
Synthesis, Spectroscopic, Structural and Quantum Chemical Studies of a New Imine Oxime and Its Palladium(II) Complex: Hydrolysis Mechanism

Abstract
:
1. Introduction
2. Results and Discussion
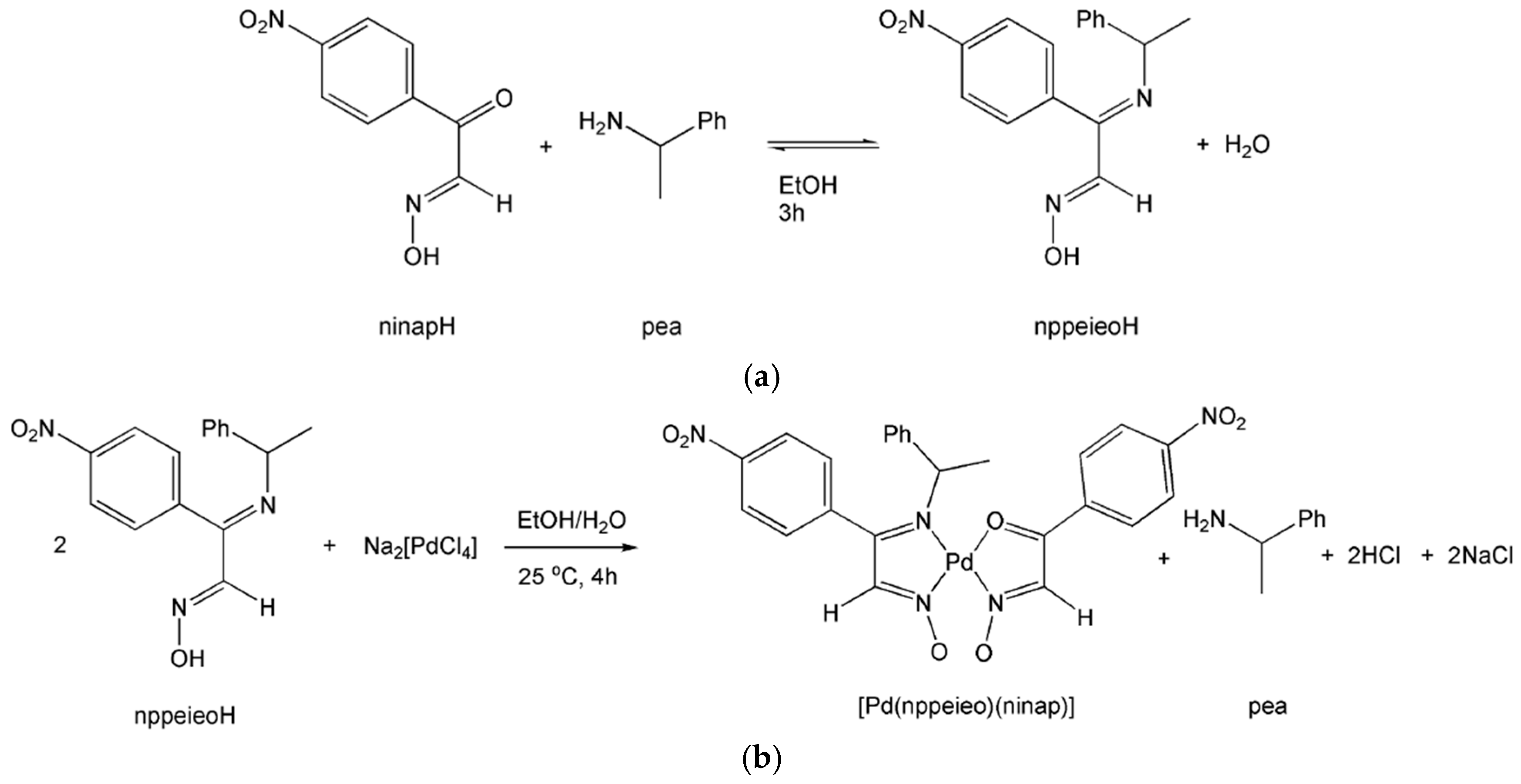
2.1. Synthesis and Characterization

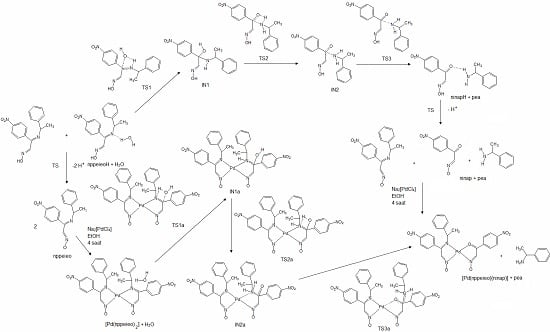
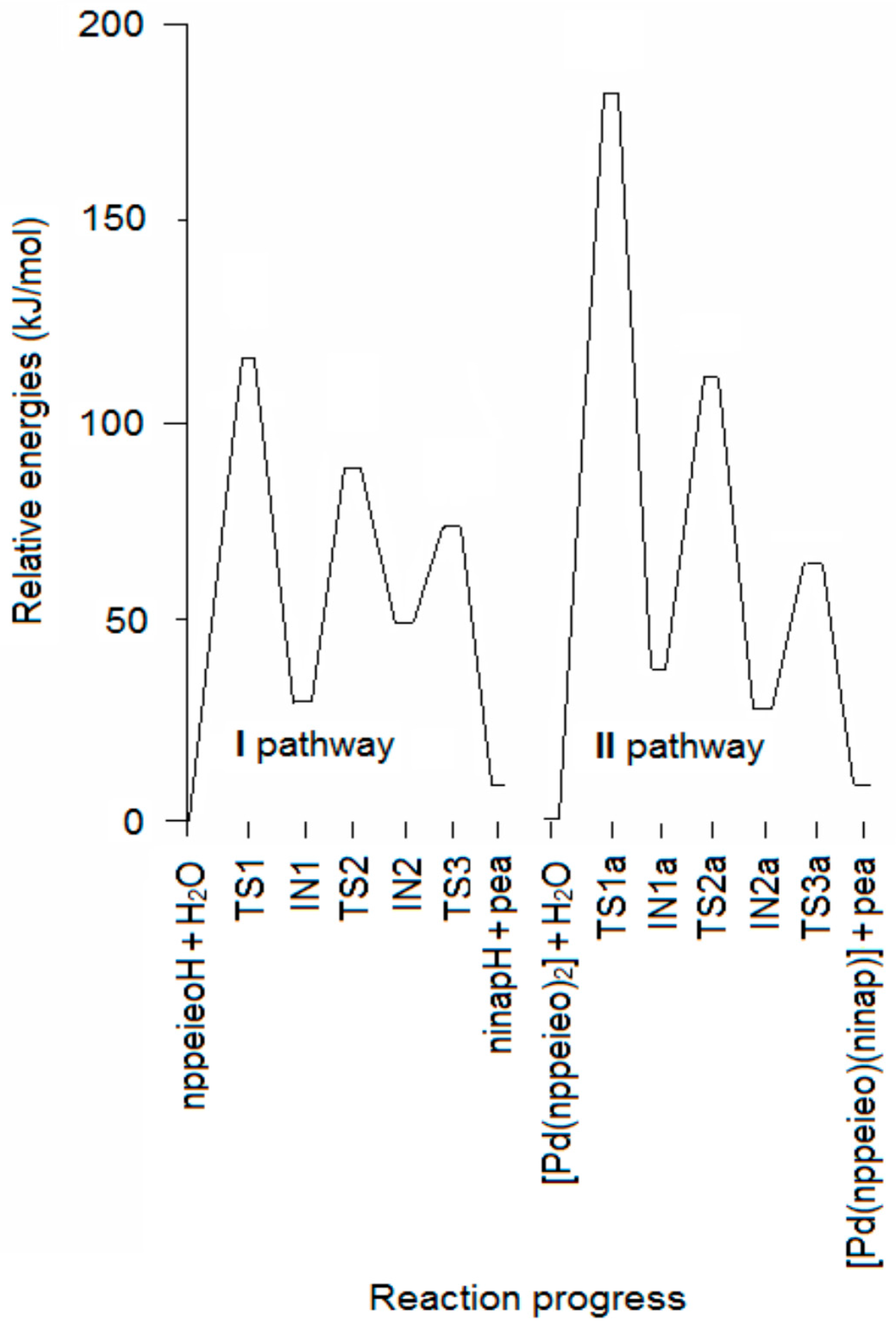
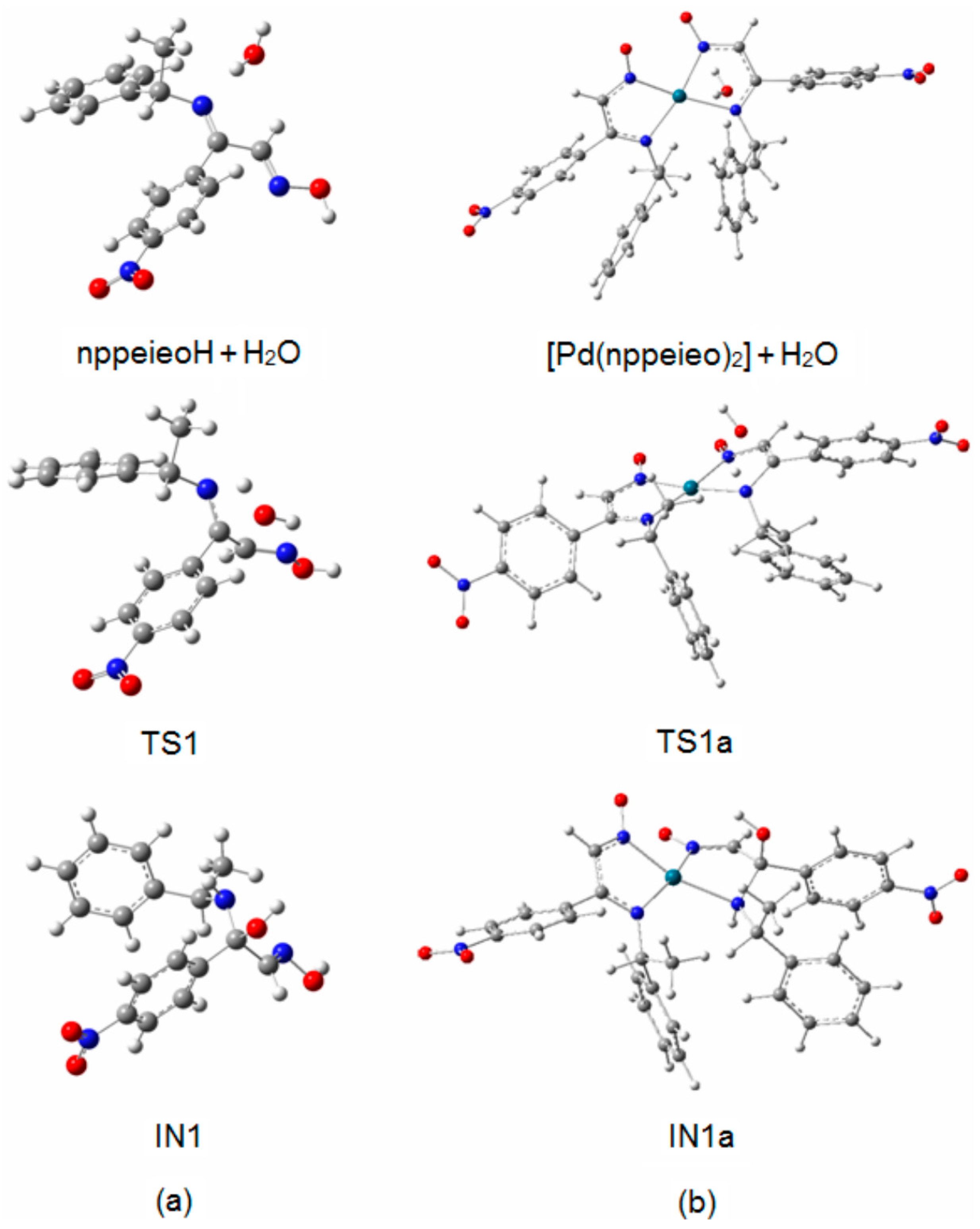
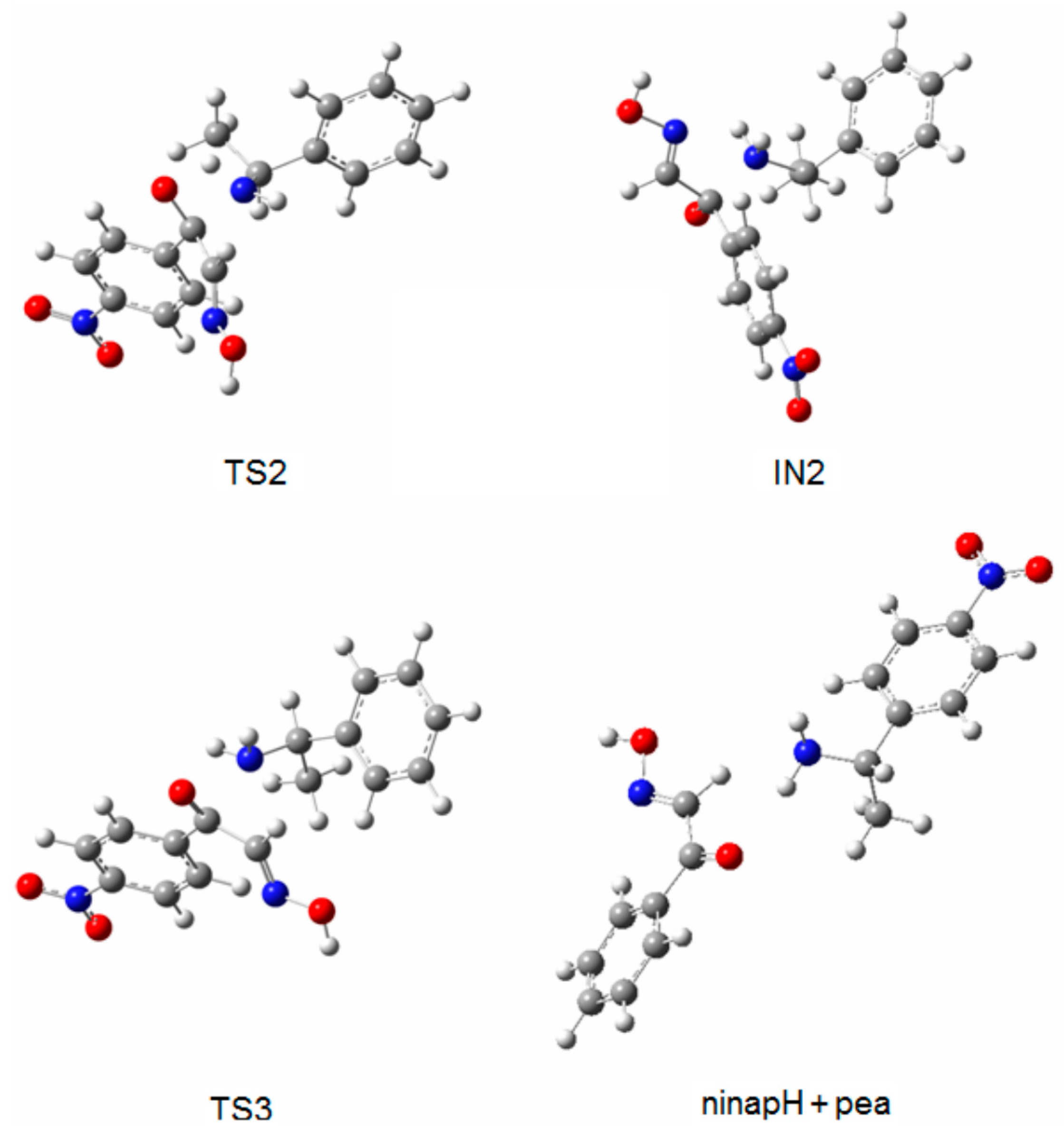
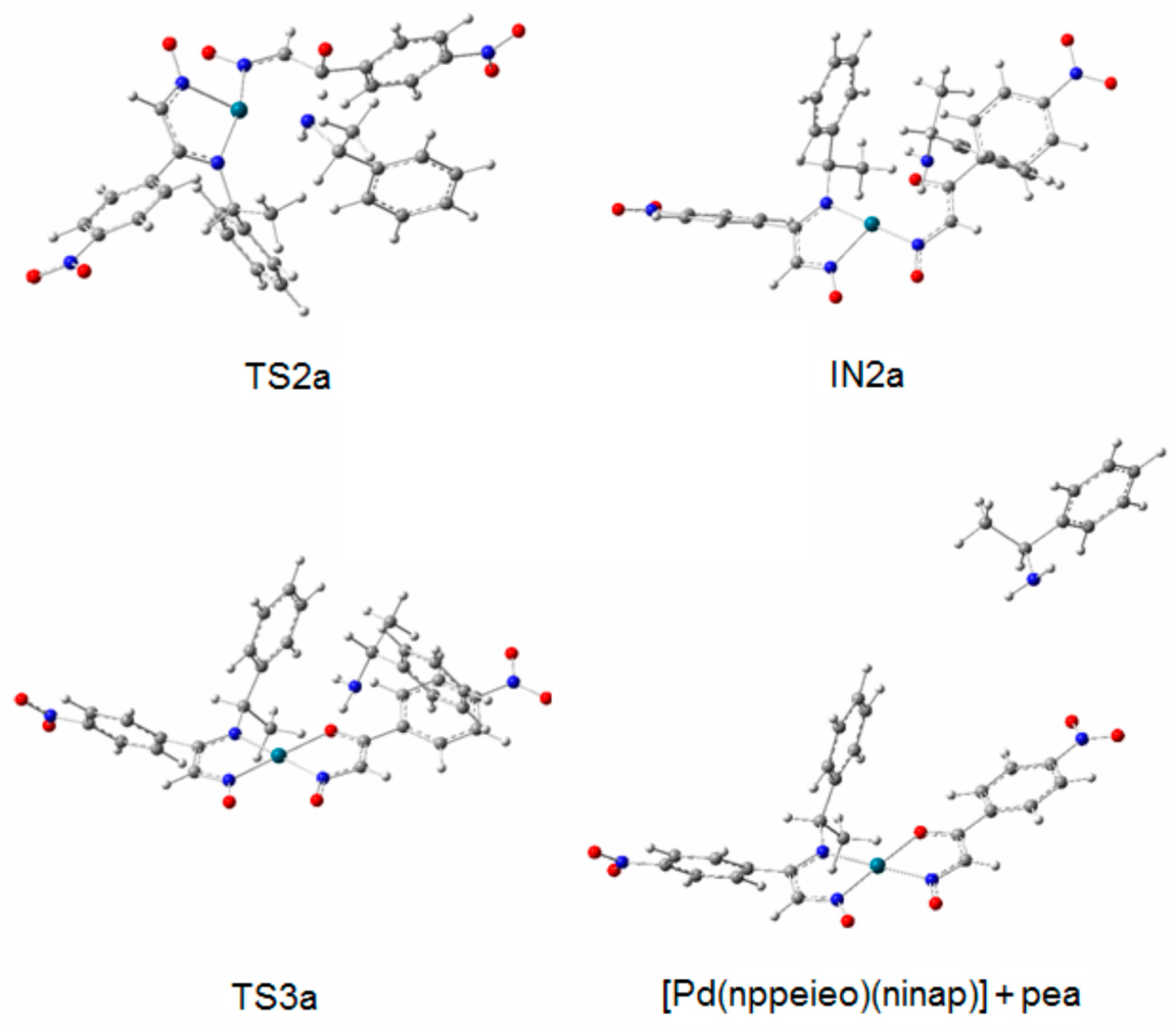
2.2. Hydrolysis Mechanism

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Molecules | Relative Energy (kJ·mol−1) | Negative Frequency (cm−1) | |
|---|---|---|---|
| 6-311G(d,p) | lanl2dz | ||
| nppeieoH | |||
| nppeieoH + H2O | 0.0 | 0.0 | - |
| TS1 | 109.9 | 112.3 | −323 |
| IN1 | 28.5 | 31.7 | - |
| TS2 | 88.0 | 88.2 | −1631 |
| IN2 | 48.6 | 49.7 | - |
| TS3 | 73.7 | 72.1 | −294 |
| ninapH + pea | 10.7 | 13.8 | - |
| [Pd(nppeieo)2] | |||
| [Pd(nppeieo)2] + H2O | 0.0 | - | |
| TS1a | 177.4 | −1361 | |
| IN1a | 38.1 | - | |
| TS2a | 108.7 | −1574 | |
| IN2a | 28.6 | - | |
| TS3a | 58.2 | −160 | |
| [Pd(nppeieo)(ninap)] + pea | 12.5 | - | |




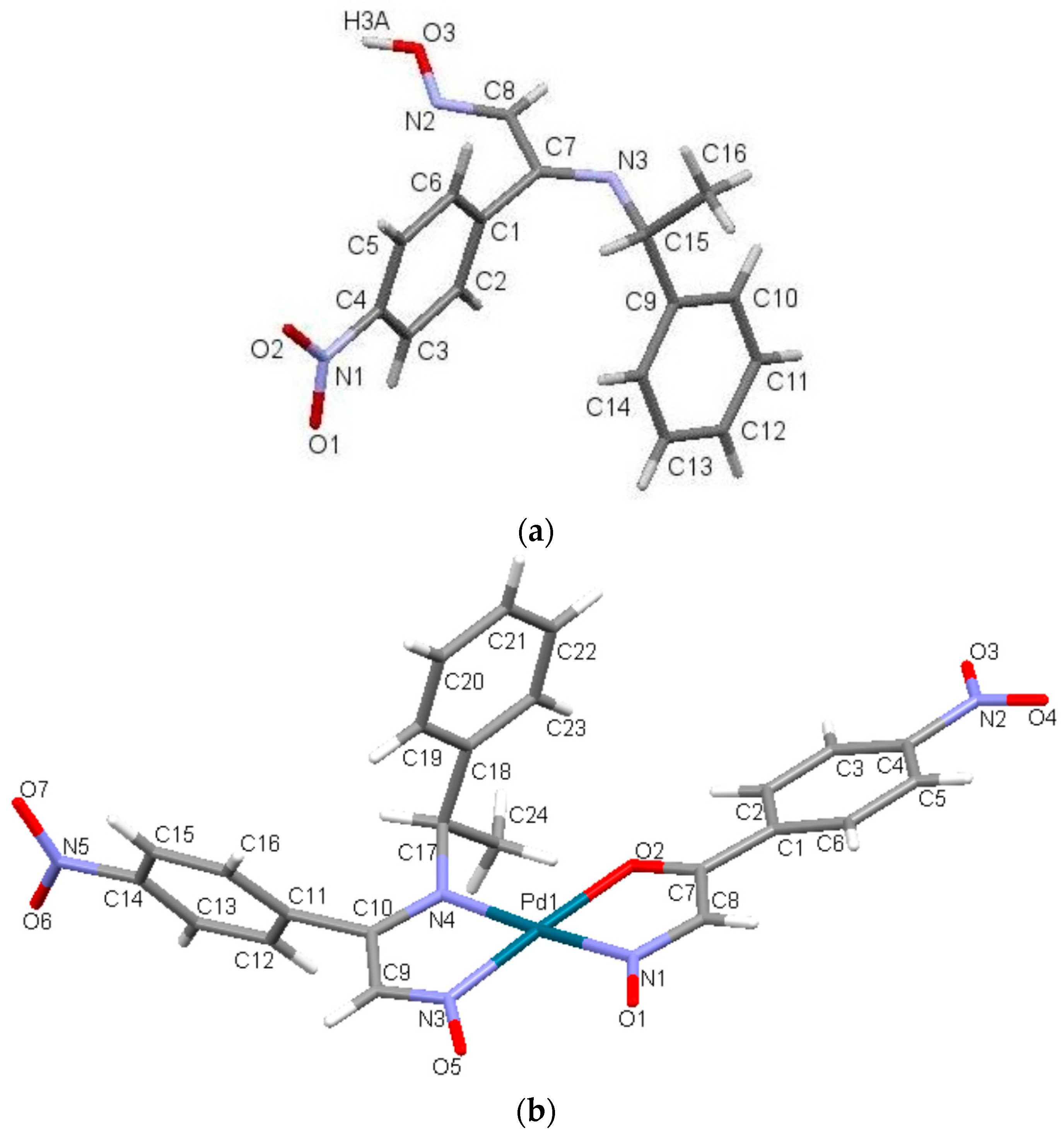
2.3. Crystal Structure

2.4. Optimized Structure
2.5. Mulliken Atomic Charges
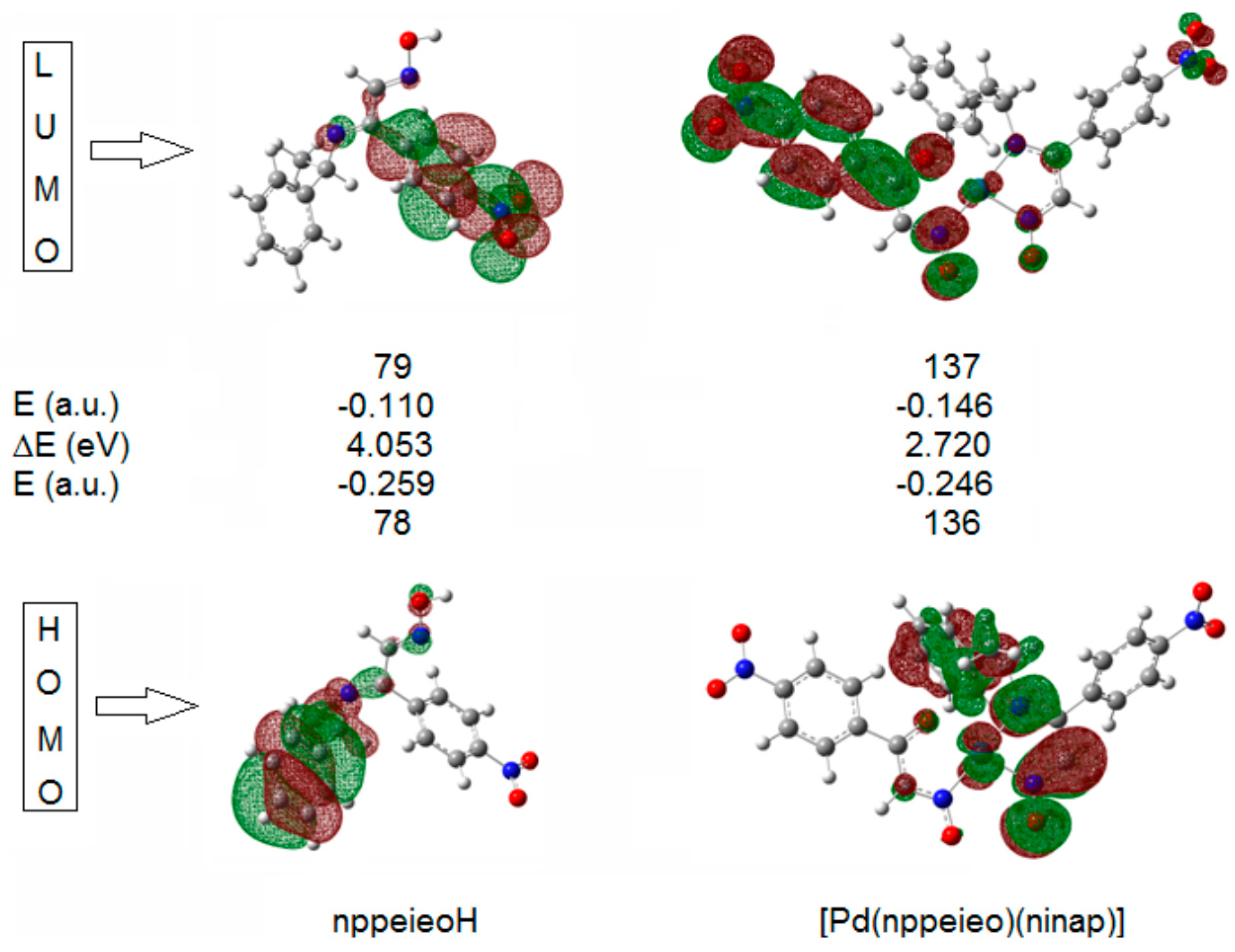
2.6. Frontier Molecular Orbitals
| Compound Global Reactivity Descriptors | nppeieoH | [Pd(nppeieo)(ninap)] |
|---|---|---|
| E (HOMO, a.u.) | −0.259 | −0.246 |
| E (LUMO, a.u.) | −0.110 | −0.146 |
| ΔE (eV) | 4.053 | 2.720 |
| χ | −5.018 | −5.372 |
| η | 2.026 | 1.360 |
| S | 0.247 | 0.368 |
| ω | 6.214 | 10.610 |

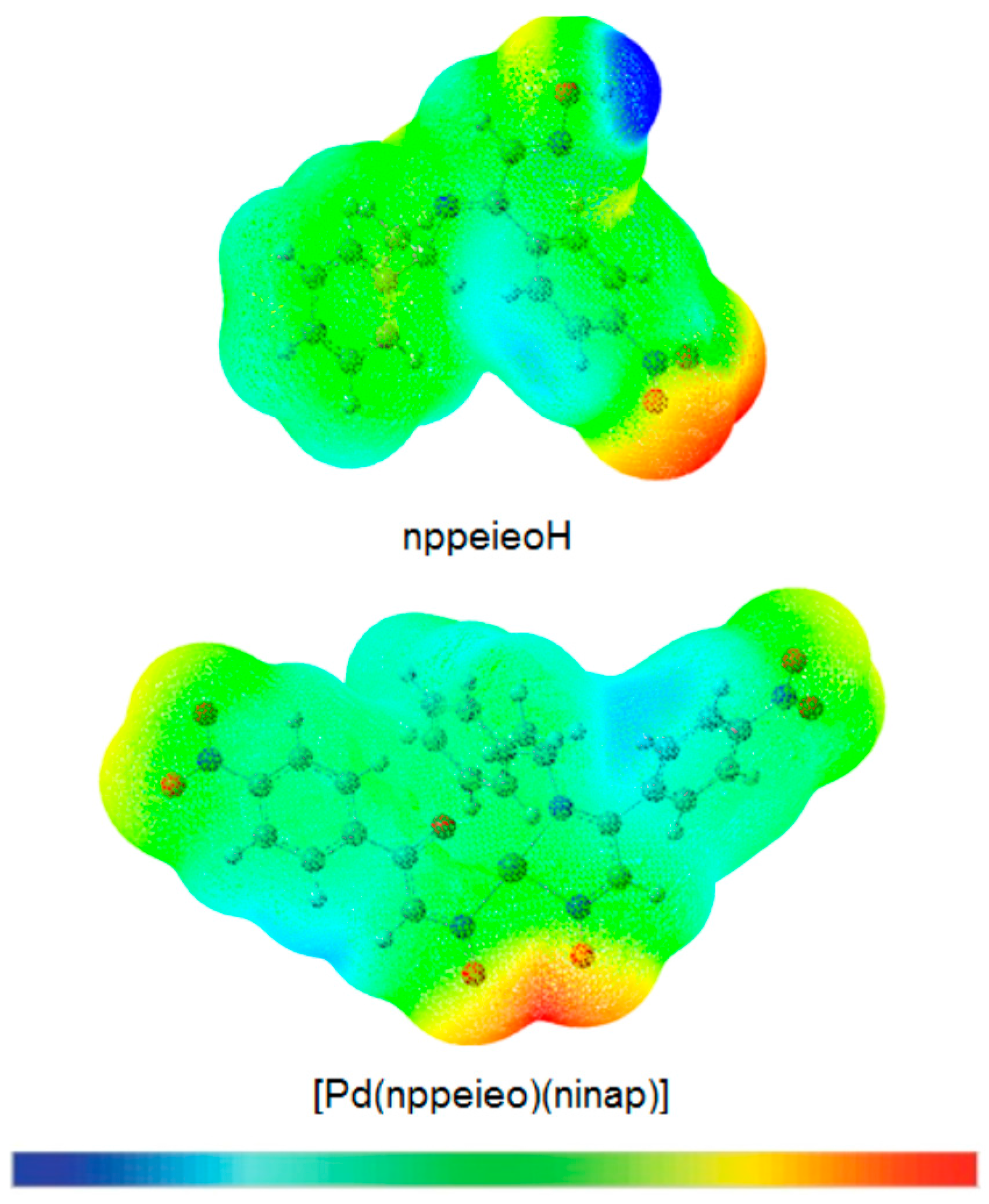
2.7. Molecular Electrostatic Potential Map

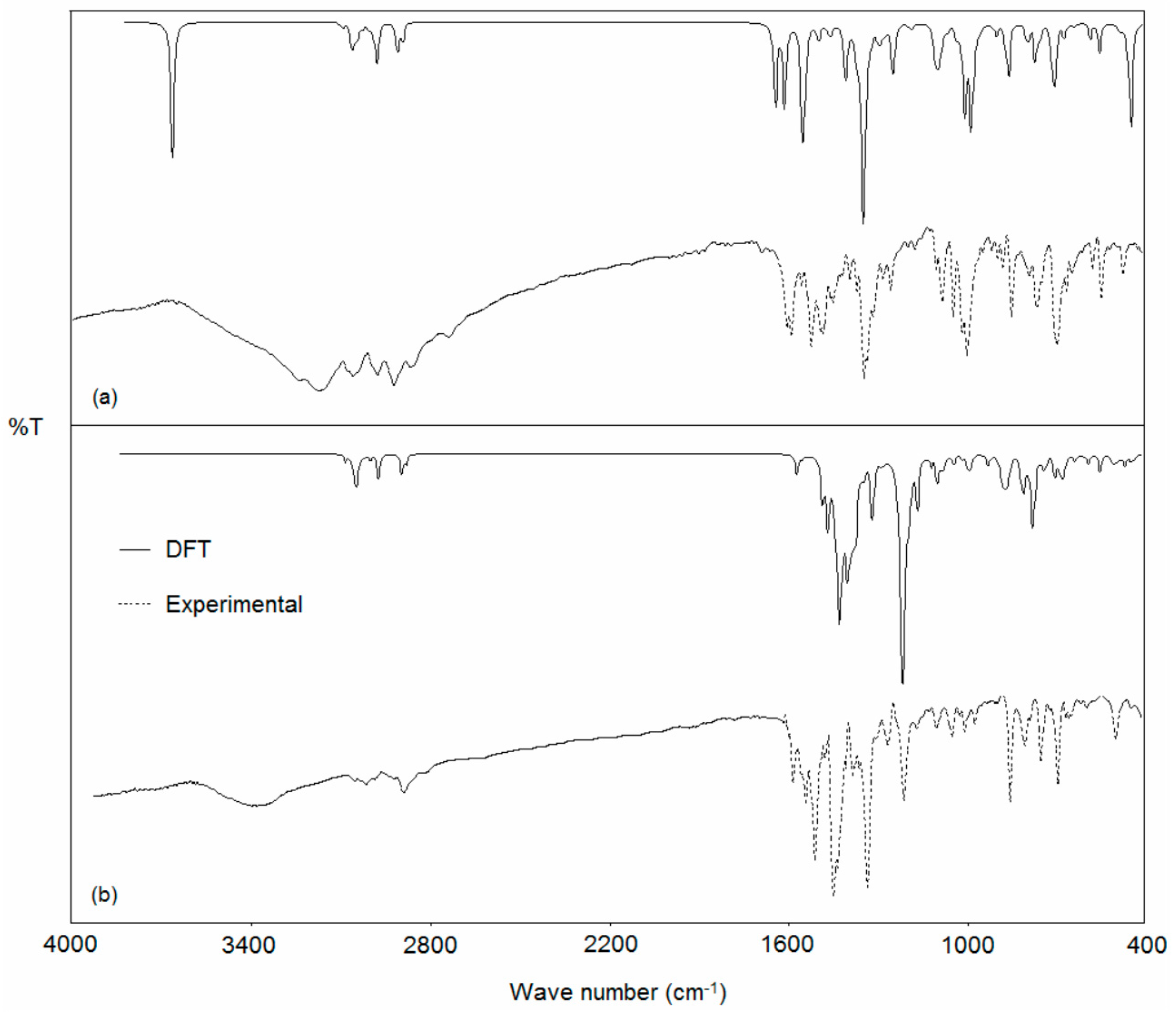
2.8. Vibrational Spectroscopy

| nppeieoH | [Pd(nppeieo)(ninap)] | ||||||
|---|---|---|---|---|---|---|---|
| Assignments (PED > 10) | Exp. | IR | I | Assignments (PED > 10) | Exp. | IR | I |
| νOH (97) | 3258br | 3659 | 158 | νCHphen. (92) | 3108w | 3097 | 31 |
| νCHphen. (95) | 3078w | 3052 | 20 | νCHphen. (97) | 3073w | 3090 | 26 |
| νCHphen. (91) | 3041 | 19 | νCHphen. (89) | 3051w | 3050 | 10 | |
| νCHmethyl (88) | 2983w | 2982 | 15 | νCHphen. (95) | 3020 | 37 | |
| νCHmethyl (90) | 2931w | 2970 | 32 | νCHmethyl (89) | 2943w | 2942 | 28 |
| νCHmethyl (87) | 2904 | 24 | νCHHCNO (98) | 2927 | 21 | ||
| νCHHCNO (99) | 2876w | 2887 | 23 | νCO (37) + νCN (27) + νCCphen. (24) | 1601m | 1587 | 24 |
| νCNimine (67) + νCNoxime (21) | 1604s | 1647 | 11 | νCN (57) + νCCphen (12) | 1562m | 1499 | 61 |
| νCNoxime (61) + νCNimine (18) | 1597s | 1633 | 76 | νCCphen. (49) | 1532s | 1482 | 129 |
| νCCphen. (54) + ™CHphen. (21) | 1605 | 58 | ™CHphen. (63) | 1474 | 13 | ||
| νCCphen (48) + ™CHphen. (34) | 1604 | 15 | ™CHphen. (71) + νCN (17) | 1468s | 1464 | 32 | |
| νCCphen. (50) + ™CHphen. (18) | 1563w | 1600 | 16 | ™CHphen. (41) + ™CHaliph. (23) | 1451s | 1446 | 351 |
| νNONO2 (51) + νCCphen. (17) | 1521s | 1541 | 207 | ™CHaliph. (33) + νCN (21) | 1436 | 341 | |
| ™CHphen. (71) | 1493s | 1490 | 13 | ™CHphen. (64) | 1419 | 26 | |
| ™OH (47) + ™CHphen. (33) | 1403w | 1400 | 36 | ™CHphen. (64) + νCO (13) + νCN (10) | 1399m | 1416 | 261 |
| ™OH (43) + ™CHphen. (39) | 1395 | 29 | ™CHphen. (38) + ™CHaliph. (16) | 1403 | 123 | ||
| ™CHHCN (46) + ™CHphen. (19) | 1356 | 25 | νCNNO2 (68) + νCN (17) | 1353s | 1389 | 222 | |
| νCNNO2 (91) | 1349s | 1340 | 373 | ™CHaliph. (53) | 1382 | 21 | |
| ™CHphen. (38) + ™CHaliph. (26) | 1264m | 1282 | 13 | ™CHphen. (48) + νCNNO2 (18) | 1371 | 18 | |
| ™OH (28) + ™CHHCN (25) | 1201w | 1239 | 62 | ™CHphen. (49) | 1355 | 21 | |
| νC–N (37) + νC–C (20) | 1092m | 1095 | 26 | ™CHaliph. (38) | 1333 | 23 | |
| νC–N (18) + νC–C(32) | 1088 | 34 | ™CHaliph. (41) + νCCphen. (12) | 1389w | 1329 | 93 | |
| ™CHaliph. (36) | 1073 | 20 | ™CHphen. (27) + ™CHaliph. (14) | 1319 | 20 | ||
| νNO(62) + ™CHaliph. (16) | 999s | 1000 | 78 | ™CHaliph. (43) | 1234 | 30 | |
| νNO(32) + ™CCphen. (28) | 994 | 33 | νCNNO2 (63) + νNO (14) | 1227 | 314 | ||
| νNO(24) + ™CHaliph. (46) | 976 | 113 | νNO (64) + νCNNO2 (23) | 1218m | 1222 | 723 | |
| ™CHaliph. (27) | 887w | 893 | 12 | ™CHaliph. (33) + ™CHphen. (24) | 1218 | 25 | |
| ™NONO2 (48) + ™CCphen. (27) | 856m | 853 | 50 | ™CHphen. (46) + νCN (16) | 1180w | 1202 | 79 |
| γCCphen. (47) + γCHaliph. (27) | 771m | 787 | 25 | ™CHphen. (48) | 1174 | 31 | |
| γCCphen (52) | 762 | 28 | ™CHphen. (54) | 1173 | 46 | ||
| γCCphen. (35) + γNONO2 (27) | 702s | 698 | 63 | ™CHphen. (26) + ™CHaliph. (14) | 1112w | 1104 | 34 |
| γCCphen (63) | 697 | 30 | ™CHphen (43) | 1099 | 13 | ||
| γCN(37) + γCCphen (25) | 652w | 662 | 14 | νC–N (47) + νC–N (12) | 1049w | 1083 | 13 |
| γCCphen. (42) | 555m | 546 | 26 | ™CCphen. (58) + ™CHphen. (10) | 984w | 995 | 25 |
| γOH (61) | 486w | 442 | 125 | νC–N (26) + ™CHaliph. (18) | 933 | 14 | |
| γCCphen (73) | 887 | 36 | |||||
| γCCphen. (68) | 856m | 871 | 72 | ||||
| ™CHaliph. (47) | 810w | 818 | 38 | ||||
| ™CHaliph. (32) | 808 | 23 | |||||
| ™CHaliph. (23) | 804 | 36 | |||||
| ™NONO2 (38) + ™CCphen. (22) | 780 | 66 | |||||
| ™NONO2 (41) + γCCphen. (27) | 756m | 774 | 103 | ||||
| γCCphen. (38) | 736 | 29 | |||||
| γCCphen. (56) | 698m | 699 | 42 | ||||
| γCCphen. (32) + γNONO2 (37) | 678 | 30 | |||||
| γCCphen. (28) + γNONO2 (21) | 671w | 677 | 27 | ||||
| νPdO (27) + νPdN (12) + δONC (12) | 575vw | 549 | 23 | ||||
| γCNCC (38) + γCCphen. (22) | 488w | 465 | 10 | ||||
| γCNCC (28) + γCCphen. (21) | 451vw | 462 | 10 | ||||
| γCCphen. (37) | 429 | 10 | |||||
| νPdN (14), γCCphen. (14), γCNCC (10) | 260w | 253 | 20 | ||||
| νPdN (21), δPdCN (15) | 244 | 18 | |||||
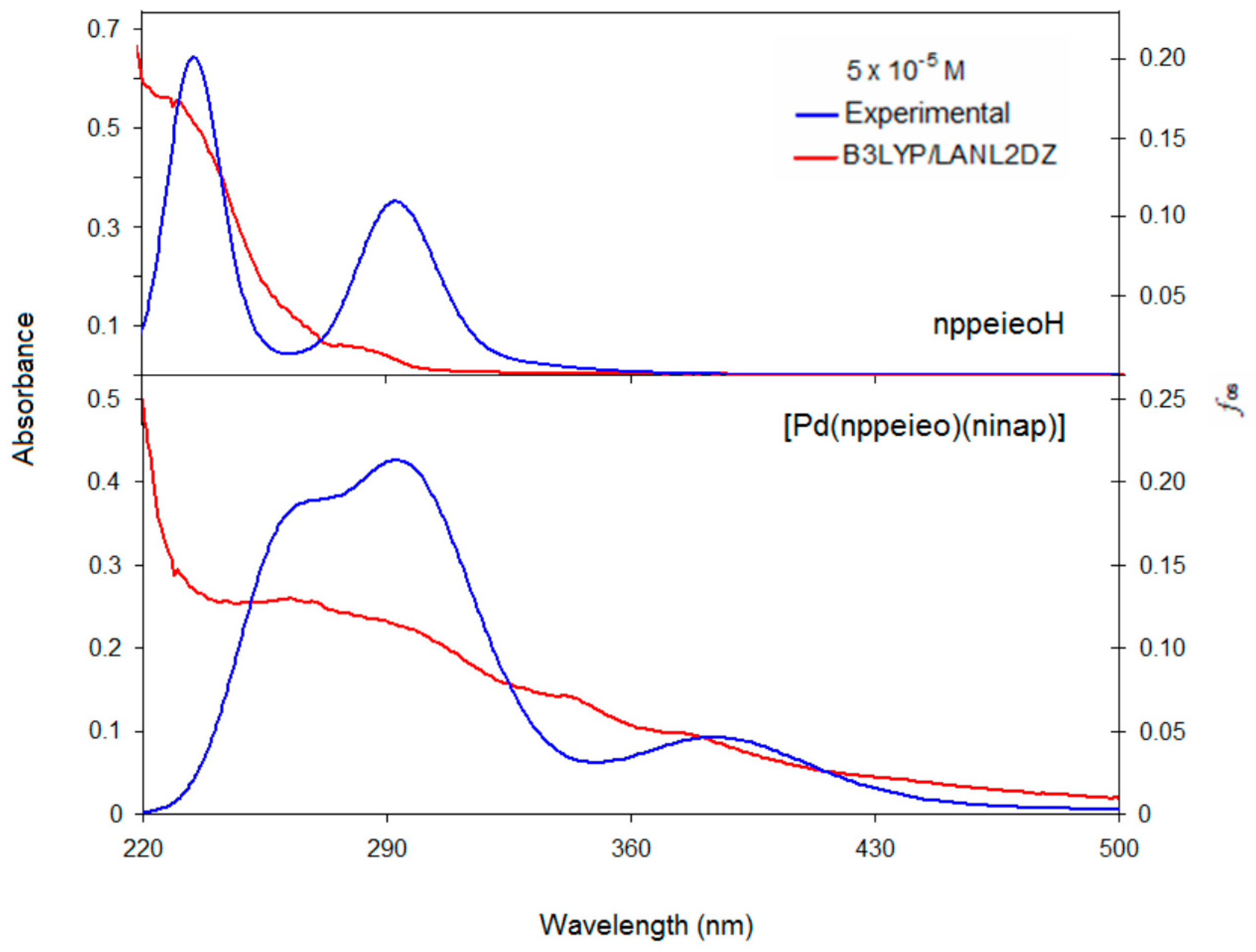
2.9. Electronic Absorption Spectra

| Exp. (nm) | ε | Calcd. (nm) | fos | Major Contribution (CI Coeff.) | Character |
|---|---|---|---|---|---|
| NppeieoH | |||||
| 282 | 0.1116 | 293 | 0.3334 | H–5 → L (87%) | π (phen.) → π* (imineoxime) |
| 282 | 0.0300 | H → L + 1 (84%) | π (phen.) → π* (phen.) | ||
| 242 | 0.0533 | H–4 → L + 1 (57%) | π (phen.) → π* (phen.) | ||
| H–5 → L + 1 (25%) | |||||
| 236 | 0.2036 | H–6 → L + 1 (38%) | π (oxime) → π* (imineoxime) | ||
| 231 | 1.0866 | 233 | 0.3864 | H–6 → L + 1 (22%) | π (phen.) → π* (phen.) |
| H–5 → L + 1 (13%) | π (imineoxime) → π* (phen.) | ||||
| [Pd(nppeieo)(ninap)] | |||||
| 376 | 0.1904 | 415 | 0.1391 | H-4 → L (74%) | d(Pd)/π (imineoxime)→π* (imineoxime) |
| 341 | 0.2839 | 346 | 0.2152 | H-7 → L (30%) | π (imineoxime) → π* (imineoxime) |
| 329 | 0.0495 | H–8 → L (32%) | π (phen.) → π* (imineoxime) | ||
| 324 | 0.1328 | H–5 → L + 2 (33%) | π (phen.) → d(Pd)/π* (imineoxime) | ||
| 295 | 0.4526 | 323 | 0.2073 | H–7 → L (18%) | π (oxime) → π* (imineoxime) |
| H–8 → L (13%) | |||||
| 296 | 0.1196 | H–7 → L + 1 (27%) | π (imineoxime) → π* (phen.) | ||
| 289 | 0.1748 | H–7 → L + 1 (23%) | π (imineoxime) → π* (phen.) | ||
| 261 | 0.5164 | 287 | 0.2047 | H−10 → L + 1 (39%) | π (phen.) → π* (phen.) |
2.10. Non-Linear Optical Properties
3. Experimental Section
3.1. General
3.2. Synthesis of nppeieoH and Its Palladium(II) Complex
3.3. X-ray Crystallography
3.4. Theoretical Calculations
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Karljiković, K.; Stanković, B.; Binenfeld, Z. Spectrophotometric behaviour of quinolinium oxime-palladium(II) complexes. Mikrochim. Acta 1985, 2, 195–202. [Google Scholar] [CrossRef]
- Kabil, M.A.; Akl, M.A.; Khalifa, M.E. Selective flotation-spectrophotometric procedure for the trace analysis of Palladium(II) in different matrices. Anal. Sci. 1999, 15, 433–438. [Google Scholar] [CrossRef]
- Sastre, A.M.; Szymanowski, J. Discussion of the physicochemical effects of modifiers on the extraction properties of hydroxyoximes. A review. Solv. Extr. Ion Exch. 2004, 22, 737–759. [Google Scholar] [CrossRef]
- Dede, B.; Karipcin, F.; Cengiz, M. Synthesis, characterization and extraction studies of N,N″-bis[1-biphenyl-2-hydroxyimino-2-(4-acetylanilino)-1-ethylidene]-diamines and their homo- and heteronuclear copper(II) complexes. J. Chem. Sci. 2009, 121, 163–171. [Google Scholar] [CrossRef]
- Shokrollahi, A.; Ghaedi, M.; Ghaedi, H. Potentiometric and spectrophotometric studies of Copper(II) complexes of some ligands in aqeuous and nonaqeuous solution. J. Chin. Chem. Soc. 2007, 54, 933–940. [Google Scholar] [CrossRef]
- Nakamura, H.; Iitaka, Y.; Sakakibara, H.; Umezawa, H. The molecular and crystal structure determination of bisanhydroal thiomycin by the X-ray diffraction method. J. Antibiot. 1974, 27, 894–896. [Google Scholar] [CrossRef] [PubMed]
- Kirst, H.A.; Szymanski, E.F.; Doman, D.E.; Occolowitz, J.L.; Jones, N.D.; Chaney, M.O.; Hamill, R.L.; Hoehn, M.M. Structure of althiomycin. J. Antibiot. 1975, 28, 286–291. [Google Scholar] [CrossRef] [PubMed]
- Ponomareva, V.V.; Dalley, N.K.; Kou, X.; Gerasimchuk, N.N.; Domasevich, K.V. Synthesis, spectra and crystal structures of complexes of ambidentate C6H5C(O)C(NO)CN−. J. Chem. Soc. Dalton Trans. 1996, 2351–2359. [Google Scholar] [CrossRef]
- Hambley, T.W.; Ling, E.C.H.; O’Mara, S.; McKeage, M.J.; Russell, P.J. Increased targeting of adenine-rich sequences by (2-amino-2-methyl-3-butanone oxime)dichloroplatinum(II) and investigations into its low cytotoxicity. J. Biol. Inorg. Chem. 2000, 5, 675–681. [Google Scholar] [CrossRef] [PubMed]
- Quiroga, A.G.; Cubo, L.; de Blas, E.; Aller, P.; Navarro-Ranninger, C.J. Trans platinum complexes design: One novel water soluble oxime derivative that contains aliphatic amines in trans configuration. Inorg. Biochem. 2007, 101, 104–110. [Google Scholar] [CrossRef] [PubMed]
- Zorbas-Seifried, S.; Jakupec, M.A.; Kukushkin, N.V.; Groessl, M.; Hartinger, C.G.; Semenova, O.; Zorbas, H.; Kukushkin, V.Y.; Kepler, B.K. Reversion of structure-activity relationships of antitumor platinum complexes by acetoxime but not hydroxylamine ligands. Mol. Pharmacol. 2007, 71, 357–365. [Google Scholar] [CrossRef] [PubMed]
- Scaffidi-Domianello, Y.Y.; Meelich, K.; Jakupec, M.A.; Arion, V.B.; Kukushkin, V.Y.; Galanski, M.; Kepler, B.K. Novel cis- and trans-configured bis(oxime)platinum(II) complexes: Synthesis, characterization, and cytotoxic activity. Inorg. Chem. 2010, 49, 5669–5678. [Google Scholar] [CrossRef] [PubMed]
- Kaya, Y.; Icsel, C.; Yilmaz, V.T.; Buyukgungor, O. A palladium(II) complex containing both carbonyl and imine oxime ligands: Crystal structure, experimental and theoretical UV-Vis, IR and NMR studies. Spectrochim. Acta A 2013, 108, 133–140. [Google Scholar] [CrossRef] [PubMed]
- Kaya, Y.; Yilmaz, V.T. Theoretical study of hydrolysis of an imine oxime in aqueous solution and crystal structure and spectroscopic characterization of a platinum(II) complex containing the hydrolysis product. Struct. Chem. 2013, 25, 231–238. [Google Scholar] [CrossRef]
- Johnson, R.W.; Stieglitz, J. The velocity of hydrolysis of stereoisomeric hydrazones and oximes. J. Am. Chem. Soc. 1934, 56, 1904–1908. [Google Scholar] [CrossRef]
- Depuy, C.H.; Ponder, B.W. Levulinic acid as a reagent for the hydrolysis of oximes and 2,4-dinitrophenylhydrazones. J. Am. Chem. Soc. 1959, 81, 4629–4631. [Google Scholar] [CrossRef]
- Mrlina, G.; Calmon, J.-P. Kinetics and mechanism of hydrolysis of insecticidal O-(methylcarbamoyl)oximes. J. Agric. Food Chem. 1980, 28, 605–609. [Google Scholar] [CrossRef]
- Attanasi, O.; Gasperoni, S.; Carletti, C. Effect of Copper(II) Ions in organic synthesis. V. Regeneration of carbonyl compounds from tosylhydrazones, phenylhydrazones, hydrazones, oximes and semicarbazones by hydrolysis in the presence of copper(II) sulphate pentahydrate. J. Prakt. Chem. 1980, 322, 1063–1066. [Google Scholar] [CrossRef]
- Lin, M.H.; Liu, H.J.; Chang, C.Y.; Lin, W.C.; Chuang, T.H. SnCl2/TiCl3-Mediated Deoximation of Oximes in an Aqueous Solvent. Molecules 2012, 17, 2464–2473. [Google Scholar] [CrossRef] [PubMed]
- Bird, J.W.; Diaper, D.G. Preparative conversion of oximes to parent carbonyl compounds by cerium(IV) oxidation. Can. J. Chem. 1969, 47, 145–150. [Google Scholar] [CrossRef]
- Yamaguchi, Y.; Yasutake, N.; Nagaoka, M. Ab initio study of noncatalytic Beckmann rearrangement and hydrolysis of cyclohexanone-oxime in subcritical and supercritical water using the polarizable continuum model. J. Mol. Struct. 2003, 639, 137–150. [Google Scholar] [CrossRef]
- Vladimirova, K.G.; Freidzon, A.Y.; Kotova, O.V.; Vaschenko, A.A.; Lepnev, L.S.; Bagatur’yants, A.A.; Vitukhnovskiy, A.G.; Stepanov, N.F.; Alfimov, M.V. Theoretical study of structure and electronic absorption spectra of some Schiff bases and their zinc complexes. Inorg. Chem. 2009, 48, 11123–11130. [Google Scholar] [CrossRef] [PubMed]
- Das, C.; Adak, P.; Mondal, S.; Sekiya, R.; Kuroda, R.; Gorelsky, S.I.; Chattopadhyay, S.K. Synthesis, characterization, X-ray crystal structure, DFT calculations, and catalytic properties of a dioxidovanadium(V) complex derived from oxamohydrazide and pyridoxal: A model complex of vanadate-dependent bromoperoxidase. Inorg. Chem. 2014, 53, 11426–11437. [Google Scholar] [CrossRef] [PubMed]
- Bingol Alpaslan, Y.; Alpaslan, G.; Alaman Agar, A.; Ocak Iskeleli, N.; Oztekin, E. Experimental and density functional theory studies on (E)-2-[(2-(hydroxymethyl)phenylimino)methyl]benzene-1,4-diol. J. Mol. Struct. 2011, 995, 58–65. [Google Scholar] [CrossRef]
- Gfeller, D.; Grosdidier, A.; Wirth, M.; Daina, A.; Michielin, O.; Zoete, V. Swiss target prediction: A web server for target prediction of bioactive small molecules. Nucl. Acids Res. 2014, 42, W32–W38. [Google Scholar] [CrossRef] [PubMed]
- Tewari, A.K.; Singh, V.P.; Yadav, P.; Gupta, G.; Singh, A.; Goel, R.K.; Shinde, P.; Mohan, C.G. Synthesis, biological evaluation and molecular modeling study of pyrazole derivatives as selective COX-2 inhibitors and anti-inflammatory agents. Bioorg. Chem. 2014, 56, 8–15. [Google Scholar] [CrossRef] [PubMed]
- Thakkar, N.V.; Haldar, B.C. Magnetic and spectral studies of complexes of isonitroso-acetophenone (HINAP) with Ni(II), Pd(II) and Pt(II). Inorg. Nucl. Chem. 1980, 42, 843–849. [Google Scholar] [CrossRef]
- Politzer, P.; Murray, J.S. The Chemistry of Hydroxylamines, Oximes and Hydroxamic Acids; Wiley: West Sussex, UK, 2009. [Google Scholar]
- Yilmaz, A.; Taner, B.; Deveci, P.; Yilmaz, O.A.; Arslan, U.; Sahin, E.; Ucan, H.I.; Ozcan, E. Novel bioactive vic-dioxime ligand containing piperazine moiety: Synthesis, X-ray crystallographic studies, 2D NMR applications and complexation with Ni(II). Polyhedron 2010, 29, 2991–2998. [Google Scholar] [CrossRef]
- Dash, A.C.; Dash, B.; Panda, D. Hydrolysis of imines. 4. Micellar effects upon the spontaneous acid, base, and copper(II) ion induced hydrolysis of N-salicylidene-2-aminothiazole and N-salicylidene-2-aminopyridine. J. Org. Chem. 1985, 50, 2905–2910. [Google Scholar] [CrossRef]
- Hall, N.E.; Smith, B.J. High-level ab initio molecular orbital calculations of imine formation. J. Phys. Chem. A 1998, 102, 4930–4938. [Google Scholar] [CrossRef]
- Hartley, J.H.; James, T.D. Saccharide accelerated hydrolysis of boronic acid imines. Tetrahedron Lett. 1999, 40, 2597–2600. [Google Scholar] [CrossRef]
- Godoy-Alcántar, C.; Yatsimirsky, A.K.; Lehn, J.-M. Structure-stability correlations for imine formation in aqueous solution. J. Phys. Org. Chem. 2005, 18, 979–985. [Google Scholar] [CrossRef]
- Colak, A.T.; Irez, G.; Mutlu, H.; Hokelek, T.; Caylak, N. A Co(III) complex with a tridentate amine-imine-oxime ligand from 1,2,3,4-tetrahydroquinazoline: synthesis, crystal structure, spectroscopic and thermal characterization. J. Coord. Chem. 2008, 62, 1005–1014. [Google Scholar] [CrossRef]
- Paris, S.I.M.; Laskay, Ü.A.; Liang, S.; Pavlyuk, O.; Tschirschwitz, S.; Lönnecke, P.; McMills, M.C.; Jackson, G.P.; Petersen, J.L.; Hey-Hawkins, E.; et al. Manganese(II) complexes of di-2-pyridinylmethylene-1,2-diimine di-Schiff base ligands: Structures and reactivity. Inorg. Chim. Acta 2010, 363, 3390–3398. [Google Scholar] [CrossRef]
- Carcelli, M.; Cozzini, P.; Marroni, R.; Pelagatti, P.; Pelizzi, C.; Sgarabotto, P. Unusual coordination mode of a 2-pyridyl ketone oxime ligand in bis(4-butylphenyl 2-pyridyl ketone oximate)palladium(II). Inorg. Chim. Acta 1999, 285, 138–141. [Google Scholar] [CrossRef]
- Audhya, A.; Bhattacharya, K.; Maity, M.; Chaudhury, M. Building metallacrown topology around a discrete [M3(μ3-O)] (M = Ni(II) and Pd(II)) core using oximato oxygen linkers: Synthesis, structures, and spectroscopic characterization of a new family of compounds with an inverse-9-MC-3 motif. Inorg. Chem. 2010, 49, 5009–5015. [Google Scholar] [CrossRef] [PubMed]
- Williams, D.E.; Wohlauer, G.; Rundle, R.E. Crystal structures of nickel and palladium dimethylglyoximes. J. Am. Chem. Soc. 1959, 81, 755–756. [Google Scholar] [CrossRef]
- Hussain, M.S.; Schlemper, E.O. A short intramolecular hydrogen bond: crystal structure of a tetradentate .α-amine oxime complex of palladium(II). Inorg. Chem. 1979, 18, 1116–1121. [Google Scholar] [CrossRef]
- Mav, M.S.; Angelici, R.J.; Powell, D.; Jacobson, R.A. Coordination chemistry of bis(δ-camphorquinone dioximato)nickel(II) and -palladium(II). Reactions and structural studies of some M3Ag3 cluster complexes of camphorquinone dioxime. Inorg. Chem. 1980, 19, 3121–3128. [Google Scholar]
- Bandyopadhyay, D.; Bandyopadhyay, P.; Chakravorty, A.; Cotton, F.A.; Falvello, L.R. Structures of trans-bis[(phenylazo)acetaldoximato]platinum(II) and -palladium(II): A case of nonplanar tetracoordination in a bis complex of palladium(II). Inorg. Chem. 1984, 23, 1785–1787. [Google Scholar] [CrossRef]
- Ryabov, A.D.; Kazankov, G.M.; Yatsimirsky, A.K.; Kuz’mina, L.G.; Burtseva, O.Y.; Dvortsova, N.V.; Polyakov, V.A. Synthesis by ligand exchange, structural characterization, and aqueous chemistry of ortho-palladated oximes. Inorg. Chem. 1992, 31, 3083–3090. [Google Scholar] [CrossRef]
- Pal, C.K.; Chattopadhyay, S.; Sinha, C.; Chakravorty, A. A bis(azo-imine)palladium(II) system with 10 ligand π electrons. Synthesis, structure, serial redox, and relationship to bis(azooximates) and other species. Inorg. Chem. 1996, 3, 2442–2447. [Google Scholar] [CrossRef]
- Selvakumar, K.; Vancheesan, S.; Varghese, B. Synthesis and characterization of cyclopalladated complexes of oximes by ligand-exchange method. Polyhedron 1997, 16, 2257–2262. [Google Scholar] [CrossRef]
- Griffith, D.M.; Bíró, L.; Platts, J.A.; Müller-Bunz, H.; Farkas, E.; Buglyó, P. Synthesis and solution behaviour of stable mono-, di- and trinuclear Pd(II) complexes of 2,5-pyridinedihydroxamic acid: X-ray crystal structure of a novel Pd(II) hydroxamato complex. Inorg. Chim. Acta 2012, 380, 291–300. [Google Scholar] [CrossRef]
- Dodoff, N.I.; Kubiak, M.; Kuduk-Jaworska, J.; Mastalarz, A.; Kochel, A.; Vassilieva, V.; Vassilev, N.; Trendafilova, N.; Georgieva, I.; Lalia-Kantouri, M.; et al. Structure, NMR spectra and cytotoxic effect of palladium(II) and platinum(II) complexes of glyoxylic acid oxime. Chemija 2009, 20, 208–217. [Google Scholar]
- Guhathakurta, B.; Biswas, C.; Naskar, J.P.; Lu, L.; Zhu, M. Synthesis and crystal structure of a complex of palladium(II) with 2-hydroxyimino-3-(2-hydrazonopyridyl)-butane. J. Chem. Crystallogr. 2011, 41, 1355–1359. [Google Scholar] [CrossRef]
- Tanga, G.-D.; Zhao, J.-Y.; Li, R.-Q.; Yuan-Cao; Zhang, Z.-C. Synthesis, characteristic and theoretical investigation of the structure, electronic properties and second-order nonlinearity of salicylaldehyde Schiff base and their derivatives. Spectrochim. Acta A 2011, 78, 449–457. [Google Scholar] [CrossRef] [PubMed]
- Elamurugu Porchelvi, E.; Muthu, S. Vibrational spectra, molecular structure, natural bond orbital, first order hyperpolarizability, thermodynamic analysis and normal coordinate analysis of Salicylaldehyde p-methylphenylthiosemicarbazone by density functional method. Spectrochim. Acta A 2015, 134, 453–464. [Google Scholar] [CrossRef] [PubMed]
- Yousef, T.A.; El-Gammal, O.A.; Ahmed, S.F.; Abu El-Reash, G.M. Synthesis, biological and comparative DFT studies on Ni(II) complexes of NO and NOS donor ligands. Spectrochim. Acta A 2015, 135, 690–703. [Google Scholar] [CrossRef] [PubMed]
- Ternavisk, R.R.; Camargo, A.J.; Machado, F.B.C.; Rocco, J.A.F.F.; Aquino, G.L.B.; Silva, V.H.C.; Napolitano, H.B. Synthesis, characterization, and computational study of a new dimethoxy-chalcone. J. Mol. Model. 2014, 20, 2526–2528. [Google Scholar] [CrossRef] [PubMed]
- Yavuz, M.; Tanak, H. Density functional modelling studies on N-2-Methoxyphenyl-2-oxo-5-nitro-1-benzylidenemethylamine. J. Mol. Struct. Theochem 2010, 961, 9–16. [Google Scholar] [CrossRef]
- Ebrahimipour, S.Y.; Abaszadeh, M.; Castro, J.; Seifi, M. Synthesis, X-ray crystal structure, DFT calculation and catalytic activity of two new oxido-vanadium(V) complexes containingONO tridentate Schiff bases. Polyhedron 2014, 79, 138–150. [Google Scholar] [CrossRef]
- Kaya, Y.; Yilmaz, V.T.; Arslan, T.; Buyukgungor, O. Experimental and theoretical DFT studies of structure, spectroscopic and fluorescence properties of a new imine oxime derivative. J. Mol. Struct. 2012, 1024, 65–72. [Google Scholar] [CrossRef]
- Grzegorzek, J.; Mielke, Z. Photochemistry of salicylaldoxime in solid argon: An experimental and theoretical study. Eur. J. Org. Chem. 2010, 2010, 5301–5309. [Google Scholar] [CrossRef]
- Bekhradnia, A.R.; Arshadi, S. Conformational analysis, infrared, and fluorescence spectra of 1-phenyl-1,2-propandione 1-oxime and related tautomers: Experimental and theoretical study. Monatshefte Chem. 2007, 138, 725–734. [Google Scholar] [CrossRef]
- Alver, O.; Kaya, M.F.; Bilge, M.; Parlak, C. Vibrational spectroscopic investigation and conformational analysis of methacrylamidoantipyrine: A comparative density functional study. J. Theor. Chem. 2013, 2013, 10. [Google Scholar] [CrossRef]
- Sohlberg, K.; Dobbs, K.D. Origin of side bands in the FTIR spectrum of acetone oxime vinyl ether. J. Mol. Struct. (Theochem) 2002, 577, 137–141. [Google Scholar] [CrossRef]
- Arjunan, V.; Mythili, C.V.; Mageswari, K.; Mohan, S. Experimental and theoretical investigations of benzamide oxime. Spectrochim. Acta A 2011, 79, 245–253. [Google Scholar] [CrossRef] [PubMed]
- Albayrak, C.; Odabasoglu, M.; Ozek, A.; Büyükgüngor, O. Synthesis, spectroscopic characterizations and quantum chemical computational studies of (Z)-4-[(E)-(4-fluorophenyl)diazenyl]-6-[(3-hydroxypropylamino)methylene]-2-methoxycyclohexa-2,4-dienone. Spectrochim. Acta A 2012, 85, 85–91. [Google Scholar] [CrossRef] [PubMed]
- Albayrak, C.; Kastas, G.; Odabasoglu, M.; Büyükgüngor, O. Probing the compound (E)-2-[(4-bromophenylimino)methyl]-6-ethoxyphenol mainly from the point of tautomerism in solvent media and the solid state by experimental and computational methods. J. Mol. Struct. 2011, 1000, 162–170. [Google Scholar] [CrossRef]
- Sheldrick, G.M. A short history of SHELX. Acta Cryst. A 2008, 64, 112–122. [Google Scholar] [CrossRef] [PubMed]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact Exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Vreven, T.; Kudin, K.N.; Burant, J.C.; Millam, J.M.; et al. Gaussian 03, Revision C.02; Gaussian, Inc.: Wallingford, CT, USA, 2004.
- Cances, E.; Mennucci, B.; Tomasi, J. A new integral equation formalism for the polarizable continuum model: Theoretical background and applications to isotropic and anisotropic dielectrics. J. Chem. Phys. 1997, 107, 3032–3041. [Google Scholar] [CrossRef]
- Mennucci, B.; Cances, E.; Tomasi, J. Evaluation of solvent effects in isotropic and anisotropic dielectrics and in ionic solutions with a unified integral equation method: Theoretical bases, computational implementation, and numerical applications. J. Phys. Chem. B 1997, 101, 10506–10517. [Google Scholar] [CrossRef]
- Mennucci, B.; Tomasi, J. Continuum solvation models: A new approach to the problem of solute’s charge distribution and cavity boundaries. J. Chem. Phys. 1997, 106, 5151–5198. [Google Scholar] [CrossRef]
- Sundaraganesan, N.; Illakiamani, S.; Saleem, H.; Wojciechowski, P.M.; Michalska, D. FT-Raman and FT-IR spectra, vibrational assignments and density functional studies of 5-bromo-2-nitropyridine. Spectrochim. Acta A 2005, 61, 2995–3001. [Google Scholar] [CrossRef] [PubMed]
- Jesus, A.J.L.; Rosado, M.T.S.; Reva, I.; Fausto, R.; Eusebio, M.E.; Redinha, J.S. Conformational study of monomeric 2,3-butanediols by matrix-isolation infrared spectroscopy and DFT calculations. J. Phys. Chem. A 2006, 110, 4169–4179. [Google Scholar] [CrossRef] [PubMed]
- Johnson, R.D., III. NIST Computational Chemistry Comparison and Benchmark Database; NIST Standart Reference Database Number 101, Release 15b; National Institute of Standards and Technology: Gaithersburg, MD, USA, 2011.
- GaussView, Version 3.07; Semichem Inc.: Shawnee Mission, KS, USA, 2003.
- Vibrational Energy Distribution Analysis VEDA 4; Jamróz, M.H. (Ed.) Publisher: Warsaw, Poland, 2004.
- O’Boyle, N.M.; Tenderholt, A.L.; Langner, K.M. A library for package-independent computational chemistry algorithms. J. Comp. Chem. 2008, 29, 839–845. [Google Scholar] [CrossRef] [PubMed]
- Sample Availability: Samples of the compounds nppeieoH and [Pd(nppeieo)(ninap)] are available from the authors.
© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kaya, Y.; Yilmaz, V.T.; Buyukgungor, O. Synthesis, Spectroscopic, Structural and Quantum Chemical Studies of a New Imine Oxime and Its Palladium(II) Complex: Hydrolysis Mechanism. Molecules 2016, 21, 52. https://doi.org/10.3390/molecules21010052
Kaya Y, Yilmaz VT, Buyukgungor O. Synthesis, Spectroscopic, Structural and Quantum Chemical Studies of a New Imine Oxime and Its Palladium(II) Complex: Hydrolysis Mechanism. Molecules. 2016; 21(1):52. https://doi.org/10.3390/molecules21010052
Chicago/Turabian StyleKaya, Yunus, Veysel T. Yilmaz, and Orhan Buyukgungor. 2016. "Synthesis, Spectroscopic, Structural and Quantum Chemical Studies of a New Imine Oxime and Its Palladium(II) Complex: Hydrolysis Mechanism" Molecules 21, no. 1: 52. https://doi.org/10.3390/molecules21010052