the human immunodeficiency virus type 1 reverse transcriptase. Antimicrob. Agents Chemother. 1995, 39, 2602–2605. [Google Scholar] [CrossRef] [PubMed]
- Young, S.D.; Britcher, S.F.; Payne, L.S.; Tran, L.O.; Lumma, W.C., Jr. Benzoxazinones as Inhibitors of HIV Reverse Transcriptase. U.S. Patent 5,519,021, 21 May 1996. [Google Scholar]
- Harms, A. Non-nucleoside HIV reverse transcriptase inhibitors. In The Art of Drug Synthesis; John Wiley and Sons: Hoboken, NJ, USA, 2007; Chapter 6; pp. 83–94. [Google Scholar]
- Thompson, A.; Corley, E.G.; Huntington, M.F.; Grabowski, E.J.J.; Julius, F.; Remenar, J.F.; Collum, D.B. Lithium ephedrate-mediated addition of a lithium acetylide to a ketone: Solution structures and relative reactivities of mixed aggregates underlying the high enantioselectivities. J. Am. Chem. Soc. 1998, 120, 2028–2038. [Google Scholar] [CrossRef]
- Corey, E.J.; Cimprich, K.A. Highly Enantioselective alkynylation of aldehydes promoted by chiral oxazaborolidine. J. Am. Chem. Soc. 1994, 116, 3151–3152. [Google Scholar] [CrossRef]
- Pierce, M.E.; Parsons, R.L., Jr.; Radesca, L.A.; Lo, Y.S.; Silverman, S.; Moore, J.R.; Islam, Q.; Choudhury, A.; Fortunak, J.M.D.; Nguyen, D.; et al. Practical asymmetric synthesis of efavirenz (DMP 266), an HIV-1 reverse transcriptase inhibitor. J. Org. Chem. 1998, 63, 8536–8543. [Google Scholar] [CrossRef]
- Chen, C.Y.; Tillyer, R.D.; Tan, L. Preparation of Chiral Tertiary Alcohols via Enantioselective Addition Reaction Using an Organozinc Reagent. Patent PCT WO 98/51676, 9 November 1998. [Google Scholar]
- Pujeri, S.S.; Khader, A.M.A.; Seetharamappa, J. Chiral separation of non-nucleoside reverse transcription inhibitor efavirenz by HPLC on cellulose-based chiral stationary phase. J. Food Drug Anal. 2013, 21, 93–100. [Google Scholar]
- Surendra-Dutt Sharma, S.D.; Gaurav Singh, G. Enantioseparation of efavirenz by ultra performance liquid chromatography. Adv. Anal. Chem. 2013, 3, 29–33. [Google Scholar]
- Corbett, J.W.; Ko, S.S.; Rodgers, J.D.; Jeffrey, S.; Bacheler, L.T.; Klabe, R.M.; Diamond, S.; Lai, C.M.; Rabel, S.R.; Saye, J.A.; et al. Expanded-spectrum nonnucleoside reverse transcriptase inhibitors inhibit clinically relevant mutant variants of human immunodeficiency virus type 1. Antimicrob. Agents Chemother. 1999, 43, 2893–2897. [Google Scholar] [PubMed]
- Corbett, J.W.; Ko, S.S.; Rodgers, J.D.; Gearhart, L.A.; Magnus, N.A.; Bacheler, L.T.; Diamond, S.; Jeffrey, S.; Klabe, R.M.; Cordova, B.C.; et al. Inhibition of clinically relevant mutant variants of HIV-1 by quinazolinone non-nucleoside reverse transcriptase inhibitors. J. Med. Chem. 2000, 43, 2019–2030. [Google Scholar] [CrossRef] [PubMed]
- Bacheler, L.; Weislow, O.; Snyder, S.; Hanna, G.; D’Aquila, R.; The Sustiva Resistance Study Team. Resistance to Efavirenz (Sustiva) in vivo. In Proceedings of the Program and abstracts of the 5th Conference on Retroviruses and Opportunistic Infections, Chicago, IL, USA, 1–5 February 1998; p. 56.
- Miller, V.; de Béthune, M.P.; Kober, A.; Stürmer, M.; Hertogs, K.; Pauwels, R.; Stoffels, P.; Staszewski, S. Patterns of resistance and cross-resistance to human immunodeficiency virus type 1 reverse transcriptase inhibitors in patients treated with the nonnucleoside reverse transcriptase inhibitor loviride. Antimicrob. Agents Chemother. 1998, 42, 3123–3129. [Google Scholar] [PubMed]
- Magnus, N.A.; Confalone, P.N.; Storace, L. A new asymmetric 1,4-addition method: Application to the synthesis of the HIV non-nucleoside reverse transcriptase inhibitor DPC 961. Tetrahedron Lett. 2000, 41, 3015–3019. [Google Scholar] [CrossRef]
- Tucker, T.J.; Lyle, T.A.; Wiscount, C.M.; Britcher, S.F.; Young, S.D.; Sanders, W.M.; Lumma, W.C.; Goldman, M.E.; O’Brien, J.A.; Ball, R.G.; et al. Synthesis of a series of 4-(arylethynyl)-6-chloro-4-cyclopropyl-3,4-dihydroquinazolin-2(1H)-ones as novel non-nucleoside HIV-1 reverse transcriptase inhibitors. J. Med. Chem. 1994, 37, 2437–2444. [Google Scholar] [CrossRef] [PubMed]
- Aubry, A.F.; Sebastian, D.S.; Williams, R.C.; Boucher, R.J. Determination of the enantiomeric purity of investigational non-nucleoside reverse transcriptase inhibitors. Chirality 2001, 13, 193–198. [Google Scholar] [CrossRef] [PubMed]
- Renit, J.; Esnouf, R.; Hopkins, A.; Ross, C.; Jones, Y.; Stammers, D.; Stuart, D. The structure of HIV-1 reverse transcriptase complexed with 9-chloro-TIBO: Lessons for inhibitor design. Structure 1995, 3, 915–926. [Google Scholar]
- Pauwels, R.; Andries, K.; Debyser, Z.; van Daele, P.; Schols, D.; Stoffels, P.; de Vreese, K.; Woestenborghsi, R.; Vandamme, A.M.; Janssen, C.G.M.; et al. Potent and highly selective human immunodeficiency virus type 1 (HIV-1) inhibition by a series of α-anilinophenylacetamide derivatives targeted at HIV-1 reverse transcriptase. Proc. Natl. Acad. Sci. USA 1993, 90, 1711–1715. [Google Scholar] [CrossRef] [PubMed]
- Ludovici, D.L.; Kukla, M.J.; Grous, P.G.; Krishnan, S.; Andries, K.; de Béthune, M.P.; Azijn, H.; Pauwels, R.; de Clercq, E.; Arnold, E.; et al. Evolution of anti-HIV drug candidates. Part 1: From α-anilinophenylacetamide (α-APA) to imidoyl thiourea (ITU). Bioorg. Med. Chem. Lett. 2001, 11, 2225–2228. [Google Scholar] [CrossRef]
- Ludovici, D.L.; Kavash, R.W.; Kukla, M.J.; Ho, C.Y.; Ye, H.; de Corte, B.L.; Andries, K.; de Béthune, M.P.; Azijn, H.; Pauwels, R.; et al. Evolution of anti-HIV drug candidates. Part 2: Diaryltriazine (DATA) analogues. Bioorg. Med. Chem. Lett. 2001, 11, 2229–2234. [Google Scholar] [CrossRef]
- Ludovici, D.L.; de Corte, B.L.; Kukla, M.J.; Ye, H.; Ho, C.Y.; Lichtenstein, M.A.; Kavash, R.W.; Andries, K.; de Béthune, M.P.; Azijn, H.; et al. Evolution of anti-HIV drug candidates. Part 3: Diarylpyrimidine (DAPY) analogues. Bioorg. Med. Chem. Lett. 2001, 11, 2235–2239. [Google Scholar] [CrossRef]
- Lewis, P.J.; de Jonge, M.; Daeyaert, F.; Koymans, L.; Vinkers, M.; Heeres, J.; Janssen, P.A.J.; Arnold, E.; Das, K.; Clark, A.D., Jr.; et al. On the detection of multiple-binding modes of ligands to proteins, from biological, structural, and modeling data. J. Comput. Aided Mol. Des. 2003, 17, 129–134. [Google Scholar] [CrossRef]
- La Regina, G.; Coluccia, A.; Silvestri, R. Looking for an active conformation of the future HIV type-1 non-nucleoside reverse transcriptase inhibitors. Antivir. Chem. Chemother. 2010, 20, 213–237. [Google Scholar] [CrossRef] [PubMed]
- Zeng, Z.S.; Liang, Y.H.; Feng, X.Q.; Chen, F.E.; Pannecouque, C.; Balzarini, J.; de Clercq, E. Lead optimization of diarylpyrimidines as non-nucleoside inhibitors of HIV-1 reverse transcriptase. ChemMedChem 2010, 5, 837–840. [Google Scholar] [CrossRef] [PubMed]
- Feng, X.Q.; Liang, Y.H.; Zeng, Z.S.; Chen, F.E.; Balzarini, J.; Pannecouque, C.; del Clercq, E. Structural modifications of DAPY analogues with potent anti-HIV-1 activity. ChemMedChem 2009, 4, 219–224. [Google Scholar] [CrossRef] [PubMed]
- Gu, S.X.; He, Q.Q.; Yang, S.Q.; Maa, X.D.; Chen, F.E.; de Clercq, E.; Balzarini, J.; Pannecouque, C. Synthesis and structure-activity relationship of novel diarylpyrimidines with hydromethyl linker (CH(OH)-DAPYs) as HIV-1 NNRTIs. Bioorg. Med. Chem. 2011, 19, 5117–5124. [Google Scholar] [CrossRef] [PubMed]
- Gu, S.X.; Li, Z.M.; Ma, X.D.; Yang, S.Q.; He, Q.Q.; Chen, F.E.; de Clercq, E.; Balzarini, J.; Pannecouque, C. Chiral resolution, absolute configuration assignment and biological activity of racemic diarylpyrimidine CH(OH)-DAPY as potent nonnucleoside HIV-1 reverse transcriptase inhibitors. Eur. J. Med. Chem. 2012, 53, 229–234. [Google Scholar] [CrossRef] [PubMed]
- Artico, M. Selected non-nucleoside reverse transcriptase inhibitors (NNRTIs): The DABOs family. Drugs Future 2002, 27, 159–175. [Google Scholar] [CrossRef]
- Rotili, D.; Tarantino, D.; Artico, M.; Nawrozkij, M.B.; Gonzalez-Ortega, E.; Clotet, B.; Samuele, A.; Esté, J.A.; Maga, G.; Mai, A. Diarylpyrimidine-dihydrobenzyloxopyrimidine hybrids: New, wide-spectrum anti-HIV-1 agents active at (sub)-nanomolar level. J. Med. Chem. 2011, 54, 3091–3096. [Google Scholar] [CrossRef] [PubMed]
- Rotili, D.; Samuele, A.; Tarantino, D.; Ragno, R.; Musmuca, I.; Ballante, F.; Botta, G.; Morera, L.; Pierini, M.; Cirilli, R.; et al. 2-(Alkyl/aryl)amino-6-benzylpyrimidin-4(3H)-ones as Inhibitors of wild-type and mutant HIV-1: Enantioselectivity studies. J. Med. Chem. 2012, 55, 3558–3562. [Google Scholar] [CrossRef] [PubMed]
- Bell, F.W.; Cantrell, A.S.; Hogberg, M.; Jaskunas, S.R.; Johansson, N.G.; Jordan, C.L.; Kinnick, M.D.; Lind, P.; Morin, J.M., Jr. Phenethylthiazolylthiourea (PETT) compounds, a new class of HIV-1 reverse transcriptase inhibitors. 1. Synthesis and basic structure–activity relationship studies of PETT analogs. J. Med. Chem. 1995, 38, 4929–4936. [Google Scholar] [CrossRef] [PubMed]
- Cantrell, A.S.; Engelhardt, P.; Hogberg, M.; Jaskunas, S.R.; Johansson, N.G.; Jordan, C.L.; Kangasmetsä, J.; Kinnick, M.D.; Lind, P.; Morin, J.M., Jr.; et al. Phenethylthiazolyl thiourea (PETT) compounds as a new class of HIV-1 reverse transcriptase inhibitors. 2. Synthesis and further structure–activity relationship studies of PETT analogs. J. Med. Chem. 1996, 39, 4261–4274. [Google Scholar] [CrossRef] [PubMed]
- Venkatachalam, T.K.; Mao, C.; Uckun, F.M. Effect of stereo and regiochemistry towards wild and multidrug resistant HIV-1 virus: Viral potency of chiral PETT derivatives. Biochem. Pharmacol. 2004, 67, 1933–1946. [Google Scholar] [CrossRef] [PubMed]
- Venkatachalam, T.K.; Mao, C.; Uckun, F. Effect of stereochemistry on the anti-HIV activity of chiral thiourea compounds. Bioorg. Med. Chem. 2004, 12, 4275–4284. [Google Scholar] [CrossRef] [PubMed]
- Williams, T.M.; Ciccarone, T.M.; MacTough, S.C.; Rooney, C.S.; Balani, S.K.; Condra, J.K.; Emini, E.A.; Goldman, M.E.; Greenlee, W.J. 5-Chloro-3-(phenylsulfonyl)indole-2-carboxamide: A novel, non-nucleoside inhibitor of HIV-1 reverse transcriptase. J. Med. Chem. 1993, 36, 1291–1294. [Google Scholar] [CrossRef] [PubMed]
- Silvestri, R.; de Martino, G.; la Regina, G.; Artico, M.; Massa, S.; Vargiu, L.; Mura, M.; Loi, A.G.; Marceddu, T.; la Colla, P. Novel indolyl aryl sulfones active against HIV-1 carrying NNRTI resistance mutations: Synthesis and SAR studies. J. Med. Chem. 2003, 46, 2418–2493. [Google Scholar] [CrossRef] [PubMed]
- Silvestri, R.; Artico, M.; de Martino, G.; la Regina, G.; Loddo, R.; la Colla, M.; la Colla, P. Simple, short peptide derivatives of a sulfonylindolecarboxamide (L-737,126) active in vitro in vitro against HIV-1 wild type and variants carrying non-nucleoside reverse transcriptase inhibitor resistance mutations. J. Med. Chem. 2004, 47, 3892–3896. [Google Scholar] [CrossRef] [PubMed]
- Piscitelli, F.; Coluccia, A.; Brancale, A.; la Regina, G.; Sansone, A.; Giordano, C.; Balzarini, J.; Maga, G.; Zanoli, S.; Samuele, A.; et al. Indolylarylsulfones bearing natural and unnatural amino acids. Discovery of potent inhibitors of HIV-1 non-nucleoside wild type and resistant mutant strains reverse transcriptase and Coxsackie B4 virus. J. Med. Chem. 2009, 52, 1922–1934. [Google Scholar] [CrossRef] [PubMed]
- Famiglini, V.; la Regina, G.; Coluccia, A.; Pelliccia, S.; Brancale, A.; Maga, G.; Crespan, E.; Badia, R.; Clotet, B.; Esté, J.A.; et al. New indolylarylsulfones as highly potent and broad spectrum HIV-1 non-nucleoside reverse transcriptase inhibitors. Eur. J. Med. Chem. 2011, 80, 101–111. [Google Scholar] [CrossRef] [PubMed]
- Famiglini, V.; la Regina, G.; Coluccia, A.; Pelliccia, S.; Brancale, A.; Maga, G.; Crespan, E.; Badia, R.; Riveira-Muñoz, E.; Esté, J.A.; et al. Indolylarylsulfones carrying a heterocyclic tail as very potent and broad spectrum HIV-1 non-nucleoside reverse transcriptase inhibitors. J. Med. Chem. 2014, 57, 9945–9957. [Google Scholar] [CrossRef] [PubMed]
- Wodak, S.J.; Janin, J. Analytical approximation to the accessible surface area of proteins. Proc. Natl. Acad. Sci. USA 1980, 77, 1736–1740. [Google Scholar] [CrossRef] [PubMed]
- Alexandre, F.A.; Amador, A.; Bot, S.; Caillet, C.; Convard, T.; Jakubik, J.; Musiu, C.; Poddesu, B.; Vargiu, L.; Liuzzi, M.; et al. Synthesis and biological evaluation of aryl-phospho-indole as novel HIV-1 non-nucleoside reverse transcriptase inhibitors. J. Med. Chem. 2011, 547, 392–395. [Google Scholar] [CrossRef] [PubMed]
- Klibano, O.M.; Kaczor, R.L. IDX-899, an aryl phosphinate-indole non-nucleoside reverse transcriptase inhibitor for the potential treatment of HIV infection. Curr. Opin. Investig. Drugs 2010, 11, 237–245. [Google Scholar]
- Storer, R.; Alexande, F.R.; Dousson, C.; Moussa, A.M.; Bridges, E. Enantiomerically Pure Phosphoindoles as HIV Inhibitors. Patent PCT WO/2008/042240, 10 April 2008. [Google Scholar]
- Afarinkia, K.; Yu, H.W. Hewitt reaction revisited. Tetrahedron Lett. 2003, 44, 781–783. [Google Scholar] [CrossRef]
- Han, X.; Ouyang, W.; Liu, B.; Wang, W.; Tien, P.; Wu, S.; Zhou, H.B. Enantioselective inhibition of reverse transcriptase (RT) of HIV-1 by non-racemic indole-based trifluoropropanoates developed by asymmetric catalysis using recyclable organocatalysts. Org. Biomol. Chem. 2013, 11, 8463–8475. [Google Scholar] [CrossRef] [PubMed]
- Han, X.; Liu, B.; Zhou, H.B.; Dong, C. Enhanced efficiency of recyclable C3-symmetric cinchonine-squaramides in the asymmetric Friedel-Crafts reaction of indoles with alkyl trifluoropyruvate. Tetrahedron Asymmetry 2012, 23, 1332–1337. [Google Scholar] [CrossRef]














© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Famiglini, V.; Silvestri, R. Focus on Chirality of HIV-1 Non-Nucleoside Reverse Transcriptase Inhibitors. Molecules 2016, 21, 221. https://doi.org/10.3390/molecules21020221
Famiglini V, Silvestri R. Focus on Chirality of HIV-1 Non-Nucleoside Reverse Transcriptase Inhibitors. Molecules. 2016; 21(2):221. https://doi.org/10.3390/molecules21020221
Chicago/Turabian StyleFamiglini, Valeria, and Romano Silvestri. 2016. "Focus on Chirality of HIV-1 Non-Nucleoside Reverse Transcriptase Inhibitors" Molecules 21, no. 2: 221. https://doi.org/10.3390/molecules21020221