
Experimental and Theoretical Studies in Hydrogen-Bonding Organocatalysis

Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
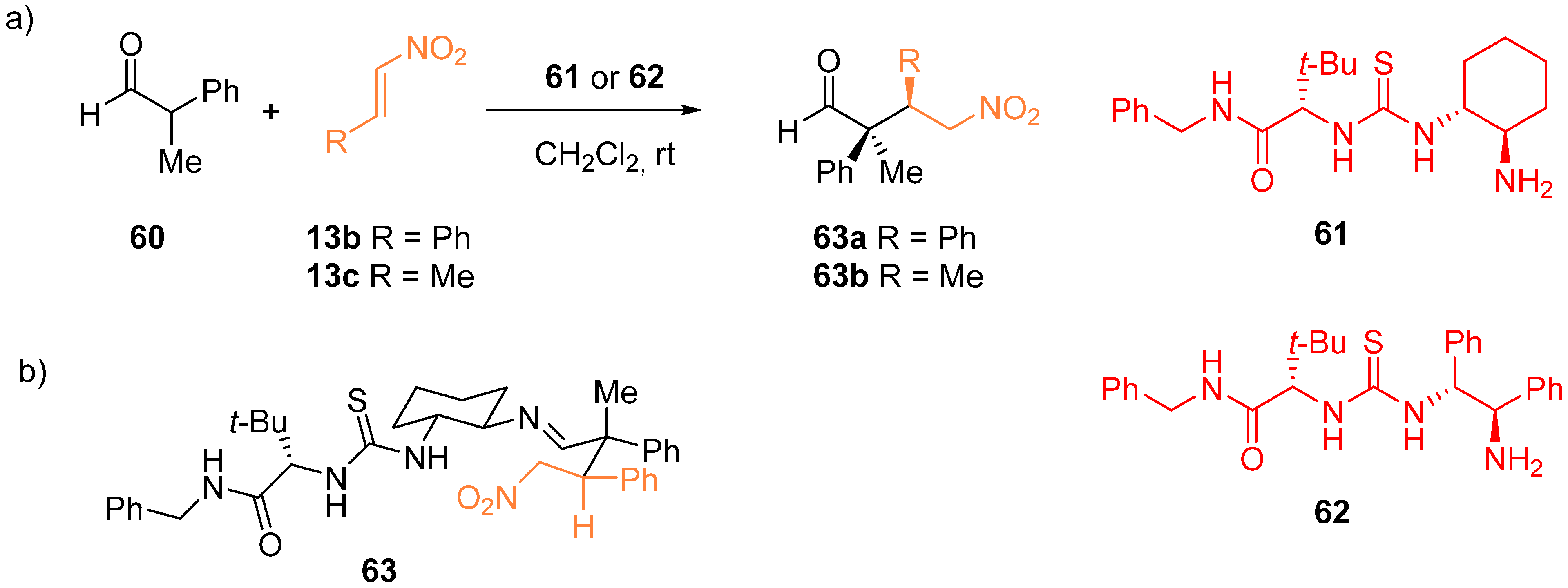{kind=link}
{kind=link}
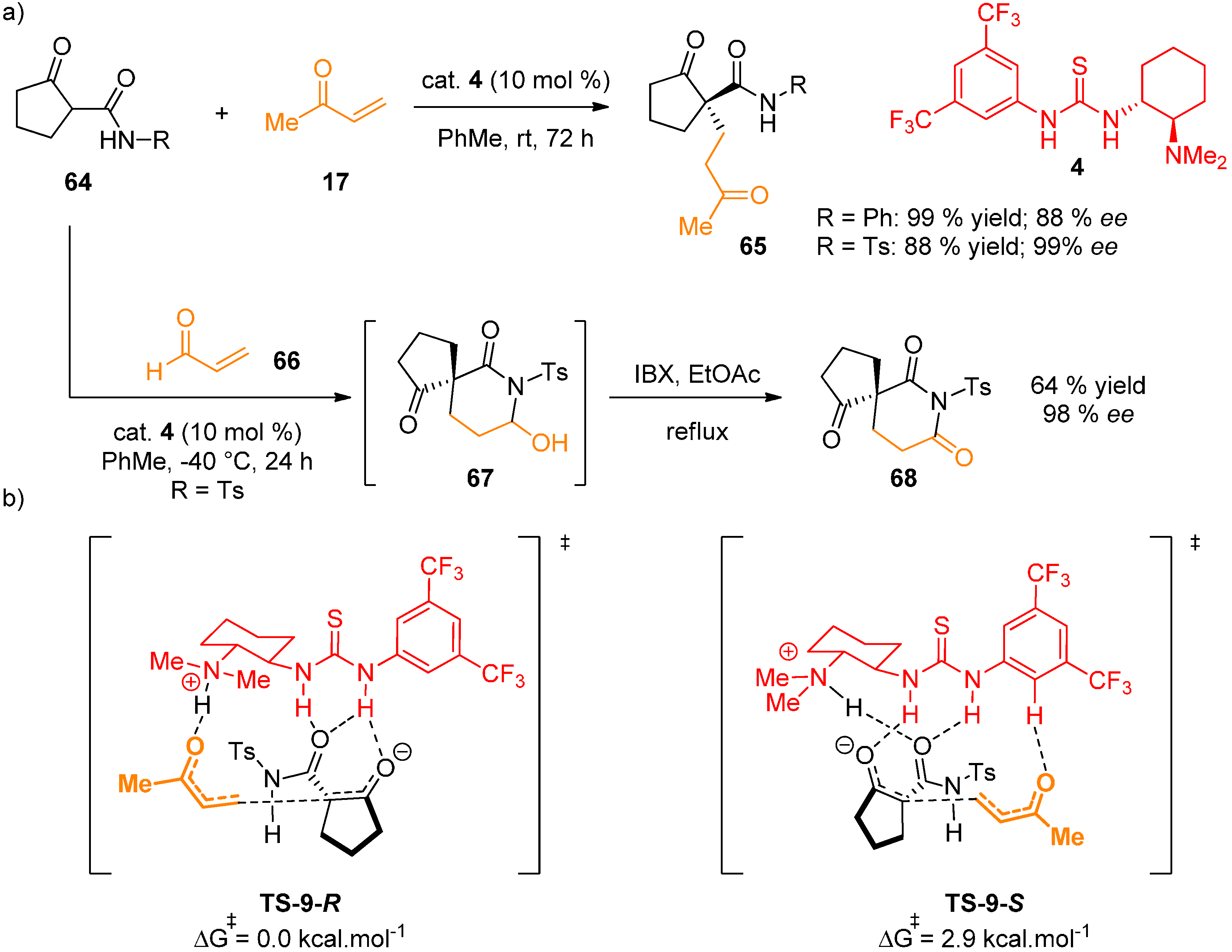{kind=link}
{kind=link}
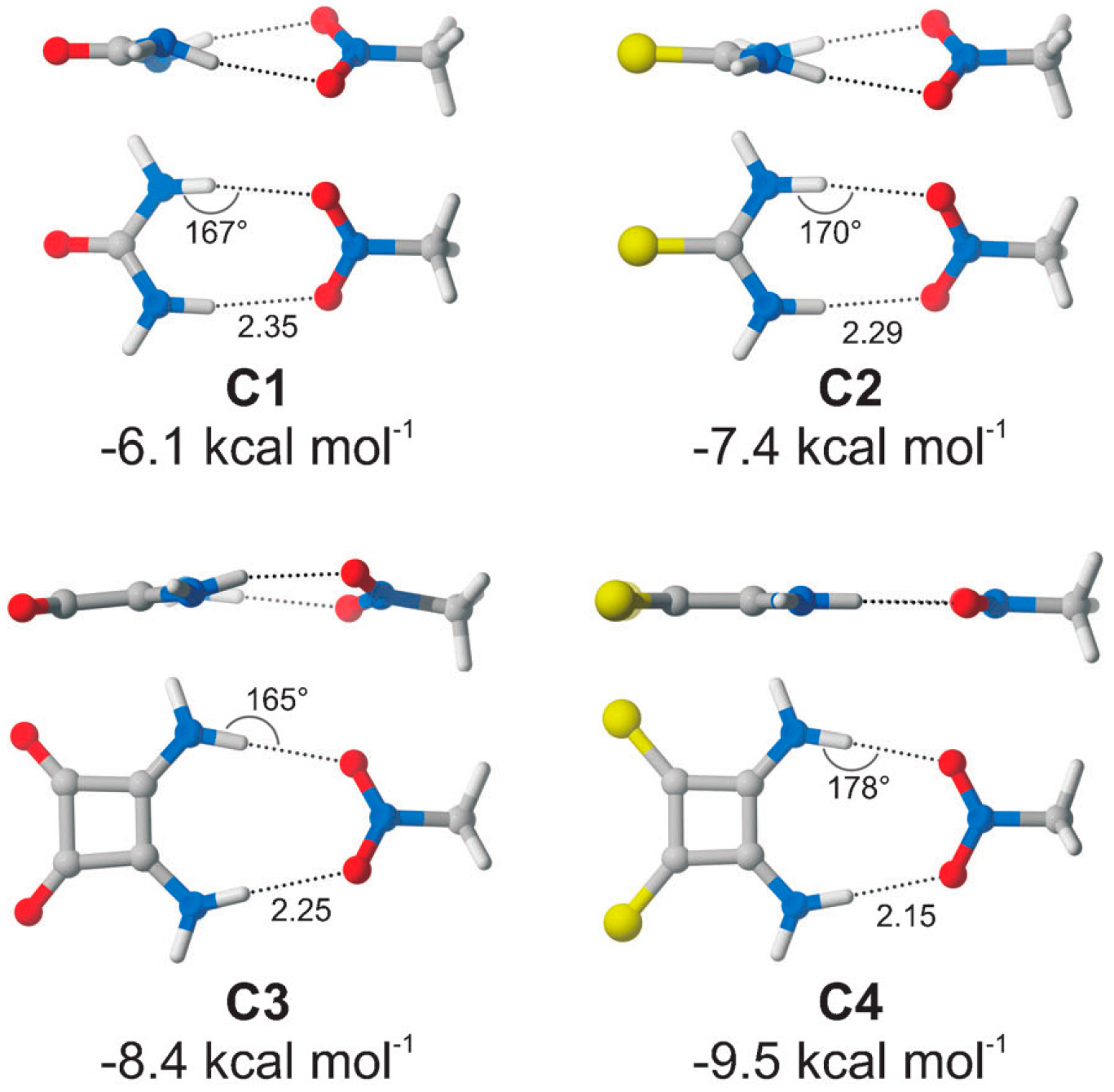{kind=link}
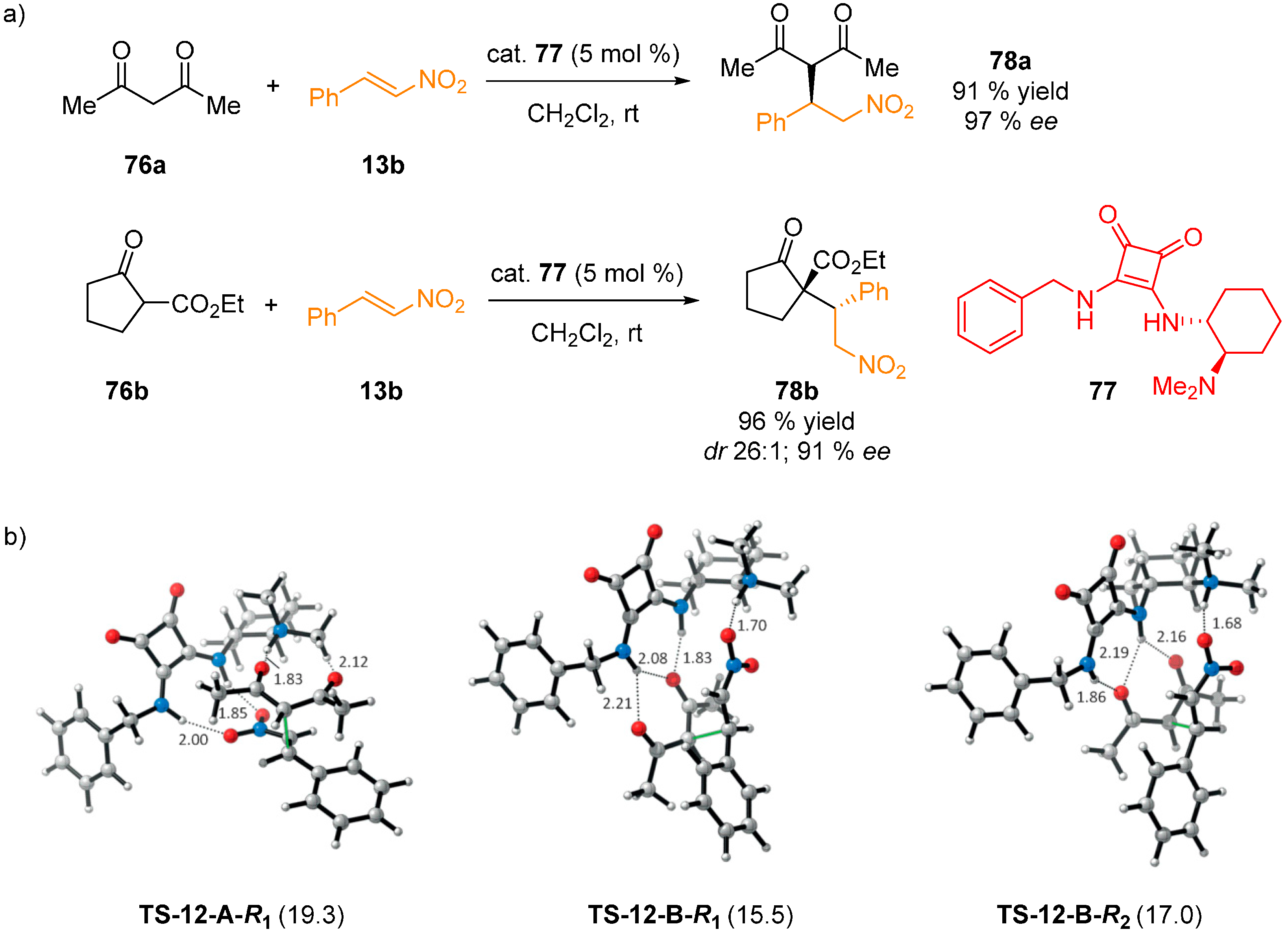{kind=link}
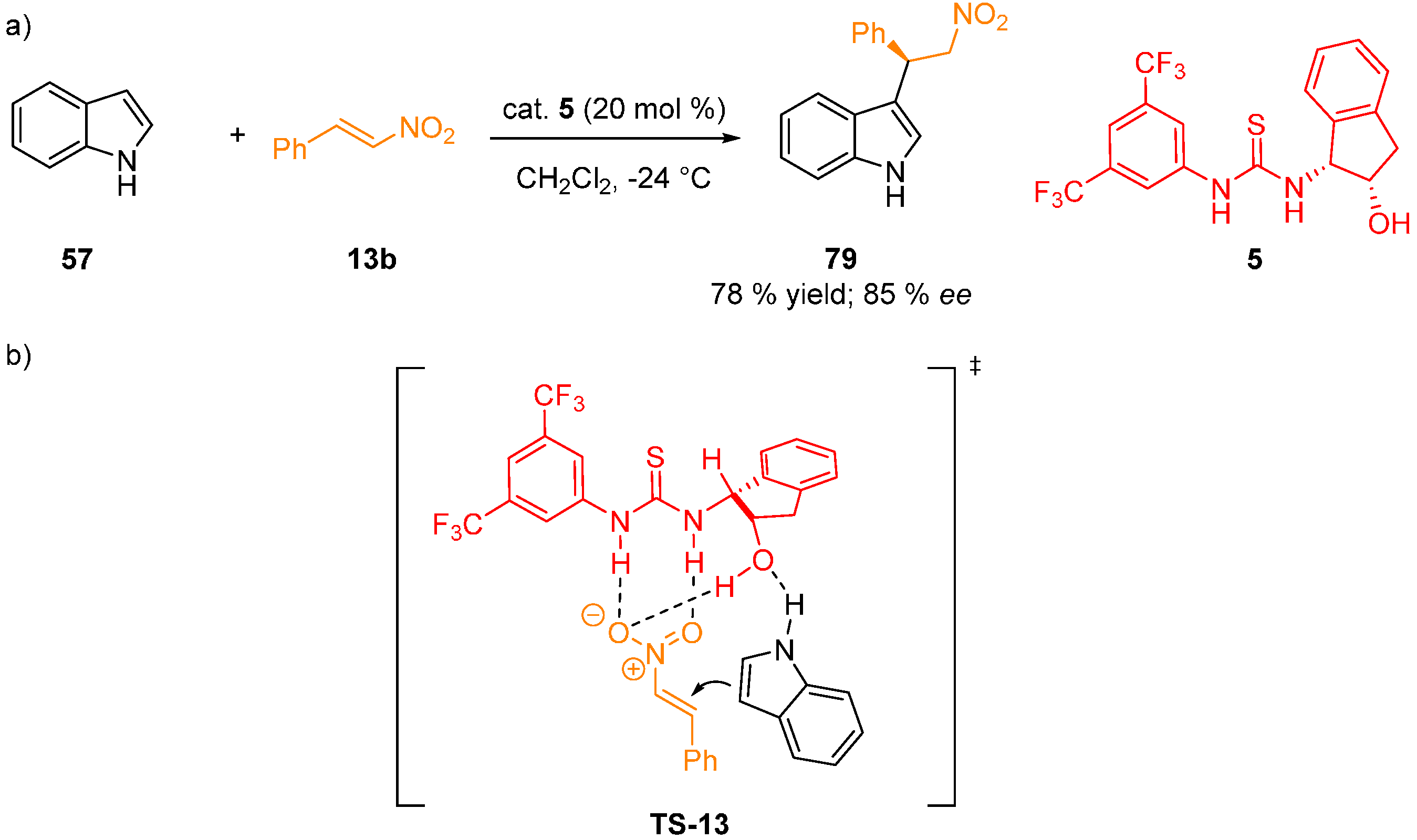{kind=link}
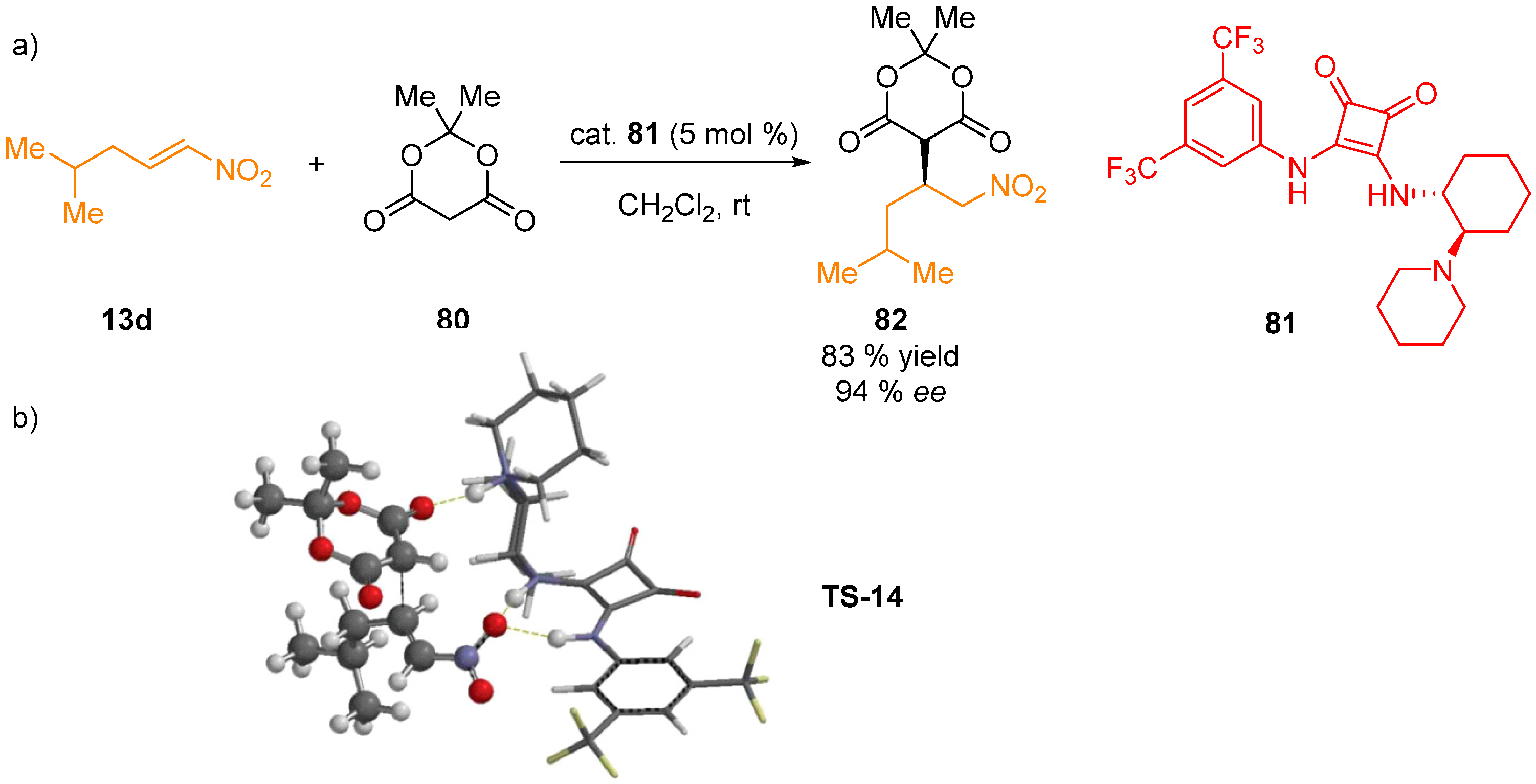{kind=link}
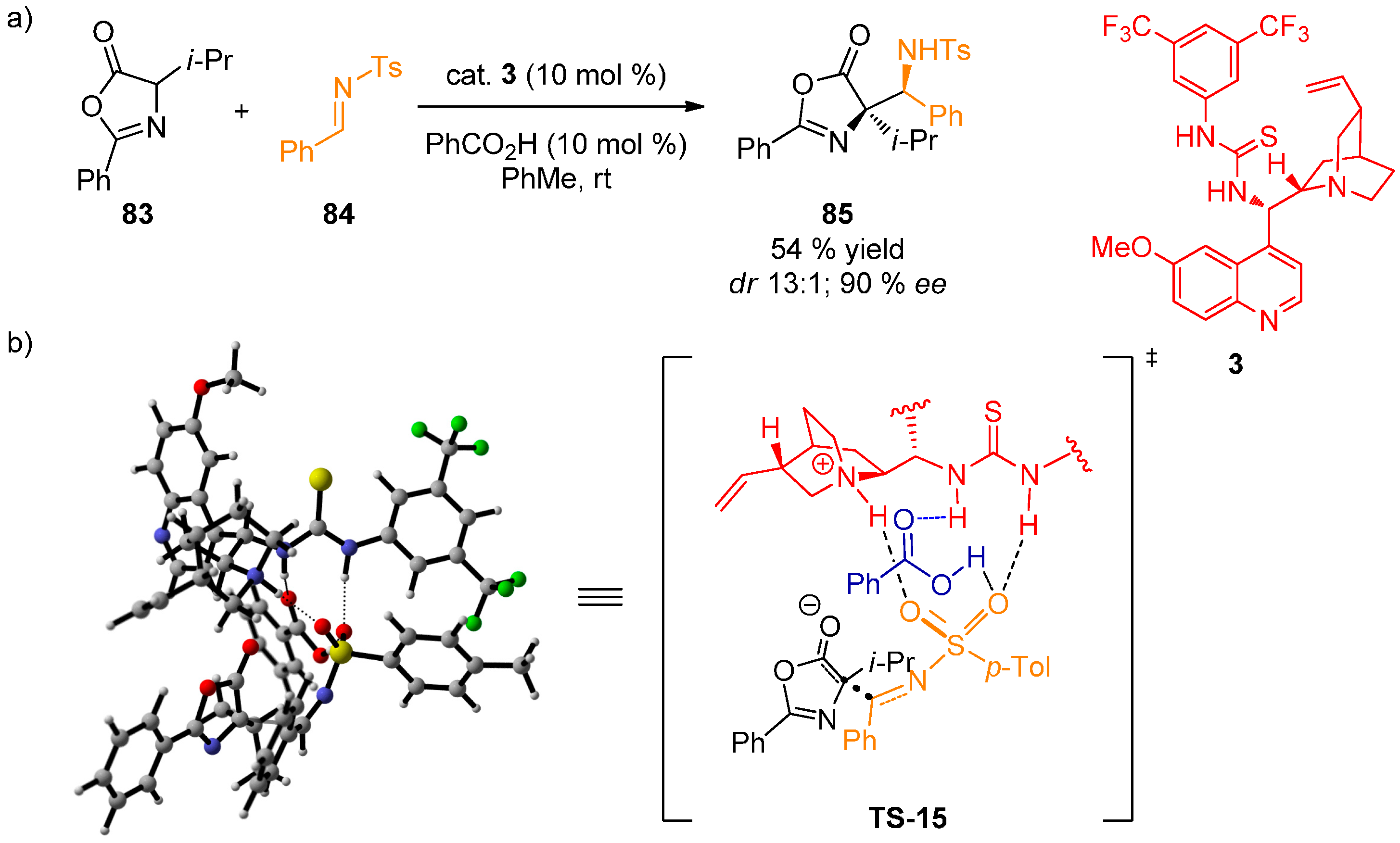{kind=link}
1. Introduction
2. Experimental Techniques
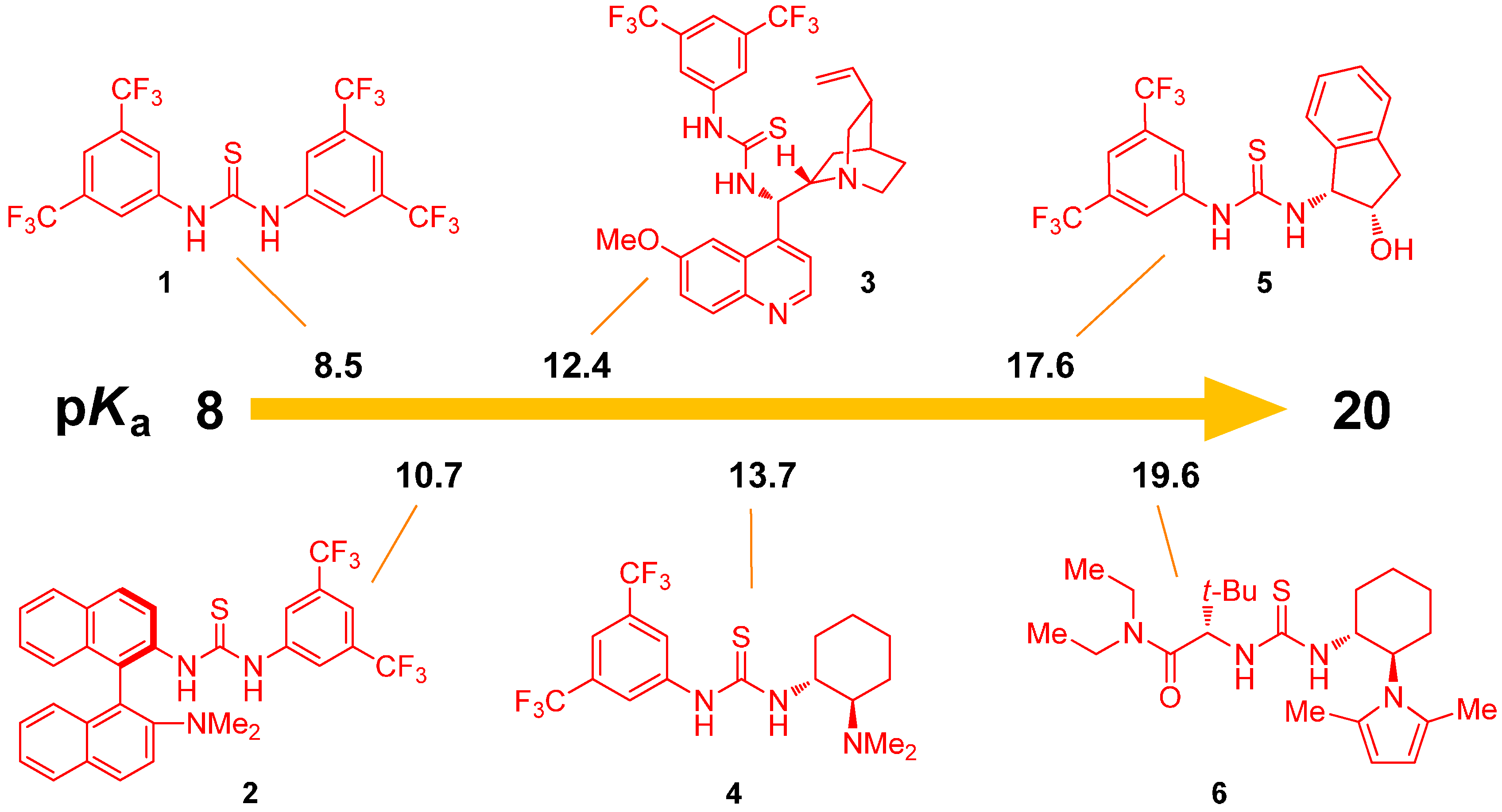
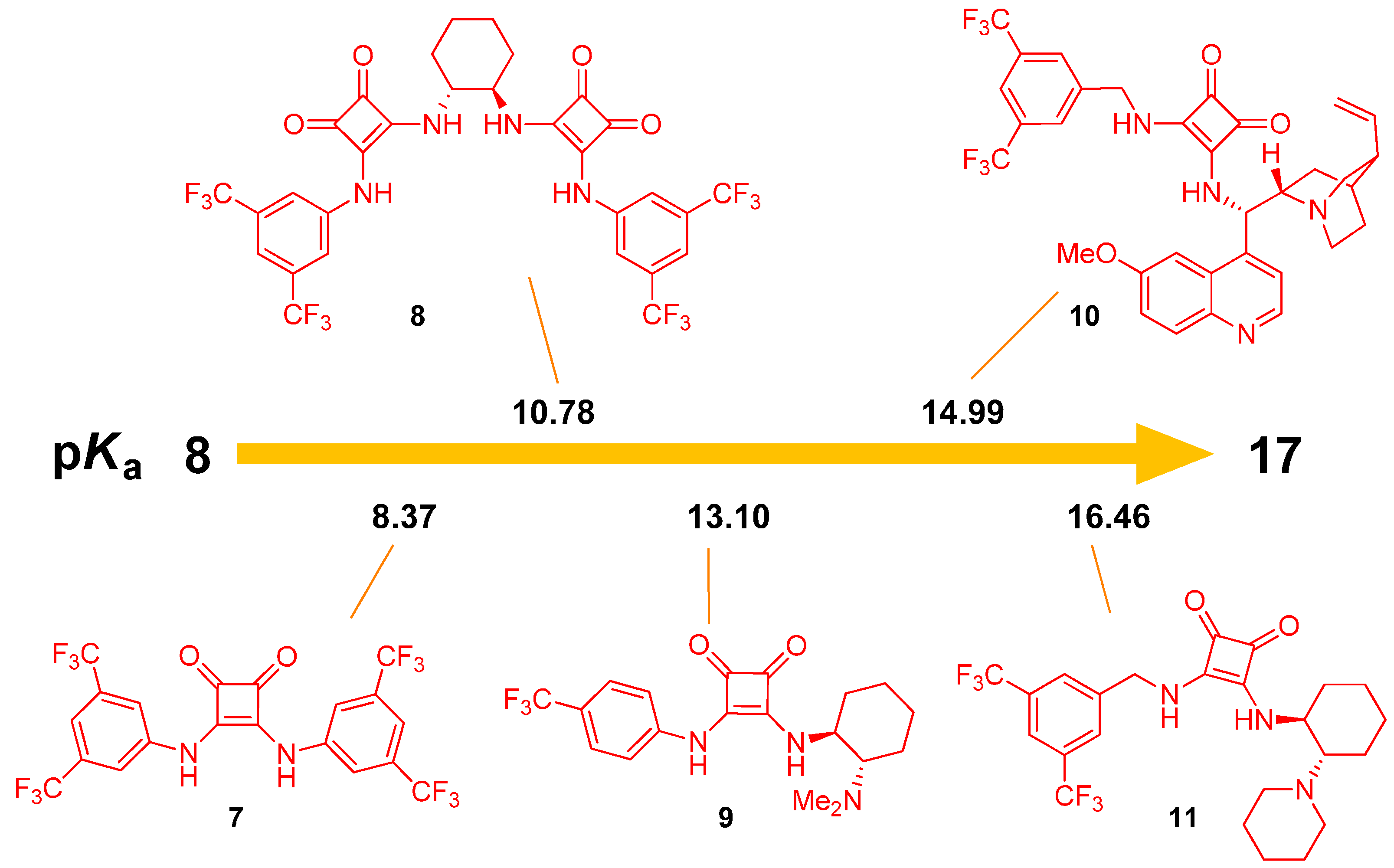
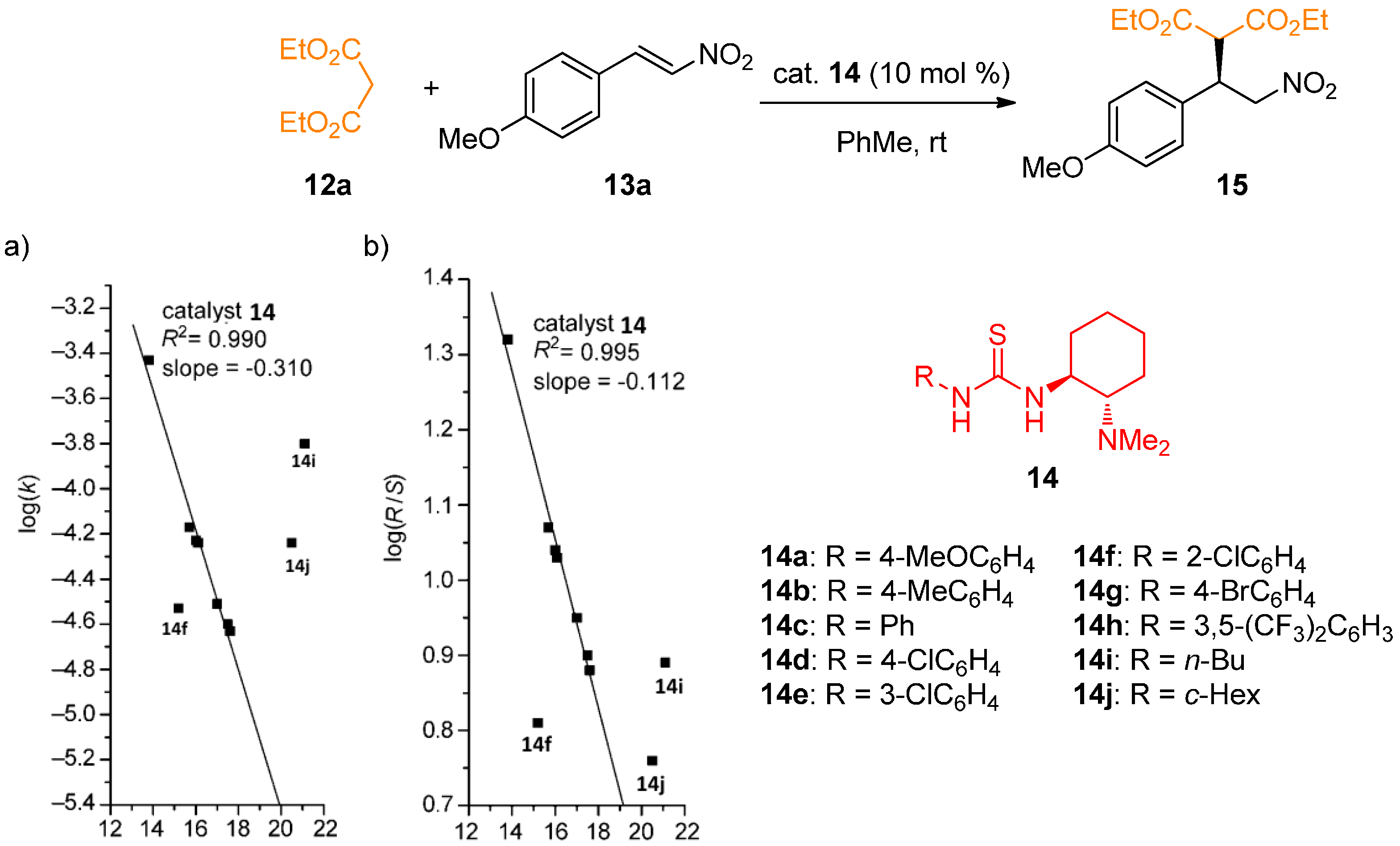
2.1. Determination of Catalyst Acidity: UV-Spectrophotometric Methods




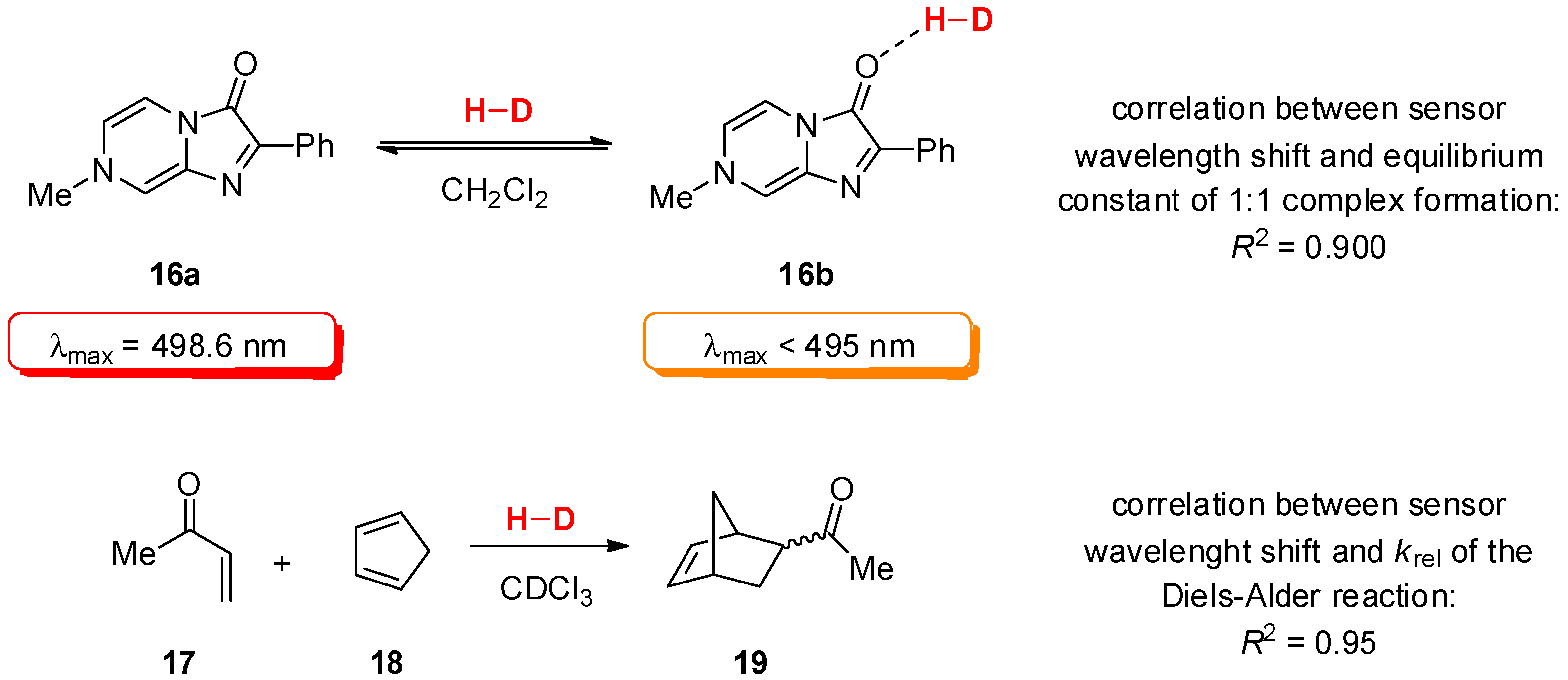
2.2. Determination of Catalyst Reactivity: Spectroscopic Methods


2.3. Combined NMR and DFT Approaches in the Mechanism Elucidation






2.4. Combined Spectroscopic and Kinetic Studies Investigations
- (1)
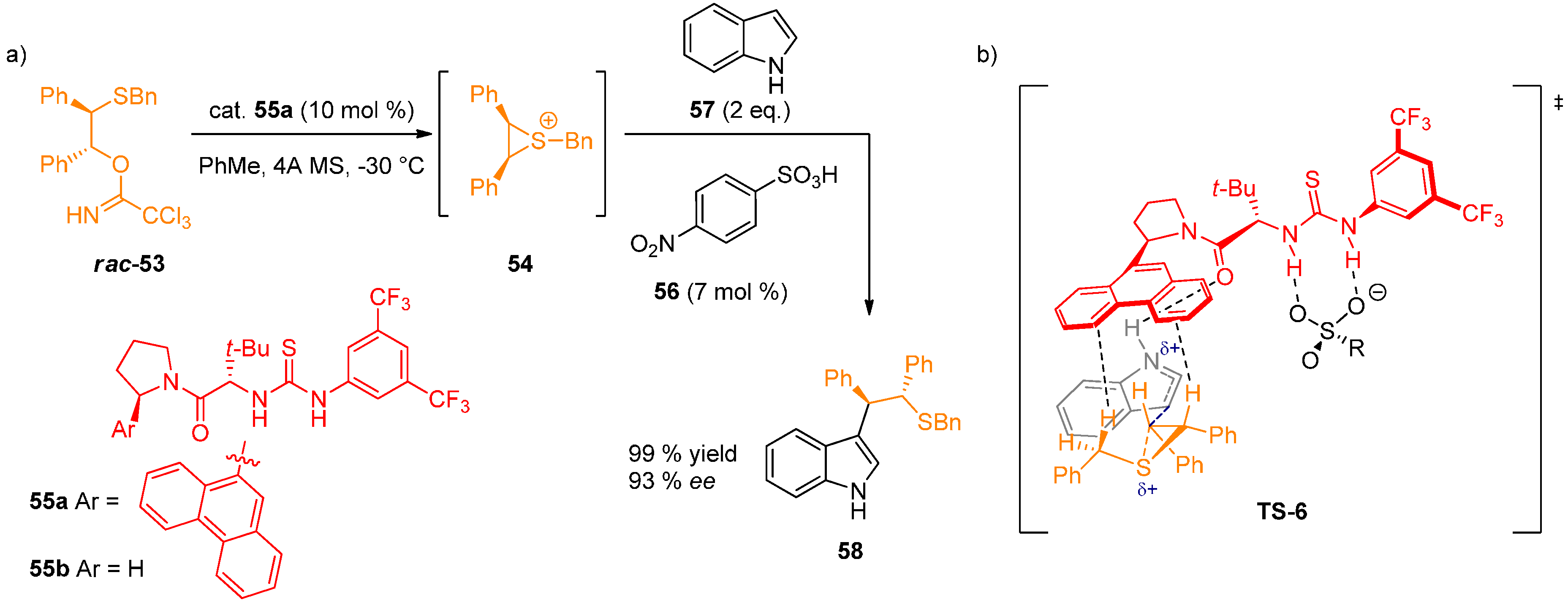
- Thiourea double H-bond interaction with the sulfonate anion: Determined by 1H-NMR titration of catalyst 55a with (Bn2SMe)+(OTf)− (59), leading to the formation of 1:1 complex accompanied by sharpening and downfield shift of N–H protons. This is supported by the observed dependence of ee on the sulfonate anion structure.
- (2)
- H-bond interaction with the indole N–H: In contrast to indole, N-methylindole and the series of π-nucleophiles without N–H group in a 1,3-relationship gave very low enantioselectivities. The acceleration rate for the asymmetric reaction log(kasym/krac) was found to be linked linearly with the pKa of 5-substituted indoles. This is in accordance with a general base activation of indole by the catalyst. After ruling out several possibilities experimentally, the authors proposed that the catalyst amide oxygen-indole N–H H-bond is responsible for this activation.
- (3)
- Stabilization of the cationic transition state by cation-π interactions: Strong correlation was observed between the enantioselectivity of the reaction and the arene unit of the catalyst. The pathway leading to minor enantiomer also shows a positive correlation, indicating stabilization of the minor transition structure, although to a lesser degree. 1H-NMR study of 1:1 complex formation between 59 and thiourea derivatives 55a and 55b was conducted. Thiourea 55a caused a upfield shift of the benzylic and methyl protons of 59 by 0.6–0.8 ppm, whereas 55b without aryl substituents had no effect on the chemical shift of the protons. These data suggest that attractive π-interactions between the arene and the sulfonium ion are responsible for the observed enantioselectivity, with more extended aromatic substituents resulting in an increase in cation-π interactions.


2.5. Computational and Kinetic Studies

2.6. Combined Spectroscopic, Kinetic and Computational Studies
3. Computational Methods







4. Conclusions
Acknowledgments
Conflicts of Interest
References
- Doyle, A.G.; Jacobsen, E.N. Small-molecule H-bond donors in asymmetric catalysis. Chem. Rev. 2007, 107, 5713–5743. [Google Scholar] [CrossRef] [PubMed]
- Connon, S.J. Asymmetric catalysis with bifunctional cinchona alkaloid-based urea and thiourea organocatalysts. Chem. Commun. 2008, 2499–2510. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Schreiner, P.R. (Thio)urea organocatalysis-what can be learnt from anion recognition? Chem. Soc. Rev. 2009, 38, 1187–1198. [Google Scholar] [CrossRef] [PubMed]
- Siau, W.Y.; Wang, J. Asymmetric organocatalytic reactions by bifunctional amine-thioureas. Catal. Sci. Technol. 2011, 1, 1298–1310. [Google Scholar] [CrossRef]
- Serdyuk, O.V.; Heckel, C.M.; Tsogoeva, S.B. Bifunctional primary amine-thioureas in asymmetric organocatalysis. Org. Biomol. Chem. 2013, 11, 7051–7071. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Bao, Z.; Xing, H. N,N[prime or minute]-bis[3,5-bis(trifluoromethyl)phenyl]thiourea: A privileged motif for catalyst development. Org. Biomol. Chem. 2014, 12, 3151–3162. [Google Scholar] [CrossRef] [PubMed]
- Chauhan, P.; Mahajan, S.; Kaya, U.; Hack, D.; Enders, D. Bifunctional amine-squaramides: Powerful hydrogen-bonding organocatalysts for asymmetric domino/cascade reactions. Adv. Synth. Catal. 2015, 357, 253–281. [Google Scholar] [CrossRef]
- Auvil, T.J.; Schafer, A.G.; Mattson, A.E. Design strategies for enhanced hydrogen-bond donor catalysts. Eur. J. Org. Chem. 2014, 2014, 2633–2646. [Google Scholar] [CrossRef]
- Türkmen, Y.E.; Zhu, Y.; Rawal, V.H. Brønsted acids. In Comprehensive Enantioselective Organocatalysis: Catalysts, Reactions, and Applications; Dalko, P.I., Ed.; Wiley-VCH: Weinheim, Germany, 2013; pp. 239–288. [Google Scholar]
- Beckendorf, S.; Asmus, S.; Mancheño, O.G. H-donor anion acceptor organocatalysis—The ionic electrophile activation approach. ChemCatChem 2012, 4, 926–936. [Google Scholar] [CrossRef]
- Brak, K.; Jacobsen, E.N. Asymmetric ion-pairing catalysis. Angew. Chem. Int. Ed. 2013, 52, 534–561. [Google Scholar] [CrossRef] [PubMed]
- Arunan, E.; Desiraju, G.R.; Klein, R.A.; Sadlej, J.; Scheiner, S.; Alkorta, I.; Clary, D.C.; Crabtree, R.H.; Dannenberg, J.J.; Hobza, P.; et al. Definition of the hydrogen bond (iupac recommendations 2011). Pure Appl. Chem. 2011, 83, 1637–1641. [Google Scholar] [CrossRef]
- Seebach, D.; Groselj, U.; Badine, D.M.; Schweizer, W.B.; Beck, A.K. Isolation and X-ray structures of reactive intermediates of organocatalysis with diphenylprolinol ethers and with imidazolidinones. Helv. Chim. Acta 2008, 91, 1999–2034. [Google Scholar] [CrossRef]
- Schmid, M.B.; Zeitler, K.; Gschwind, R.M. The elusive enamine intermediate in proline-catalyzed aldol reactions: Nmr detection, formation pathway, and stabilization trends. Angew. Chem. Int. Ed. 2010, 49, 4997–5003. [Google Scholar] [CrossRef] [PubMed]
- Burés, J.; Armstrong, A.; Blackmond, D.G. Mechanistic rationalization of organocatalyzed conjugate addition of linear aldehydes to nitro-olefins. J. Am. Chem. Soc. 2011, 133, 8822–8825. [Google Scholar] [CrossRef] [PubMed]
- Seebach, D.; Sun, X.; Ebert, M.O.; Schweizer, W.B.; Purkayastha, N.; Beck, A.K.; Duschmalé, J.; Wennemers, H.; Mukaiyama, T.; Benohoud, M.; et al. Stoichiometric reactions of enamines derived from diphenylprolinol silyl ethers with nitro olefins and lessons for the corresponding organocatalytic conversions—A survey. Helv. Chim. Acta 2013, 96, 799–852. [Google Scholar] [CrossRef]
- Holland, M.C.; Gilmour, R. Deconstructing covalent organocatalysis. Angew. Chem. Int. Ed. 2015, 54, 3862–3871. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, M.; Worgull, D.; Zweifel, T.; Gschwend, B.; Bertelsen, S.; Jorgensen, K.A. Mechanisms in aminocatalysis. Chem. Commun. 2011, 47, 632–649. [Google Scholar] [CrossRef] [PubMed]
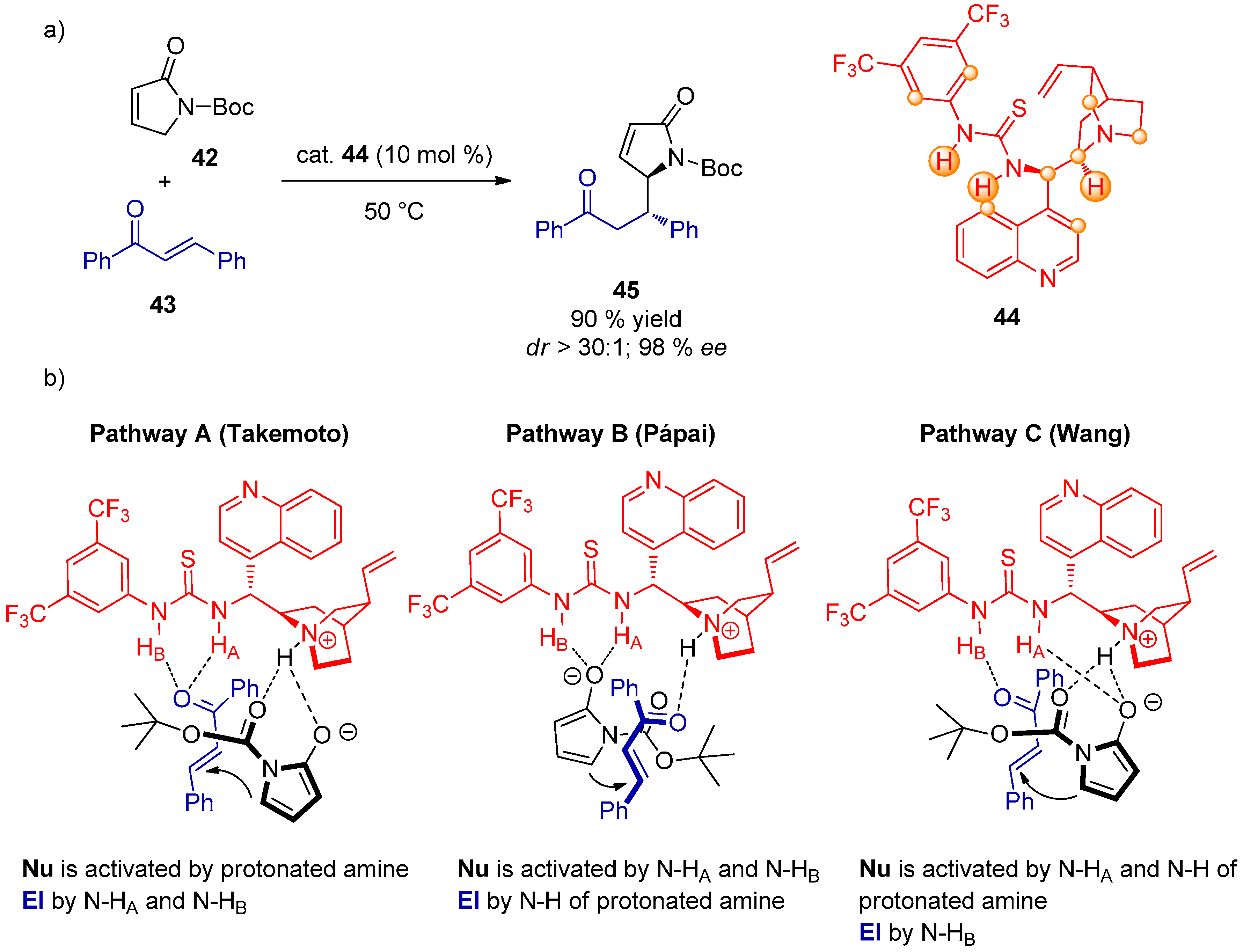
- Azuma, T.; Kobayashi, Y.; Sakata, K.; Sasamori, T.; Tokitoh, N.; Takemoto, Y. Synthesis and characterization of binary-complex models of ureas and 1,3-dicarbonyl compounds: Deeper insights into reaction mechanisms using snap-shot structural analysis. J. Org. Chem. 2014, 79, 1805–1817. [Google Scholar] [CrossRef] [PubMed]
- Selig, P. Guanidine organocatalysis. Synthesis 2013, 45, 703–718. [Google Scholar] [CrossRef]
- Nagasawa, K.; Sohtome, Y. Brønsted bases: Chiral guanidine and amidine organocatalysts. In Asymmetric Organocatalysis 2: Brønsted Base and Acid Catalysts, and Additional Topics; Maruoka, K., Ed.; Georg Thieme Verlag: Stuttgart, Germany, 2012; Volume 2, pp. 1–40. [Google Scholar]
- Klussmann, M. Mechanism in organocatalysis. In Asymmetric Organocatalysis 2: Brønsted Base and Acid Catalysts, and Additional Topic; Maruoka, K., Ed.; Georg Thieme Verlag: Stuttgart, Germany, 2012; Volume 2, pp. 633–671. [Google Scholar]
- Fleischmann, M.; Drettwan, D.; Sugiono, E.; Rueping, M.; Gschwind, R.M. Brønsted acid catalysis: Hydrogen bonding versus ion pairing in imine activation. Angew. Chem. Int. Ed. 2011, 50, 6364–6369. [Google Scholar] [CrossRef] [PubMed]
- Jakab, G.; Tancon, C.; Zhang, Z.; Lippert, K.M.; Schreiner, P.R. (Thio)urea organocatalyst equilibrium acidities in dmso. Org. Lett. 2012, 14, 1724–1727. [Google Scholar] [CrossRef] [PubMed]
- Malerich, J.P.; Hagihara, K.; Rawal, V.H. Chiral squaramide derivatives are excellent hydrogen bond donor catalysts. J. Am. Chem. Soc. 2008, 130, 14416–14417. [Google Scholar] [CrossRef] [PubMed]
- Ni, X.; Li, X.; Wang, Z.; Cheng, J.P. Squaramide equilibrium acidities in dmso. Org. Lett. 2014, 16, 1786–1789. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Deng, H.; Zhang, B.; Li, J.; Zhang, L.; Luo, S.; Cheng, J.P. Physical organic study of structure-activity-enantioselectivity relationships in asymmetric bifunctional thiourea catalysis: Hints for the design of new organocatalysts. Chem. Eur. J. 2010, 16, 450–455. [Google Scholar] [CrossRef] [PubMed]
- Walvoord, R.R.; Huynh, P.N.H.; Kozlowski, M.C. Quantification of electrophilic activation by hydrogen-bonding organocatalysts. J. Am. Chem. Soc. 2014, 136, 16055–16065. [Google Scholar] [CrossRef] [PubMed]
- Nödling, A.R.; Jakab, G.; Schreiner, P.R.; Hilt, G. 31P-NMR spectroscopically quantified hydrogen-bonding strength of thioureas and their catalytic activity in Diels-Alder reactions. Eur. J. Org. Chem. 2014, 2014, 6394–6398. [Google Scholar] [CrossRef]
- Lippert, K.M.; Hof, K.; Gerbig, D.; Ley, D.; Hausmann, H.; Guenther, S.; Schreiner, P.R. Hydrogen-bonding thiourea organocatalysts: The privileged 3,5-bis(trifluoromethyl)phenyl group. Eur. J. Org. Chem. 2012, 5919–5927. [Google Scholar] [CrossRef]
- Johnston, R.C.; Cheong, P.H.Y. C-HO non-classical hydrogen bonding in the stereomechanics of organic transformations: Theory and recognition. Org. Biomol. Chem. 2013, 11, 5057–5064. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.; An, J.; Lu, L.Q.; Luo, L.; Wang, Z.Y.; Chen, J.R.; Xiao, W.J. Asymmetric cyclopropanation of β,γ-unsaturated α-ketoesters with stabilized sulfur ylides catalyzed by C2-symmetric ureas. J. Org. Chem. 2011, 76, 281–284. [Google Scholar] [CrossRef] [PubMed]
- Hamza, A.; Schubert, G.; Soós, T.; Pápai, I. Theoretical studies on the bifunctionality of chiral thiourea-based organocatalysts: Competing routes to C–C bond formation. J. Am. Chem. Soc. 2006, 128, 13151–13160. [Google Scholar] [CrossRef] [PubMed]
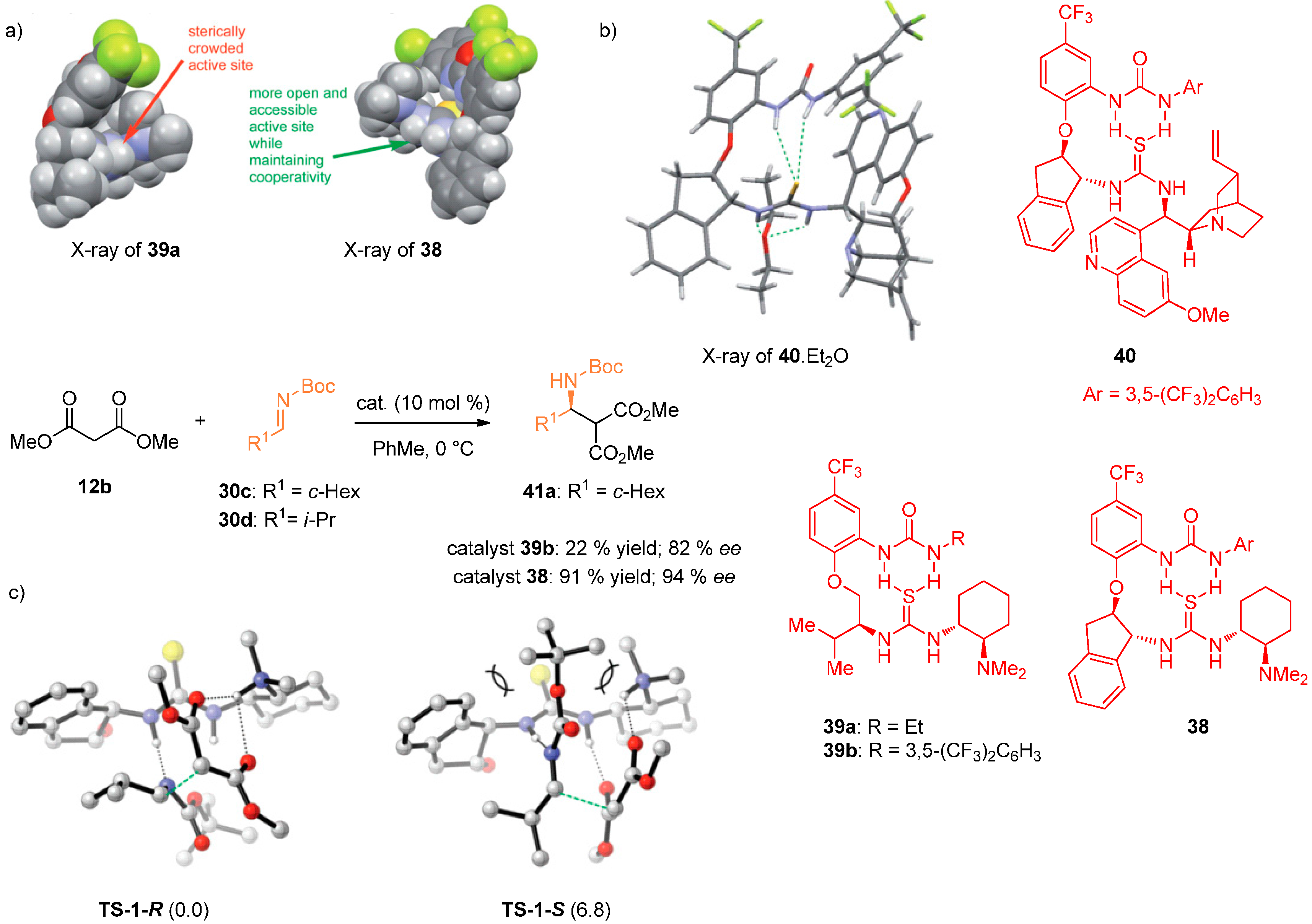
- Jones, C.R.; Dan Pantoş, G.; Morrison, A.J.; Smith, M.D. Plagiarizing proteins: Enhancing efficiency in asymmetric hydrogen-bonding catalysis through positive cooperativity. Angew. Chem. Int. Ed. 2009, 48, 7391–7394. [Google Scholar] [CrossRef] [PubMed]
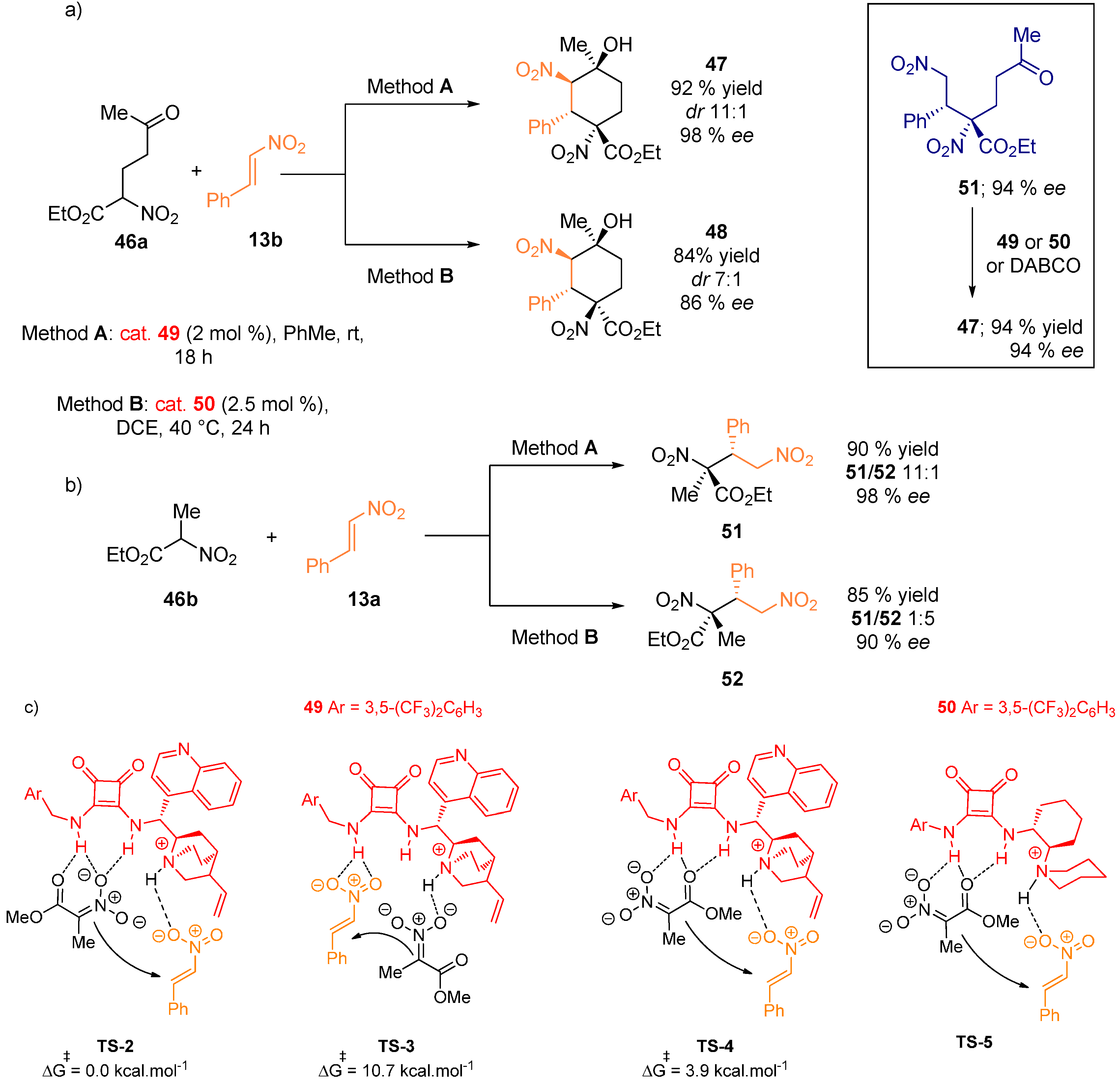
- Probst, N.; Madarász, Á.; Valkonen, A.; Pápai, I.; Rissanen, K.; Neuvonen, A.; Pihko, P.M. Cooperative assistance in bifunctional organocatalysis: Enantioselective mannich reactions with aliphatic and aromatic imines. Angew. Chem. Int. Ed. 2012, 51, 8495–8499. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.L.; Zhang, Y.; Liu, C.; Zheng, A.M.; Wang, W. Insights into the dual activation mechanism involving bifunctional cinchona alkaloid thiourea organocatalysts: An NMR and DFT study. J. Org. Chem. 2012, 77, 9813–9825. [Google Scholar] [CrossRef] [PubMed]
- Martínez, J.I.; Villar, L.; Uria, U.; Carrillo, L.; Reyes, E.; Vicario, J.L. Bifunctional squaramide catalysts with the same absolute chirality for the diastereodivergent access to densely functionalised cyclohexanes through enantioselective domino reactions. Synthesis and mechanistic studies. Adv. Synth. Catal. 2014, 356, 3627–3648. [Google Scholar] [CrossRef]
- Lin, S.; Jacobsen, E.N. Thiourea-catalysed ring opening of episulfonium ions with indole derivatives by means of stabilizing non-covalent interactions. Nat. Chem. 2012, 4, 817–824. [Google Scholar] [CrossRef] [PubMed]
- Knowles, R.R.; Jacobsen, E.N. Attractive noncovalent interactions in asymmetric catalysis: Links between enzymes and small molecule catalysts. Proc. Natl. Acad. Sci. USA 2010, 107, 20678–20685. [Google Scholar] [CrossRef] [PubMed]
- Knowles, R.R.; Lin, S.; Jacobsen, E.N. Enantioselective thiourea-catalyzed cationic polycyclizations. J. Am. Chem. Soc. 2010, 132, 5030–5032. [Google Scholar] [CrossRef] [PubMed]
- Harper, K.C.; Sigman, M.S. Using physical organic parameters to correlate asymmetric catalyst performance. J. Org. Chem. 2013, 78, 2813–2818. [Google Scholar] [CrossRef] [PubMed]
- Ji, Y.; Blackmond, D.G. The role of reversibility in the enantioselective conjugate addition of α,α-disubstituted aldehydes to nitro-olefins catalyzed by primary amine thioureas. Catal. Sci. Technol. 2014, 4, 3505–3509. [Google Scholar] [CrossRef]
- Zuend, S.J.; Jacobsen, E.N. Cooperative catalysis by tertiary amino-thioureas: Mechanism and basis for enantioselectivity of ketone cyanosilylation. J. Am. Chem. Soc. 2007, 129, 15872–15883. [Google Scholar] [CrossRef] [PubMed]
- Zuend, S.J.; Jacobsen, E.N. Mechanism of amido-thiourea catalyzed enantioselective imine hydrocyanation: Transition state stabilization via multiple non-covalent interactions. J. Am. Chem. Soc. 2009, 131, 15358–15374. [Google Scholar] [CrossRef] [PubMed]
- Quintard, A.; Cheshmedzhieva, D.; Sanchez Duque, M.D.M.; Gaudel-Siri, A.; Naubron, J.V.; Génisson, Y.; Plaquevent, J.C.; Bugaut, X.; Rodriguez, J.; Constantieux, T. Origin of the enantioselectivity in organocatalytic michael additions of β-ketoamides to α,β-unsaturated carbonyls: A combined experimental, spectroscopic and theoretical study. Chem. Eur. J. 2015, 21, 778–790. [Google Scholar] [CrossRef] [PubMed]
- Pápai, I. Computational and theoretical studies. In Asymmetric Organocatalysis 2: Brønsted Base and Acid Catalysts, and Additional Topics; Maruoka, K., Ed.; Georg Thieme Verlag: Stuttgart, Germany, 2012; Volume 2, pp. 601–632. [Google Scholar]
- Cheong, P.H.Y.; Legault, C.Y.; Um, J.M.; Çelebi-Ölçüm, N.; Houk, K.N. Quantum mechanical investigations of organocatalysis: Mechanisms, reactivities, and selectivities. Chem. Rev. 2011, 111, 5042–5137. [Google Scholar] [CrossRef] [PubMed]
- Schenker, S.; Schneider, C.; Tsogoeva, S.B.; Clark, T. Assessment of popular dft and semiempirical molecular orbital techniques for calculating relative transition state energies and kinetic product distributions in enantioselective organocatalytic reactions. J. Chem. Theory Comput. 2011, 7, 3586–3595. [Google Scholar] [CrossRef]
- Etzenbach-Effers, K.; Berkessel, A. Noncovalent organocatalysis based on hydrogen bonding: Elucidation of reaction paths by computational methods. Top. Curr. Chem. 2010, 291, 1–27. [Google Scholar] [PubMed]
- Lu, T.; Wheeler, S.E. Origin of the superior performance of (thio)squaramides over (thio)ureas in organocatalysis. Chem. Eur. J. 2013, 19, 15141–15147. [Google Scholar] [CrossRef] [PubMed]
- Kótai, B.; Kardos, G.; Hamza, A.; Farkas, V.; Pápai, I.; Soós, T. On the mechanism of bifunctional squaramide-catalyzed organocatalytic michael addition: A protonated catalyst as an oxyanion hole. Chem. Eur. J. 2014, 20, 5631–5639. [Google Scholar] [CrossRef] [PubMed]
- Roca-Lopez, D.; Marques-Lopez, E.; Alcaine, A.; Merino, P.; Herrera, R.P. A friedel-crafts alkylation mechanism using an aminoindanol-derived thiourea catalyst. Org. Biomol. Chem. 2014, 12, 4503–4510. [Google Scholar] [CrossRef] [PubMed]
- Baran, R.; Veverková, E.; Škvorcová, A.; Šebesta, R. Enantioselective michael addition of 1,3-dicarbonyl compounds to a nitroalkene catalyzed by chiral squaramides—A key step in the synthesis of pregabalin. Org. Biomol. Chem. 2013, 11, 7705–7711. [Google Scholar] [CrossRef] [PubMed]
- Žabka, M.; Malastová, A.; Šebesta, R. Enantioselective addition of oxazolones to N-protected imines catalysed by chiral thioureas. RSC Adv. 2015, 5, 12890–12893. [Google Scholar] [CrossRef]
- Breugst, M.; Houk, K.N. Computational analysis of cyclophane-based bisthiourea-catalyzed henry reactions. J. Org. Chem. 2014, 79, 6302–6309. [Google Scholar] [CrossRef] [PubMed]
- Mittal, N.; Lippert, K.M.; De, C.K.; Klauber, E.G.; Emge, T.J.; Schreiner, P.R.; Seidel, D. A dual-catalysis anion-binding approach to the kinetic resolution of amines: Insights into the mechanism via a combined experimental and computational study. J. Am. Chem. Soc. 2015, 137, 5748–5758. [Google Scholar] [CrossRef] [PubMed]
- Rahaman, H.; Madarász, Á.; Pápai, I.; Pihko, P.M. Dual hydrogen-bond/enamine catalysis enables a direct enantioselective three-component domino reaction. Angew. Chem. Int. Ed. 2011, 50, 6123–6127. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Huang, D.; Lv, Y. Theoretical study on the mechanism and stereochemistry of the cinchona-thiourea organocatalytic hydrophosphonylation of an α-ketoester. Org. Biomol. Chem. 2013, 11, 7497–7506. [Google Scholar] [CrossRef] [PubMed]
- Badiola, E.; Fiser, B.; Gómez-Bengoa, E.; Mielgo, A.; Olaizola, I.; Urruzuno, I.; García, J.M.; Odriozola, J.M.; Razkin, J.; Oiarbide, M.; et al. Enantioselective construction of tetrasubstituted stereogenic carbons through brønsted base catalyzed michael reactions: Α′-hydroxy enones as key enoate equivalent. J. Am. Chem. Soc. 2014, 136, 17869–17881. [Google Scholar] [CrossRef] [PubMed]
- Albrecht, Ł.; Dickmeiss, G.; Acosta, F.C.; Rodríguez-Escrich, C.; Davis, R.L.; Jørgensen, K.A. Asymmetric organocatalytic formal [2 + 2]-cycloadditions via bifunctional H-bond directing dienamine catalysis. J. Am. Chem. Soc. 2012, 134, 2543–2546. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Qi, Z.H.; Zhang, S.Y.; Kong, L.P.; Wang, Y.; Wang, X.W. Enantioselective construction of functionalized thiopyrano-indole annulated heterocycles via a formal thio [3 + 3]-cyclization. Org. Lett. 2015, 17, 42–45. [Google Scholar] [CrossRef] [PubMed]
- Yalalov, D.A.; Tsogoeva, S.B.; Shubina, T.E.; Martynova, I.M.; Clark, T. Evidence for an enol mechanism in a highly enantioselective mannich-type reaction catalyzed by primary amine-thiourea. Angew. Chem. Int. Ed. 2008, 47, 6624–6628. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Žabka, M.; Šebesta, R. Experimental and Theoretical Studies in Hydrogen-Bonding Organocatalysis. Molecules 2015, 20, 15500-15524. https://doi.org/10.3390/molecules200915500
Žabka M, Šebesta R. Experimental and Theoretical Studies in Hydrogen-Bonding Organocatalysis. Molecules. 2015; 20(9):15500-15524. https://doi.org/10.3390/molecules200915500
Chicago/Turabian StyleŽabka, Matej, and Radovan Šebesta. 2015. "Experimental and Theoretical Studies in Hydrogen-Bonding Organocatalysis" Molecules 20, no. 9: 15500-15524. https://doi.org/10.3390/molecules200915500