
Unique Reactivity of Transition Metal Atoms Embedded in Graphene to CO, NO, O2 and O Adsorption: A First-Principles Investigation


Abstract
:1. Introduction
2. Results and Discussion

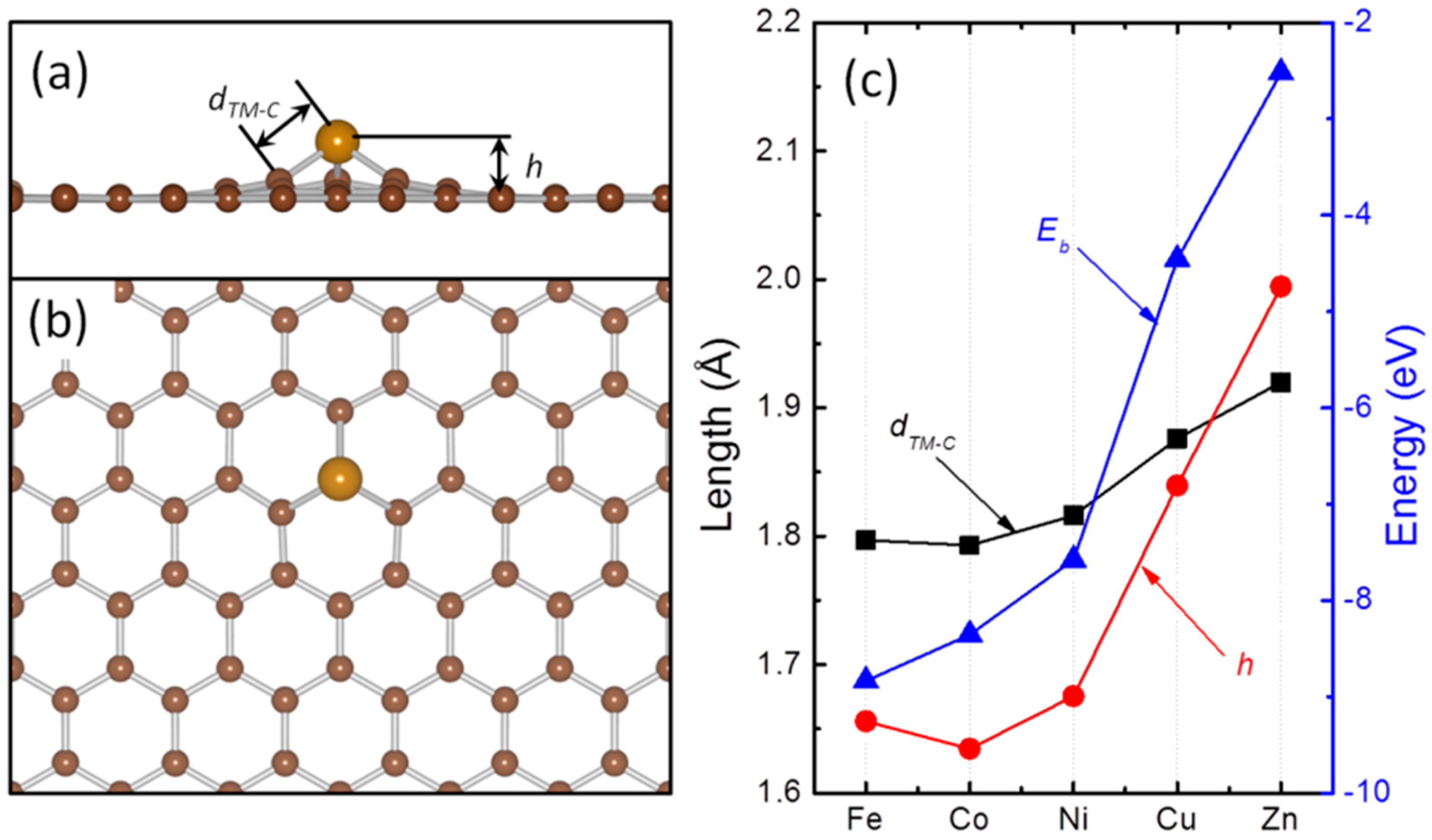{kind=link}
{kind=link}
{kind=link}
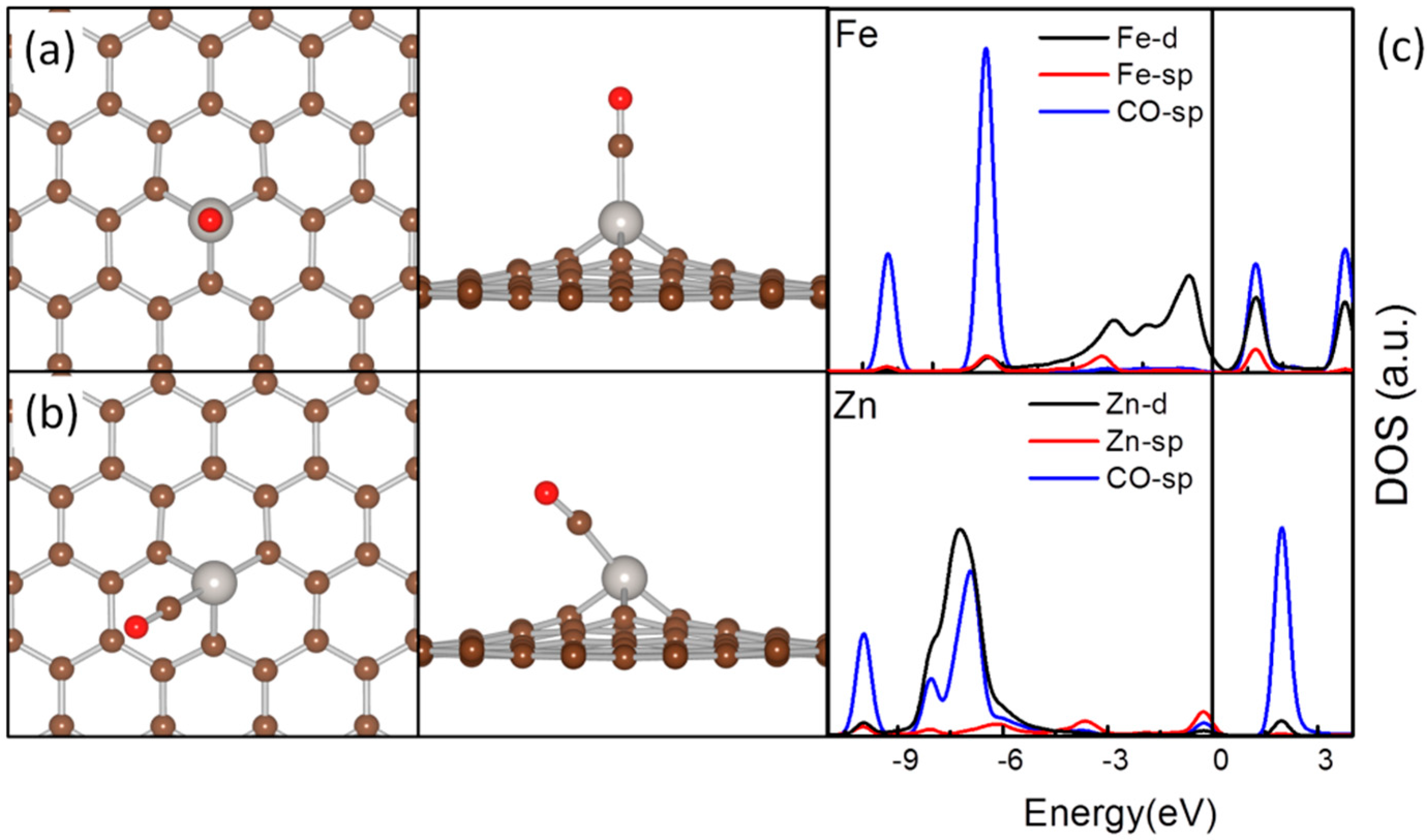{kind=link}
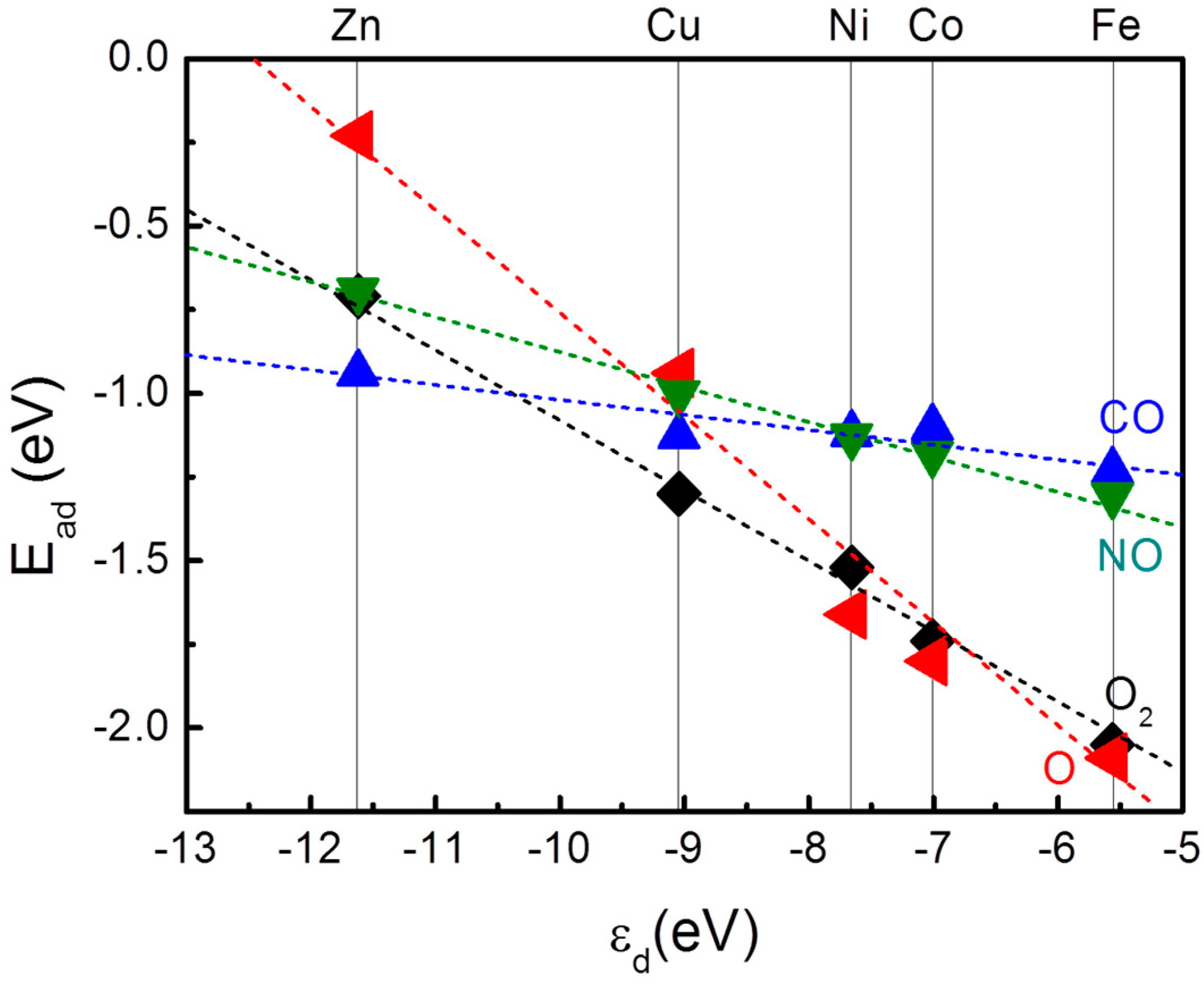{kind=link}
| TM | Eb (eV) a | Ea (eV) b | dTM-C (Å) c | h (Å) d |
|---|---|---|---|---|
| Fe | −0.92 (Hol) | 0.42 | 2.12 | 1.56 |
| Co | −1.44 (Hol) | 0.44 | 2.10 | 1.53 |
| Ni | −1.51 (Hol) | 0.21 | 2.12 | 1.56 |
| Cu | −0.23 (Atop) | 0.03 | 2.20 | 2.04 |
| Zn | −0.02 (Hol) | 0.01 | 3.02 | 2.78 |


| TM | O2 | O | ||||||
|---|---|---|---|---|---|---|---|---|
| Ead a (eV) | dO-TM b (Å) | dO-O c (Å) | dC-TM d (Å) | C.E. e | Ead a (eV) | dO-TM b (Å) | dC-TM d (Å) | |
| Fe | −2.05 | 1.85 | 1.39 | 1.82 | Tetr | −2.09 | 1.62 | 1.82 |
| Co | −1.74 | 1.90 | 1.37 | 1.80 | Octa | −1.80 | 1.64 | 1.80 |
| Ni | −1.52 | 1.94 | 1.37 | 1.84 | Octa | −1.66 | 1.66 | 1.82 |
| Cu | −1.30 | 1.92 | 1.36 | 1.89 | Octa | −0.94 | 1.72 | 1.92 |
| Zn | −0.71 | 2.02 | 1.35 | 1.97 | Octa | −0.23 | 1.79 | 2.00 |

| TM | CO | NO | ||||
|---|---|---|---|---|---|---|
| Ead a (eV) | dC-TM b (Å) | dC-O c (Å) | Ead a (eV) | dN-TM d (Å) | dN-O e (Å) | |
| Fe | −1.23 | 1.91 | 1.16 | −1.30 | 1.77 | 1.19 |
| Co | −1.10 | 1.89 | 1.16 | −1.18 | 1.77 | 1.19 |
| Ni | −1.12 | 1.87 | 1.16 | −1.13 | 1.74 | 1.19 |
| Cu | −1.13 | 1.87 | 1.16 | −1.00 | 1.79 | 1.18 |
| Zn | −0.94 | 1.94 | 1.16 | −0.69 | 1.99 | 1.19 |


3. Theoretical Methods
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Castro Neto, A.H.; Guinea, F.; Peres, N.M.R.; Novoselov, K.S.; Geim, A.K. The electronic properties of graphene. Rev. Mod. Phys. 2009, 81, 109–162. [Google Scholar] [CrossRef]
- Novoselov, K.S.; Geim, A.K.; Morozov, S.V.; Jiang, D.; Zhang, Y.; Dubonos, S.V.; Grigorieva, I.V.; Firsov, A.A. Electric field effect in atomically thin carbon films. Science 2004, 306, 666–669. [Google Scholar] [CrossRef] [PubMed]
- Schwierz, F. Graphene transistors. Nat. Nanotechnol. 2010, 5, 487–496. [Google Scholar] [CrossRef] [PubMed]
- Avouris, P.; Chen, Z.H.; Perebeinos, V. Carbon-based electronics. Nat. Nanotechnol. 2007, 2, 605–615. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.W.; Murali, S.; Stoller, M.D.; Ganesh, K.J.; Cai, W.W.; Ferreira, P.J.; Pirkle, A.; Wallace, R.M.; Cychosz, K.A.; Thommes, M.; et al. Carbon-based supercapacitors produced by activation of graphene. Science 2011, 332, 1537–1541. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Shi, Z.Q.; Huang, Y.; Ma, Y.F.; Wang, C.Y.; Chen, M.M.; Chen, Y.S. Supercapacitor devices based on graphene materials. J. Phys. Chem. C 2009, 113, 13103–13107. [Google Scholar] [CrossRef]
- Shao, Y.Y.; Wang, J.; Wu, H.; Liu, J.; Aksay, I.A.; Lin, Y.H. Graphene based electrochemical sensors and biosensors: A review. Electroanalysis 2010, 22, 1027–1036. [Google Scholar] [CrossRef]
- Huang, X.; Zeng, Z.Y.; Fan, Z.X.; Liu, J.Q.; Zhang, H. Graphene-based electrodes. Adv. Mater. 2012, 24, 5979–6004. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Yin, Z.Y.; Wu, S.X.; Qi, X.Y.; He, Q.Y.; Zhang, Q.C.; Yan, Q.Y.; Boey, F.; Zhang, H. Graphene-based materials: Synthesis, characterization, properties, and applications. Small 2011, 7, 1876–1902. [Google Scholar] [CrossRef] [PubMed]
- Julkapli, N.M.; Bagheri, S. Graphene supported heterogeneous catalysts: An overview. Int. J. Hydrog. Energy 2015, 40, 948–979. [Google Scholar] [CrossRef]
- Cheng, Y.; Fan, Y.; Pei, Y.; Qiao, M. Graphene-supported metal/metal oxide nanohybrids: Synthesis and applications in heterogeneous catalysis. Catal. Sci. Technol. 2015, 5, 3903–3916. [Google Scholar] [CrossRef]
- Fan, X.B.; Zhang, G.L.; Zhang, F.B. Multiple roles of graphene in heterogeneous catalysis. Chem. Soc. Rev. 2015, 44, 3023–3035. [Google Scholar] [CrossRef] [PubMed]
- Yang, F.; Deng, D.H.; Pan, X.L.; Fu, Q.; Bao, X.H. Understanding nano effects in catalysis. Natl. Sci. Rev. 2015, 2, 183–201. [Google Scholar] [CrossRef]
- Yang, X.F.; Wang, A.Q.; Qiao, B.T.; Li, J.; Liu, J.Y.; Zhang, T. Single-atom catalysts: A new frontier in heterogeneous catalysis. Acc. Chem. Res. 2013, 46, 1740–1748. [Google Scholar] [CrossRef] [PubMed]
- Tang, Q.; Zhou, Z.; Chen, Z.F. Graphene-related nanomaterials: Tuning properties by functionalization. Nanoscale 2013, 5, 4541–4583. [Google Scholar] [CrossRef] [PubMed]
- Chan, K.T.; Neaton, J.B.; Cohen, M.L. First-principles study of metal adatom adsorption on graphene. Phys. Rev. B 2008, 77. [Google Scholar] [CrossRef]
- Krasheninnikov, A.V.; Lehtinen, P.O.; Foster, A.S.; Pyykko, P.; Nieminen, R.M. Embedding transition-metal atoms in graphene: Structure, bonding, and magnetism. Phys. Rev. Lett. 2009, 102. [Google Scholar] [CrossRef]
- Hummers, W.S.; Offeman, R.E. Preparation of graphitic oxide. J. Am. Chem. Soc. 1958, 80, 1339–1340. [Google Scholar] [CrossRef]
- Meyer, J.C.; Kisielowski, C.; Erni, R.; Rossell, M.D.; Crommie, M.F.; Zettl, A. Direct imaging of lattice atoms and topological defects in graphene membranes. Nano Lett. 2008, 8, 3582–3586. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Zhou, Y.G.; Bang, J.; Prange, M.P.; Zhang, S.B.; Gao, F. Modification of defect structures in graphene by electron irradiation: Ab initio molecular dynamics simulations. J. Phys. Chem. C 2012, 116, 16070–16079. [Google Scholar] [CrossRef]
- Liu, X.; Yao, K.X.; Meng, C.G.; Han, Y. Graphene substrate-mediated catalytic performance enhancement of Ru nanoparticles: A first-principles study. Dalton Trans. 2012, 41, 1289–1296. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Sui, Y.; Meng, C.; Han, Y. Tuning the reactivity of Ru nanoparticles by defect engineering of the reduced graphene oxide support. RSC Adv. 2014, 4, 22230–22240. [Google Scholar] [CrossRef]
- Liu, X.; Li, L.; Meng, C.; Han, Y. Palladium Nanoparticles/defective graphene composites as oxygen reduction electrocatalysts: A first-principles study. J. Phys. Chem. C 2012, 116, 2710–2719. [Google Scholar] [CrossRef]
- Liu, X.; Meng, C.G.; Han, Y. Defective graphene supported MPd12 (M = Fe, Co, Ni, Cu, Zn, Pd) nanoparticles as potential oxygen reduction electrocatalysts: A first-principles study. J. Phys. Chem. C 2013, 117, 1350–1357. [Google Scholar] [CrossRef]
- Liu, X.; Meng, C.G.; Han, Y. Substrate-mediated enhanced activity of Ru nanoparticles in catalytic hydrogenation of benzene. Nanoscale 2012, 4, 2288–2295. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Meng, C.; Han, Y. Unique reactivity of Fe nanoparticles-defective graphene composites toward NHx (x = 0, 1, 2, 3) adsorption: A first-principles study. Phys. Chem. Chem. Phys. 2012, 14, 15036–15045. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Sui, Y.; Duan, T.; Meng, C.; Han, Y. CO oxidation catalyzed by Pt-embedded graphene: A first-principles investigation. Phys. Chem. Chem. Phys. 2014, 16, 23584–23593. [Google Scholar] [CrossRef] [PubMed]
- Yao, K.X.; Liu, X.; Li, Z.; Li, C.C.; Zeng, H.C.; Han, Y. Preparation of Ru nanoparticles/defective graphene composite as a highly efficient arene hydrogenation catalyst. ChemCatChem 2012, 4, 1938–1942. [Google Scholar] [CrossRef]
- Liu, X.; Sui, Y.; Duan, T.; Meng, C.; Han, Y. Monodispersed Pt atoms anchored on N-doped graphene as efficient catalysts for CO oxidation: A first-principles investigation. Catal. Sci. Technol. 2015, 5, 1658–1667. [Google Scholar] [CrossRef]
- Qiao, B.; Wang, A.; Yang, X.; Allard, L.F.; Jiang, Z.; Cui, Y.; Liu, J.; Li, J.; Zhang, T. Single-atom catalysis of CO oxidation using Pt1/FeOx. Nat. Chem. 2011, 3, 634–641. [Google Scholar] [CrossRef] [PubMed]
- Wei, H.S.; Liu, X.Y.; Wang, A.Q.; Zhang, L.L.; Qiao, B.T.; Yang, X.F.; Huang, Y.Q.; Miao, S.; Liu, J.Y.; Zhang, T. FeOx-supported platinum single-atom and pseudo-single-atom catalysts for chemoselective hydrogenation of functionalized nitroarenes. Nat. Commun. 2014, 5. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.; Fang, G.; Li, G.; Ma, H.; Fan, H.; Yu, L.; Ma, C.; Wu, X.; Deng, D.; Wei, M.; et al. Direct, nonoxidative conversion of methane to ethylene, aromatics, and hydrogen. Science 2014, 344, 616–619. [Google Scholar] [CrossRef] [PubMed]
- Manadé, M.; Viñes, F.; Illas, F. Transition metal adatoms on graphene: A systematic density functional study. Carbon 2015, 95, 525–534. [Google Scholar]
- Valencia, H.; Gil, A.; Frapper, G. Trends in the hydrogen activation and storage by adsorbed 3d transition metal atoms onto graphene and nanotube surfaces: A DFT study and molecular orbital analysis. J. Phys. Chem. C 2015, 119, 5506–5522. [Google Scholar] [CrossRef]
- Lu, Y.H.; Zhou, M.; Zhang, C.; Feng, Y.P. Metal-embedded graphene: A possible catalyst with high activity. J. Phys. Chem. C 2009, 113, 20156–20160. [Google Scholar]
- Song, E.H.; Wen, Z.; Jiang, Q. CO catalytic oxidation on copper-embedded graphene. J. Phys. Chem. C 2011, 115, 3678–3683. [Google Scholar] [CrossRef]
- Li, Y.F.; Zhou, Z.; Yu, G.T.; Chen, W.; Chen, Z.F. CO catalytic oxidation on iron-embedded graphene: Computational quest for low-cost nanocatalysts. J. Phys. Chem. C 2010, 114, 6250–6254. [Google Scholar] [CrossRef]
- Sevincli, H.; Topsakal, M.; Durgun, E.; Ciraci, S. Electronic and magnetic properties of 3d transition-metal atom adsorbed graphene and graphene nanoribbons. Phys. Rev. B 2008, 77. [Google Scholar] [CrossRef]
- Valencia, H.; Gil, A.; Frapper, G. Trends in the adsorption of 3d transition metal atoms onto graphene and nanotube surfaces: A DFT study and molecular orbital analysis. J. Phys. Chem. C 2010, 114, 14141–14153. [Google Scholar] [CrossRef]
- Liu, X.; Duan, T.; Meng, C.; Han, Y. Pt atoms stabilized on hexagonal boron nitride as efficient single-atom catalysts for CO oxidation: A first-principles investigation. RSC Adv. 2015, 5, 10452–10459. [Google Scholar] [CrossRef]
- Zhou, M.; Lu, Y.H.; Cai, Y.Q.; Zhang, C.; Feng, Y.P. Adsorption of gas molecules on transition metal embedded graphene: A search for high-performance graphene-based catalysts and gas sensors. Nanotechnology 2011, 22. [Google Scholar] [CrossRef] [PubMed]
- Leenaerts, O.; Partoens, B.; Peeters, F.M. Adsorption of H2O, NH3, CO, NO2, and NO on graphene: A first-principles study. Phys. Rev. B 2008, 77. [Google Scholar] [CrossRef]
- Zhang, Y.-H.; Chen, Y.-B.; Zhou, K.-G.; Liu, C.-H.; Zeng, J.; Zhang, H.-L.; Peng, Y. Improving gas sensing properties of graphene by introducing dopants and defects: A first-principles study. Nanotechnology 2009, 20. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.; Liu, E.Z.; Jiang, J.Z. Magnetic behavior of graphene absorbed with N, O, and F atoms: A first-principles study. Appl. Phys. Lett. 2008, 93. [Google Scholar] [CrossRef]
- Fukui, K.; Yonezawa, T.; Shingu, H. A molecular orbital theory of reactivity in aromatic hydrocarbons. J. Chem. Phys. 1952, 20, 722–725. [Google Scholar] [CrossRef]
- Hammer, B.; Norskov, J.K. Electronic factors determining the reactivity of metal surfaces. Surf. Sci. 1995, 343, 211–220. [Google Scholar]
- Delley, B. An all-electron numerical-method for solving the local density functional for polyatomic-molecules. J. Chem. Phys. 1990, 92, 508–517. [Google Scholar] [CrossRef]
- Delley, B. From molecules to solids with the DMol(3) approach. J. Chem. Phys. 2000, 113, 7756–7764. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [PubMed]
- Delley, B. Hardness conserving semilocal pseudopotentials. Phys. Rev. B 2002, 66. [Google Scholar] [CrossRef]
- Liu, X.; Meng, C.G.; Liu, C.H. Molecular dynamics study on superheating of Pd at high heating rates. Phase Transit. 2006, 79, 249–259. [Google Scholar] [CrossRef]
- Liu, X.; Meng, C.G.; Liu, C.H. Melting and superheating of Ag at high heating rate. Acta Phys. -Chim. Sin. 2004, 20, 280–284. [Google Scholar]
- Monkhorst, H.J.; Pack, J.D. Special points for Brillouin-zone integrations. Phys. Rev. B 1976, 13, 5188–5192. [Google Scholar] [CrossRef]
- Govind, N.; Petersen, M.; Fitzgerald, G.; King-Smith, D.; Andzelm, J. A generalized synchronous transit method for transition state location. Comput. Mater. Sci. 2003, 28, 250–258. [Google Scholar] [CrossRef]
- Gajdos, M.; Eichler, A.; Hafner, J. CO adsorption on close-packed transition and noble metal surfaces: Trends from ab initio calculations. J. Phys. -Condens. Matter 2004, 16, 1141–1164. [Google Scholar] [CrossRef]
- Sample Availability: Not available.
© 2015 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chu, M.; Liu, X.; Sui, Y.; Luo, J.; Meng, C. Unique Reactivity of Transition Metal Atoms Embedded in Graphene to CO, NO, O2 and O Adsorption: A First-Principles Investigation. Molecules 2015, 20, 19540-19553. https://doi.org/10.3390/molecules201019540
Chu M, Liu X, Sui Y, Luo J, Meng C. Unique Reactivity of Transition Metal Atoms Embedded in Graphene to CO, NO, O2 and O Adsorption: A First-Principles Investigation. Molecules. 2015; 20(10):19540-19553. https://doi.org/10.3390/molecules201019540
Chicago/Turabian StyleChu, Minmin, Xin Liu, Yanhui Sui, Jie Luo, and Changgong Meng. 2015. "Unique Reactivity of Transition Metal Atoms Embedded in Graphene to CO, NO, O2 and O Adsorption: A First-Principles Investigation" Molecules 20, no. 10: 19540-19553. https://doi.org/10.3390/molecules201019540