Ionization States, Cellular Toxicity and Molecular Modeling Studies of Midazolam Complexed with Trimethyl-β-Cyclodextrin

 ,
, 

1. Introduction
2. Results and Discussion
 (pred)), CR (
(pred)), CR (  (pred)), and degree of dissociation for these forms of midazolam (Figure 1A) were calculated by using MarvinSketch software (ChemAxon, Budapest, Hungary). The net charges for midazolam forms at different pH were calculated from the Henderson-Hasselbalch equation [19] as follows:
(pred)), and degree of dissociation for these forms of midazolam (Figure 1A) were calculated by using MarvinSketch software (ChemAxon, Budapest, Hungary). The net charges for midazolam forms at different pH were calculated from the Henderson-Hasselbalch equation [19] as follows:


 (exp) = 7.0, (pred) = 8.52) is affected by the pH-dependent reaction. Hence, the diazepine ring of 1,4-benzodiazepine moiety was formed with (exp) of 2.4 [15,20] and (pred) of 3.48. In addition, the basic nitrogen in position 2 of the imidazole ring, which belongs to the imidazobenzodiazepine moiety, allows the active elements of midazolam to form water-soluble salts with acids [21]. The imidazole ring of midazolam also accounts for its stability in solution and rapid metabolism [22]. Consequently, the calculated net charges for both molecular forms were found to be positive (+0.9) for the OR form of midazolam and benzodiazepines (+0.3) or neutral for the CR form at physiological pH (Figure 1B) and Supplementary material 2.
(exp) = 7.0, (pred) = 8.52) is affected by the pH-dependent reaction. Hence, the diazepine ring of 1,4-benzodiazepine moiety was formed with (exp) of 2.4 [15,20] and (pred) of 3.48. In addition, the basic nitrogen in position 2 of the imidazole ring, which belongs to the imidazobenzodiazepine moiety, allows the active elements of midazolam to form water-soluble salts with acids [21]. The imidazole ring of midazolam also accounts for its stability in solution and rapid metabolism [22]. Consequently, the calculated net charges for both molecular forms were found to be positive (+0.9) for the OR form of midazolam and benzodiazepines (+0.3) or neutral for the CR form at physiological pH (Figure 1B) and Supplementary material 2. = −9.98 kcal·mol−1) compared to the OR form of midazolam with the SES (SAS) value of 296.68 (548.07) Å2, ClogP (ionic species) of 0.08, and
= −9.98 kcal·mol−1) compared to the OR form of midazolam with the SES (SAS) value of 296.68 (548.07) Å2, ClogP (ionic species) of 0.08, and  of −67.01 kcal·mol−1. For the OR/CR-TRIMEB inclusion complex, the optimal pH value to be easily dissolved is in the range from 3.5 to 3.7, which might imply some difficulties on its intravenous applications (unpublished data). Moreover, the further pH elevation in the solution (up to 7.0) might increase the risk of a suspension at physiological pH or even precipitate formation at basic pH value. and ) for midazolam open-ring (OR) and closed-ring (CR) forms (A) and reversible pH-dependent OR-to-CR conversion reaction (B) occurring through the formation of the ionized transition state intermediate (TS).
and ) for midazolam open-ring (OR) and closed-ring (CR) forms (A) and reversible pH-dependent OR-to-CR conversion reaction (B) occurring through the formation of the ionized transition state intermediate (TS).
of −67.01 kcal·mol−1. For the OR/CR-TRIMEB inclusion complex, the optimal pH value to be easily dissolved is in the range from 3.5 to 3.7, which might imply some difficulties on its intravenous applications (unpublished data). Moreover, the further pH elevation in the solution (up to 7.0) might increase the risk of a suspension at physiological pH or even precipitate formation at basic pH value. and ) for midazolam open-ring (OR) and closed-ring (CR) forms (A) and reversible pH-dependent OR-to-CR conversion reaction (B) occurring through the formation of the ionized transition state intermediate (TS).
and ) for midazolam open-ring (OR) and closed-ring (CR) forms (A) and reversible pH-dependent OR-to-CR conversion reaction (B) occurring through the formation of the ionized transition state intermediate (TS).



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Average Value | SD | |||||||
|---|---|---|---|---|---|---|---|---|
| Form | ΔGbind * | LElig | Kb (μM) | pKb | ΔGbind * | LElig | Kb (μM) | pKb |
| OR | −5.57 | −0.21 | 79.89 | 4.09 | 0.02 | 0.001 | 2.706 | 0.015 |
| TS | −5.15 | −0.19 | 162.66 | 3.79 | - | - | - | - |
| CR | −4.66 | −0.2 | 434.05 | 3.43 | 0.427 | 0.018 | 235.034 | 0.308 |


3. Experimental Section



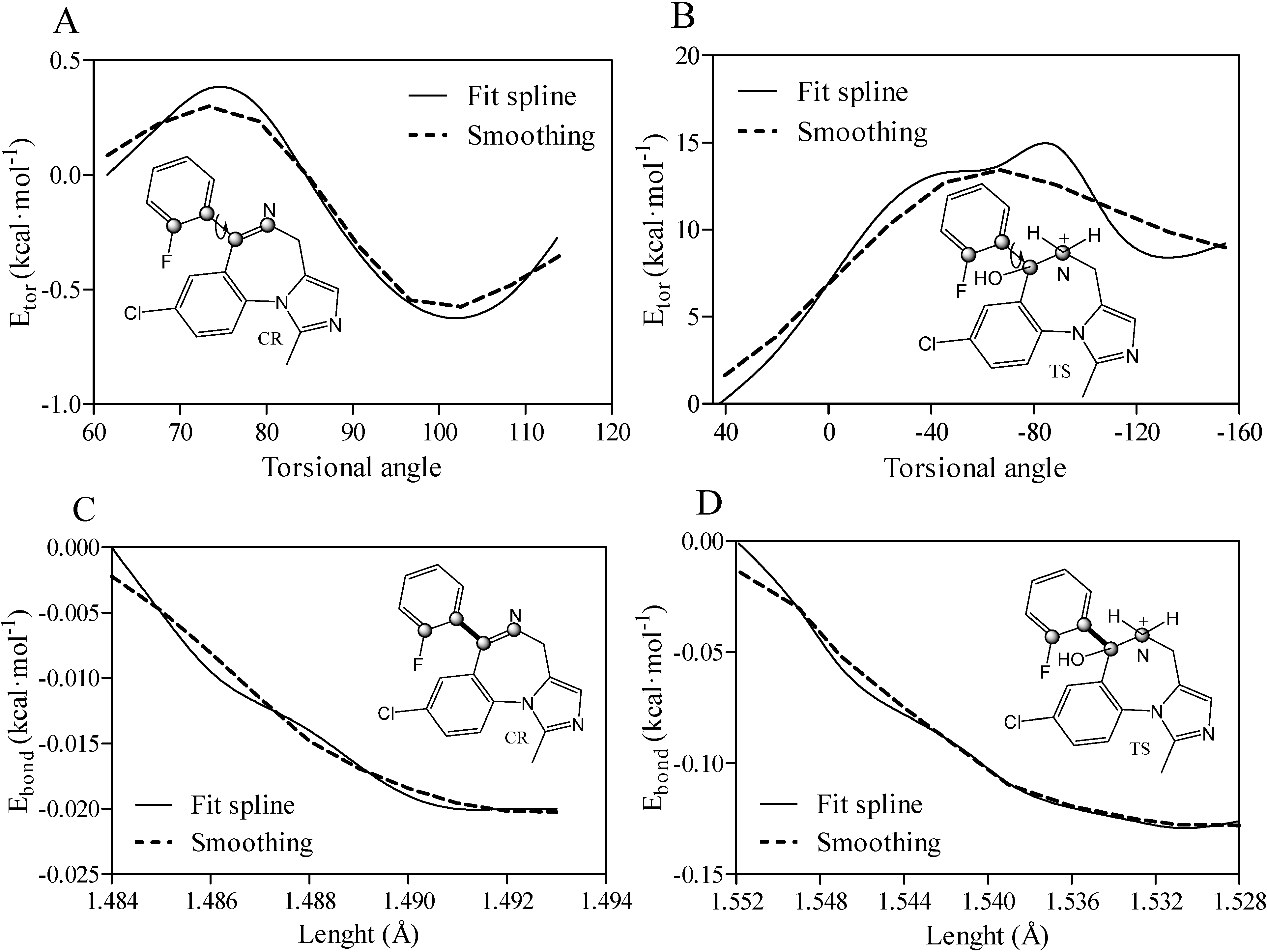
 , ), relative torsional (Etor), and bond length (Ebond) energies. Relative torsional energy Etor was calculated from a truncated Fourier series using the equation in the form shown below:
, ), relative torsional (Etor), and bond length (Ebond) energies. Relative torsional energy Etor was calculated from a truncated Fourier series using the equation in the form shown below:
4. Conclusions
= −9.98 kcal·mol−1) than for the more hydrophilic OR form with a minimal of −67.01 kcal·mol−1 during the OR-to-CR conversion that occurred through the formation of ionized TS intermediate. The absence of TRIMEB and OR/CR-TRIMEB toxicity in the cEND cells after 24 h of incubation (in either Dulbecco’s Modified Eagles Medium or heat-inactivated human serum) was confirmed by a CellTiter-Glo® (Promega) luminescent cell viability assay. Despite the lack of cellular toxicity, the native pH value required to dissolve the OR/CR-TRIMEB complex is in the range of 3.5–3.7, limiting its usage to clinical applications. The molecular docking method detected that the more flexible OR form (Ntor = 5) of midazolam may serve as a better binder to trimethyl-β-cyclodextrin with the fluorophenyl ring introduced inside the amphiphilic cavity of the TRIMEB. The optimal OR binding affinity was verified by a minimal ΔGbind value of −5.57 ± 0.02 kcal·mol−1, an equilibrium binding constant (Kb) of 79.89 ± 2.706 μM, and a ligand efficiency index (LElig) of −0.21 ± 0.001. A decrease in the torsional energy (Etor = −0.25 kcal·mol−1) for the active CR form was detected in order to reach the lowest energy orientation during the conformational sampling. Therefore, it is important to improve the clinical applications of midazolam via its complexation with trimethyl-β-cyclodextrin in order to increase its overall aqueous solubility concerning the different forms and ionization states of this anaesthetic.Supplementary Materials
Supplementary Files
Supplementary File 1Acknowledgments
Author Contributions
Conflicts of Interest
References
- Szejtli, J. Introduction and general overview of cyclodextrin chemistry. Chem. Rev. 1998, 98, 1743–1753. [Google Scholar] [CrossRef] [PubMed]
- Correa, D.H.A.; Melo, P.S.; de Carvalho, C.A.; de Azevedo, M.B.; Duran, N.; Haun, M. Dehydrocrotonin and its beta-cyclodextrin complex: Cytotoxicity in V79 fibroblasts and rat cultured hepatocytes. Eur. J. Pharmacol. 2005, 510, 17–24. [Google Scholar] [CrossRef] [PubMed]
- Ehteda, A.; Galettis, P.; Chu, S.W.; Pillai, K.; Morris, D.L. Complexation of albendazole with hydroxypropyl-beta-cyclodextrin significantly improves its pharmacokinetic profile, cell cytotoxicity and antitumor efficacy in nude mice. Anticancer Res. 2012, 32, 3659–3666. [Google Scholar]
- Felton, L.A.; Popescu, C.; Wiley, C.; Esposito, E.X.; Lefevre, P.; Hopfinger, A.J. Experimental and computational studies of physicochemical properties influence NSAID-cyclodextrin complexation. AAPS PharmSciTech 2014, 15, 872–881. [Google Scholar] [CrossRef] [PubMed]
- Upadhyay, S.K.; Kumar, G. NMR and molecular modelling studies on the interaction of fluconazole with beta-cyclodextrin. Chem. Cent. J. 2009, 3. [Google Scholar] [CrossRef] [PubMed]
- Upadhyay, S.K.; Ali, S.M. Solution structure of loperamide and beta-cyclodextrin inclusion complexes using NMR spectroscopy. J. Chem. Sci. 2009, 121, 521–527. [Google Scholar] [CrossRef]
- Jain, A.S.; Date, A.A.; Pissurlenkar, R.R.; Coutinho, E.C.; Nagarsenker, M.S. Sulfobutyl ether(7) beta-cyclodextrin (SBE(7) beta-CD) carbamazepine complex: Preparation, characterization, molecular modeling, and evaluation of in vivo anti-epileptic activity. AAPS PharmSciTech 2011, 12, 1163–1175. [Google Scholar] [CrossRef] [PubMed]
- Shityakov, S.; Förster, C. Pharmacokinetic delivery and metabolizing rate of nicardipine incorporated in hydrophilic and hydrophobic cyclodextrins using two-compartment mathematical model. Sci. World J. 2013. [Google Scholar] [CrossRef]
- Sohajda, T.; Beni, S.; Varga, E.; Ivanyi, R.; Racz, A.; Szente, L.; Noszal, B. Characterization of aspartame-cyclodextrin complexation. J. Pharm. Biomed. Anal. 2009, 50, 737–745. [Google Scholar] [CrossRef] [PubMed]
- Luger, T.; Hayashi, T.; Weiss, C.G.; Hill, H.F. The spinal potentiating effect and the supraspinal inhibitory effect of midazolam on opioid-induced analgesia in rats. Eur. J. Pharmacol. 1995, 275, 153–162. [Google Scholar] [CrossRef] [PubMed]
- Agrawal, A.; Pergadia, M.L.; Saccone, S.F.; Hinrichs, A.L.; Lessov-Schlaggar, C.N.; Saccone, N.L.; Neuman, R.J.; Breslau, N.; Johnson, E.; Hatsukami, D.; et al. Gamma-aminobutyric acid receptor genes and nicotine dependence: Evidence for association from a case-control study. Addiction 2008, 103, 1027–1038. [Google Scholar] [CrossRef]
- Agrawal, A.; Pergadia, M.L.; Balasubramanian, S.; Saccone, S.F.; Hinrichs, A.L.; Saccone, N.L.; Breslau, N.; Johnson, E.O.; Hatsukami, D.; Martin, N.G.; et al. Further evidence for an association between the gamma-aminobutyric acid receptor A, subunit 4 genes on chromosome 4 and fagerstrom test for nicotine dependence. Addiction 2009, 104, 471–477. [Google Scholar] [CrossRef]
- Andersin, R. Solubility and acid-base behaviour of midazolam in media of different pH, studied by ultraviolet spectrophotometry with multicomponent software. J. Pharm. Biomed. Anal. 1991, 9, 451–455. [Google Scholar] [CrossRef] [PubMed]
- Andersin, R.; Tammilehto, S. Photochemical decomposition of midazolam. 4. Study of pH-dependent stability by high-performance liquid-chromatography. Int. J. Pharm. 1995, 123, 229–235. [Google Scholar]
- Loftsson, T.; Gudmundsdóttir, H.; Sigurjónsdóttir, J.F.; Sigurdsson, H.H.; Sigfússon, S.D.; Másson, M.; Stefánsson, E. Cyclodextrin solubilization of benzodiazepines: formulation of midazolam nasal spray. Int. J. Pharm. 2001, 212, 29–40. [Google Scholar] [CrossRef] [PubMed]
- Mathiron, D.; Marçon, F.; Dubaele, J.M.; Cailleu, D.; Pilard, S.; Djedaïni-Pilard, F. Benefits of methylated cyclodextrins in the development of midazolam pharmaceutical formulations. J. Pharm. Sci. 2013, 102, 2102–2111. [Google Scholar] [CrossRef] [PubMed]
- Kurono, Y.; Kuwayama, T.; Kamiya, K.; Yashiro, T.; Ikeda, K. The behavior of 1,4-benzodiazepine drugs in acidic media. II. Kinetics and mechanism of the acid-base equilibrium reaction of oxazolam. Chem. Pharm. Bull. 1985, 33, 1633–1640. [Google Scholar] [CrossRef] [PubMed]
- Kaartama, R.; Turunen, E.; Toljamo, K.; Kokki, H.; Lehtonen, M.; Ranta, V.P.; Savolainen, J.; Järvinen, K.; Jarho, P. The effect of hydroxypropyl-beta-cyclodextrin and sucrose on the sublingual absorption of midazolam in rabbits. Eur. J. Pharm. Biopharm. 2012, 81, 178–183. [Google Scholar] [CrossRef] [PubMed]
- Hills, A.G. pH and the Henderson-Hasselbalch equation. Am. J. Med. 1973, 55, 131–133. [Google Scholar] [CrossRef] [PubMed]
- Cho, M.J.; Scahill, T.A.; Hester, J.B., Jr. Kinetic and equilibrium of the reversible alprazolam ring-opening reaction. J. Pharm. Sci. 1983, 72, 356–362. [Google Scholar] [CrossRef] [PubMed]
- Saari, T.I.; Uusi-Oukari, M.; Ahonen, J.; Olkkola, K.T. Enhancement of GABAergic activity: Neuropharmacological effects of benzodiazepines and therapeutic use in anesthesiology. Pharmacol. Rev. 2011, 63, 243–267. [Google Scholar] [CrossRef] [PubMed]
- Arendt, R.M.; Greenblatt, D.J.; de Jong, R.H.; Bonin, J.D.; Abernethy, D.R.; Ehrenberg, B.L.; Giles, H.G.; Sellers, E.M.; Shader, R.I. In vitro correlates of benzodiazepine cerebrospinal fluid uptake, pharmacodynamic action and peripheral distribution. J. Pharmacol. Exp. Ther. 1983, 227, 98–106. [Google Scholar] [PubMed]
- Cosconati, S.; Forli, S.; Perryman, A.L.; Harris, R.; Goodsell, D.S.; Olson, A.J. Virtual screening with AutoDock: Theory and practice. Expert. Opin. Drug. Discov. 2010, 5, 597–607. [Google Scholar] [CrossRef] [PubMed]
- Steffen, A.; Thiele, C.; Tietze, S.; Strassnig, C.; Kämper, A.; Lengauer, T.; Wenz, G.; Apostolakis, J. Improved cyclodextrin-based receptors for camptothecin by inverse virtual screening. Chemistry 2007, 13, 6801–6809. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Lai, W.P.; Zhang, Z.; Li, W.K.; Cheung, H.Y. Computational study on the molecular inclusion of andrographolide by cyclodextrin. J. Comput. Aided Mol. Des. 2009, 23, 153–162. [Google Scholar] [CrossRef] [PubMed]
- Shityakov, S.; Broscheit, J.; Förster, C. Alpha-cyclodextrin dimer complexes of dopamine and levodopa derivatives to assess drug delivery to the central nervous system: ADME and molecular docking studies. Int. J. Nanomed. 2012, 7, 3211–3219. [Google Scholar] [CrossRef]
- Harata, K.; Hirayama, F.; Arima, H.; Uekama, K.; Miyaji, T. Crystal structure of heptakis(2,3,6-tri-O-methyl)-β-cyclodextrin complexes with m-iodophenol and 4-biphenylacetic acid. Guest-induced conformational change of a pyranose ring. J. Chem. Soc. Perkin Trans. 2 1992, 1159–1166. [Google Scholar]
- Li, W.S.; Wang, S.C.; Hwang, T.S.; Chao, I. Substituent effect on the structural behavior of modified cyclodextrin: A molecular dynamics study on methylated beta-CDs. J. Phys. Chem. B 2012, 116, 3477–3489. [Google Scholar] [CrossRef] [PubMed]
- Brown, G.R.; Caira, M.R.; Nassimbeni, L.R.; van Oudtshoorn, B. Inclusion of ibuprofen by heptakis(2,3,6-tri-O-methyl)-beta-cyclodextrin: An X-ray diffraction and thermal analysis study. J. Incl. Phenom. Macrocycl. Chem. 1996, 26, 281–294. [Google Scholar] [CrossRef]
- Steiner, T.; Saenger, W. Covalent bond lengthening in hydroxyl-groups involved in 3-center and in cooperative hydrogen-bonds-analysis of low-temperature neutron-diffraction data. J. Am. Chem. Soc. 1992, 114, 7123–7126. [Google Scholar] [CrossRef]
- Caira, M.R.; Griffith, V.J.; Nassimbeni, L.R.; van Oudtshoorn, B. X-ray structure and thermal analysis of a 1:1 complex between (S)-naproxen and heptakis(2,3,6-tri-O-methyl)-beta-cyclodextrin. J. Inclusion Phenom. Mol. Recognit. Chem. 1994, 20, 277–290. [Google Scholar] [CrossRef]
- Pacifici, G.M. Clinical pharmacology of midazolam in neonates and children: Effect of disease—A review. Int. J. Pediatr. 2014. [Google Scholar] [CrossRef]
- Szejtli, J.; Liptak, A.; Jodal, I.; Neszelyi, A. Synthesis and C-13-Nmr spectroscopy of methylated beta-cyclodextrins. Starke 1980, 32, 165–169. [Google Scholar]
- Gasteiger, J.; Marsili, M. Iterative partial equalization of orbital electronegativity—A rapid access to atomic charges. Tetrahedron 1980, 36, 3219–3228. [Google Scholar] [CrossRef]
- Hopkins, A.L.; Keserü, G.M.; Leeson, P.D.; Rees, D.C.; Reynolds, C.H. The role of ligand efficiency metrics in drug discovery. Nat. Rev. Drug. Discov. 2014, 13, 105–121. [Google Scholar] [CrossRef] [PubMed]
- Murray, C.W.; Erlanson, D.A.; Hopkins, A.L.; Keserü, G.M.; Leeson, P.D.; Rees, D.C.; Reynolds, C.H.; Richmond, N.J. Validity of ligand efficiency metrics. ACS Med. Chem. Lett. 2014, 5, 616–618. [Google Scholar] [CrossRef] [PubMed]
- Förster, C.; Silwedel, C.; Golenhofen, N.; Burek, M.; Kietz, S.; Mankertz, J.; Drenckhahn, D. Occludin as direct target for glucocorticoid-induced improvement of blood-brain barrier properties in a murine in vitro system. J. Physiol. 2005, 565, 475–486. [Google Scholar] [CrossRef] [PubMed]
- Silwedel, C.; Forster, C. Differential susceptibility of cerebral and cerebellar murine brain microvascular endothelial cells to loss of barrier properties in response to inflammatory stimuli. J. Neuroimmunol. 2006, 179, 37–45. [Google Scholar] [CrossRef] [PubMed]
- Golenhofen, N.; Ness, W.; Wawrousek, E.F.; Drenckhahn, D. Expression and induction of the stress protein alpha-B-crystallin in vascular endothelial cells. Histochem. Cell Biol. 2002, 117, 203–209. [Google Scholar] [CrossRef] [PubMed]
- Tarini, M.; Cignoni, P.; Montani, C. Ambient occlusion and edge cueing to enhance real time molecular visualization. IEEE Trans. Vis. Comput. Graph. 2006, 12, 1237–1244. [Google Scholar] [CrossRef] [PubMed]
- Sample Availability: Not available.
© 2014 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shityakov, S.; Sohajda, T.; Puskás, I.; Roewer, N.; Förster, C.; Broscheit, J.-A. Ionization States, Cellular Toxicity and Molecular Modeling Studies of Midazolam Complexed with Trimethyl-β-Cyclodextrin. Molecules 2014, 19, 16861-16876. https://doi.org/10.3390/molecules191016861
Shityakov S, Sohajda T, Puskás I, Roewer N, Förster C, Broscheit J-A. Ionization States, Cellular Toxicity and Molecular Modeling Studies of Midazolam Complexed with Trimethyl-β-Cyclodextrin. Molecules. 2014; 19(10):16861-16876. https://doi.org/10.3390/molecules191016861
Chicago/Turabian StyleShityakov, Sergey, Tamás Sohajda, István Puskás, Norbert Roewer, Carola Förster, and Jens-Albert Broscheit. 2014. "Ionization States, Cellular Toxicity and Molecular Modeling Studies of Midazolam Complexed with Trimethyl-β-Cyclodextrin" Molecules 19, no. 10: 16861-16876. https://doi.org/10.3390/molecules191016861