Isotope Effects in ESR Spectroscopy

Abstract
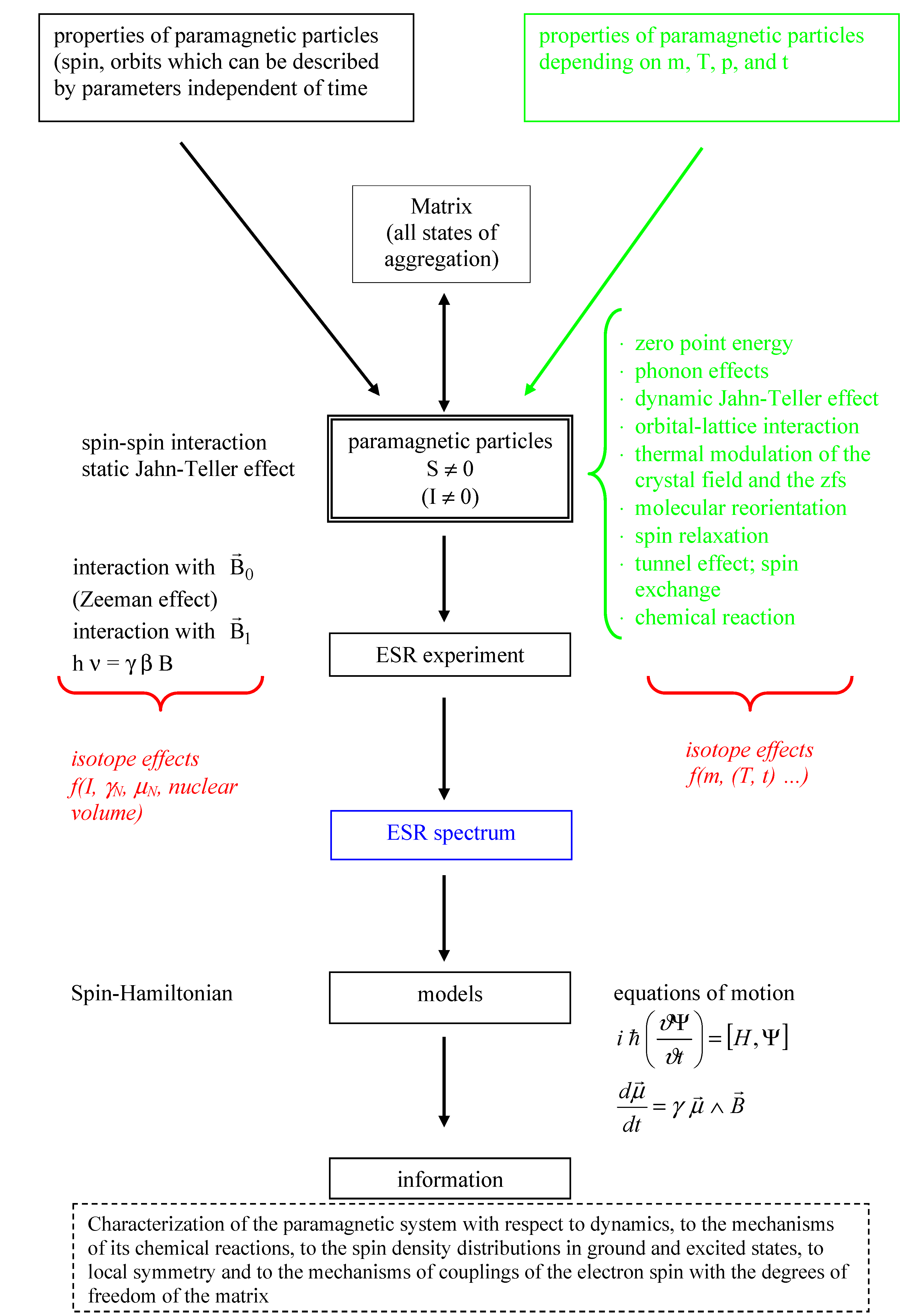
:1. Introduction
2. Electron Spin Resonance (ESR) and Relations to Isotope Effects
2.1. Basic Principle of ESR
 is perpendicular to the static field
is perpendicular to the static field  . According to the resonance condition:
. According to the resonance condition:
 or ω = γ
or ω = γ 
 , a mismatching of the resonator and hence, and a part of the microwave radiation will be registered at the microwave diode.
, a mismatching of the resonator and hence, and a part of the microwave radiation will be registered at the microwave diode.

 and nuclear spins
and nuclear spins  ) can be taken into account. To grasp the full spectrum of isotope effects further models (see Figure 1 and Section 2.9.) are necessary. The electron and nuclear spin (
) can be taken into account. To grasp the full spectrum of isotope effects further models (see Figure 1 and Section 2.9.) are necessary. The electron and nuclear spin (  and
and  ) as well as the magnetic induction
) as well as the magnetic induction  (and their combinations; see Figure 1) are represented by operators connected by spin coupling coefficients [17,18], which depend on structure and properties of the examined system (see e.g., [23,24]). Favourably the arrangement of the Spin-Hamilton terms will be selected in such a way that they transform as the irreducible presentation of the point groups of the system [25]. Next to the experimental possibility to determine the components of the Spin-Hamiltonian (see e.g., [3,17,18,22]), they can be calculated by quantum mechanical means on using electronic state (or wave) functions. As mentioned above the Spin-Hamiltonian described in such a way does not depend on m, T, p, n (n: Amount of substance), …, phonons, etc. It is assumed, that on applying that spin-Hamilton model the atomic nuclei are “fixed”. On doing so, vibronic effects cannot be considered explicitly, despite the fact that they may change experimental spectra considerably. This is true, e.g., for the Jahn-Teller effect [25], which in ESR spectroscopy is manifested in vibronic reduction, tunnel splitting, random-strain, and relaxation effects, and causes e.g., a distinctive dependency of fine and hyperfine structure on the temperature. These phenomena get eye-catching e.g., on observing the un-common paramagnetic resonance properties of Cu(II) compounds [25,26]. A qualitatively correct interpretation of the respective spectra can be achieved as a function of the Jahn-Teller dynamics of the nuclear configuration (and therefore the nuclear masses, too [25]).
(and their combinations; see Figure 1) are represented by operators connected by spin coupling coefficients [17,18], which depend on structure and properties of the examined system (see e.g., [23,24]). Favourably the arrangement of the Spin-Hamilton terms will be selected in such a way that they transform as the irreducible presentation of the point groups of the system [25]. Next to the experimental possibility to determine the components of the Spin-Hamiltonian (see e.g., [3,17,18,22]), they can be calculated by quantum mechanical means on using electronic state (or wave) functions. As mentioned above the Spin-Hamiltonian described in such a way does not depend on m, T, p, n (n: Amount of substance), …, phonons, etc. It is assumed, that on applying that spin-Hamilton model the atomic nuclei are “fixed”. On doing so, vibronic effects cannot be considered explicitly, despite the fact that they may change experimental spectra considerably. This is true, e.g., for the Jahn-Teller effect [25], which in ESR spectroscopy is manifested in vibronic reduction, tunnel splitting, random-strain, and relaxation effects, and causes e.g., a distinctive dependency of fine and hyperfine structure on the temperature. These phenomena get eye-catching e.g., on observing the un-common paramagnetic resonance properties of Cu(II) compounds [25,26]. A qualitatively correct interpretation of the respective spectra can be achieved as a function of the Jahn-Teller dynamics of the nuclear configuration (and therefore the nuclear masses, too [25]). , …, see [3,27]), important information about structure and dynamics of the paramagnetic species can be gained on using further models (see Section 2.8. and Section 2.9. and Figure 1). Simplified the spin can be considered to be a further indicator of the total state (wave) function [17,18]. Contrary to the indistinguishable electrons having only one spin quantum number S = ± ½ (see also [17,28]), the nuclei including the isotopes are described by a considerable number of nuclear spins and the corresponding magnetic dipole and electric quadrupole moments. Together with this a large number of applications exist which are based on nuclear moments. To name only the most important one: nuclear magnetic resonance (NMR). Which effects or processes explicitly involve spins or which observables are produced by the spin? There are two more items to be named:
, …, see [3,27]), important information about structure and dynamics of the paramagnetic species can be gained on using further models (see Section 2.8. and Section 2.9. and Figure 1). Simplified the spin can be considered to be a further indicator of the total state (wave) function [17,18]. Contrary to the indistinguishable electrons having only one spin quantum number S = ± ½ (see also [17,28]), the nuclei including the isotopes are described by a considerable number of nuclear spins and the corresponding magnetic dipole and electric quadrupole moments. Together with this a large number of applications exist which are based on nuclear moments. To name only the most important one: nuclear magnetic resonance (NMR). Which effects or processes explicitly involve spins or which observables are produced by the spin? There are two more items to be named:2.2. Parameters of the Spin-Hamiltonian
2.2.1. The ĝ Tensor, the Interaction of Spin and Orbital Moments with the External Magnetic Induction ![Molecules 18 06679 i012]()

 and electron spin
and electron spin  [see Equation (2)]. With respect to its transformation behaviour that coupling element (coupling matrix) is a tensor object. It mainly determines the Zeeman term:
[see Equation (2)]. With respect to its transformation behaviour that coupling element (coupling matrix) is a tensor object. It mainly determines the Zeeman term:

 (see quenching of orbital moments in molecular systems [3,17,18,23,24]) as well as the kind and energy levels of the excited electronic terms.
(see quenching of orbital moments in molecular systems [3,17,18,23,24]) as well as the kind and energy levels of the excited electronic terms.2.2.2. The Hyperfine Interaction
2.2.2.1. Nuclear Spin, Nuclear Magnetic Moment, Hyperfine Structure, and General Relations to Isotope Effects
 the nuclear magnetic moment plays a role on splitting the ground state of paramagnetic systems. At the nucleus that field should have a magnitude of some 10,000 mT and should cause a hyperfine splitting larger than 0.01 cm−1. Penrose [59] delivered the first successful experimental proof of the ESR hyperfine structure. The coupling constant of the hyperfine structure of paramagnetic systems is one of the most important parameters in ESR and from the very beginning it is connected to the different action of isotopes (e.g., 63Cu and 65Cu) [59,60,61,62]. The hyperfine structure of paramagnetic systems allows the determination of the nuclear spin simply and directly, as well as the relation of the magnetic moments of two isotopes of the same atom.
the nuclear magnetic moment plays a role on splitting the ground state of paramagnetic systems. At the nucleus that field should have a magnitude of some 10,000 mT and should cause a hyperfine splitting larger than 0.01 cm−1. Penrose [59] delivered the first successful experimental proof of the ESR hyperfine structure. The coupling constant of the hyperfine structure of paramagnetic systems is one of the most important parameters in ESR and from the very beginning it is connected to the different action of isotopes (e.g., 63Cu and 65Cu) [59,60,61,62]. The hyperfine structure of paramagnetic systems allows the determination of the nuclear spin simply and directly, as well as the relation of the magnetic moments of two isotopes of the same atom.
 instead of the real mass (9.109 × 10−31 kg). In a descriptive way this can be explained by a joint movement of the nucleus (proton mass: 1.672·× 10−27 kg, muonium mass: 9.106·× 10−31 kg.
instead of the real mass (9.109 × 10−31 kg). In a descriptive way this can be explained by a joint movement of the nucleus (proton mass: 1.672·× 10−27 kg, muonium mass: 9.106·× 10−31 kg.2.2.2.1.1. Phenomena of Magnetic Hyperfine Structure Exemplified on the Pair of Isotopomers 1H and 2H
 and nuclear spin
and nuclear spin  energy levels of the electron split.
energy levels of the electron split.

{kind=link}
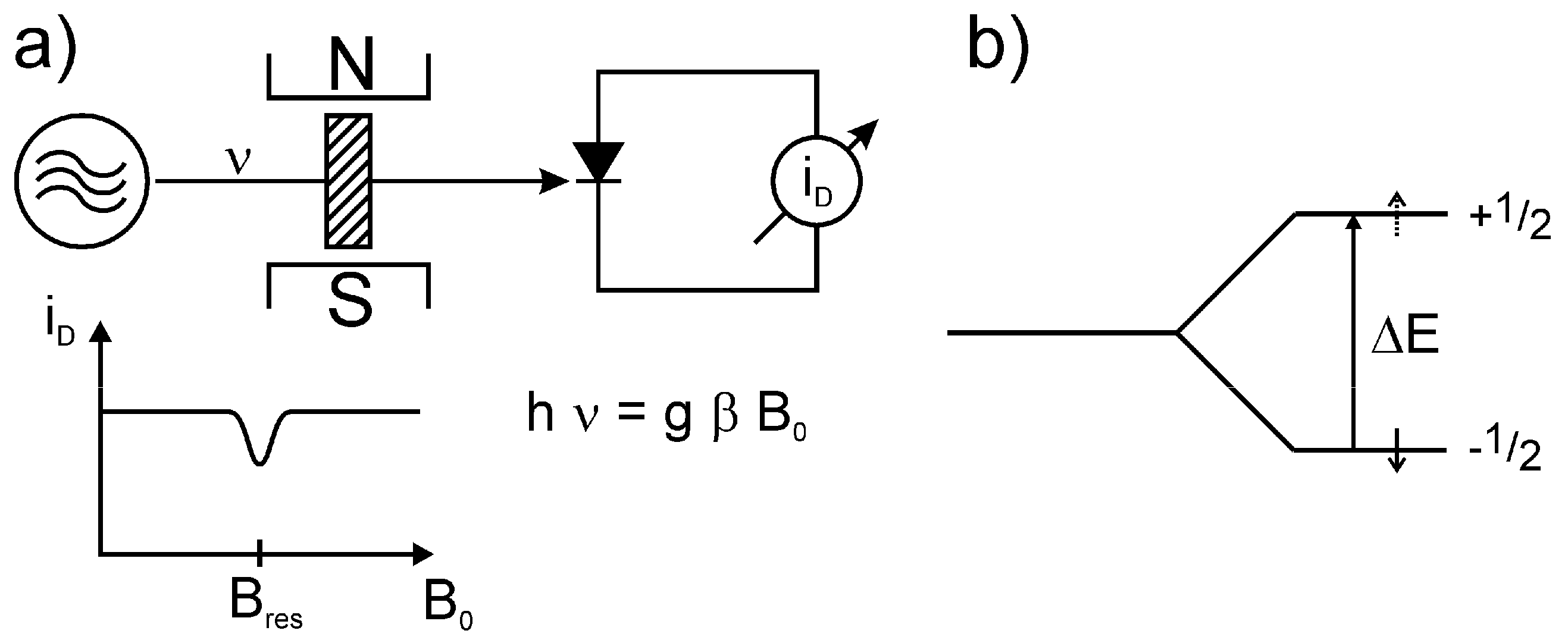{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 1H | 2H | |
|---|---|---|
| nuclear magnetic moment | μH = gN βN I = 1.4106 × 10−26 J T−1 | μD = gN βN I = 1.4106·× 10−26 J T−1 |
| nuclear spin | I = ½ | I = 1 |
| nuclear g factor | gH = 5.5856 | gD = 0.8574 |
| gyromagnetic ratio | γH = μH/I h = 2.6752 × 108 s−1 T−1 | γD = μD/I h = 4.1067 × 107 s−1 T−1 |
2.2.2.1.2. Static and Dynamic Aspects of the Hyperfine Interaction
 and
and  [30,34,35], spin exchange [35], dynamic Jahn-Teller effect [25], tunnel effect [33,80], vibronic coupling, and others. Under a dynamic aspect the isotope effects act upon the zero-point energies by vibrational averaging with amplitudes depending on isotopes. Morton and Preston used this effect for investigating the hyperfine structure of F2NO [81].
[30,34,35], spin exchange [35], dynamic Jahn-Teller effect [25], tunnel effect [33,80], vibronic coupling, and others. Under a dynamic aspect the isotope effects act upon the zero-point energies by vibrational averaging with amplitudes depending on isotopes. Morton and Preston used this effect for investigating the hyperfine structure of F2NO [81].2.3. The Zero-Field Splitting

 , (collecting magnetic dipolar and exchange spin-spin interactions) which is usually used for the parametrization of the ESR fine structure [Equation (5)], does not contain a mass depending term. Thus, for the description of the respective influence of isotopes the interaction with phonons have to be considered, e.g., in form of the spin-phonon operators [29,83] as it was shown by Shrivastava [84] for the system 52Cr, 53Cr in the system MgO:Cr3+.
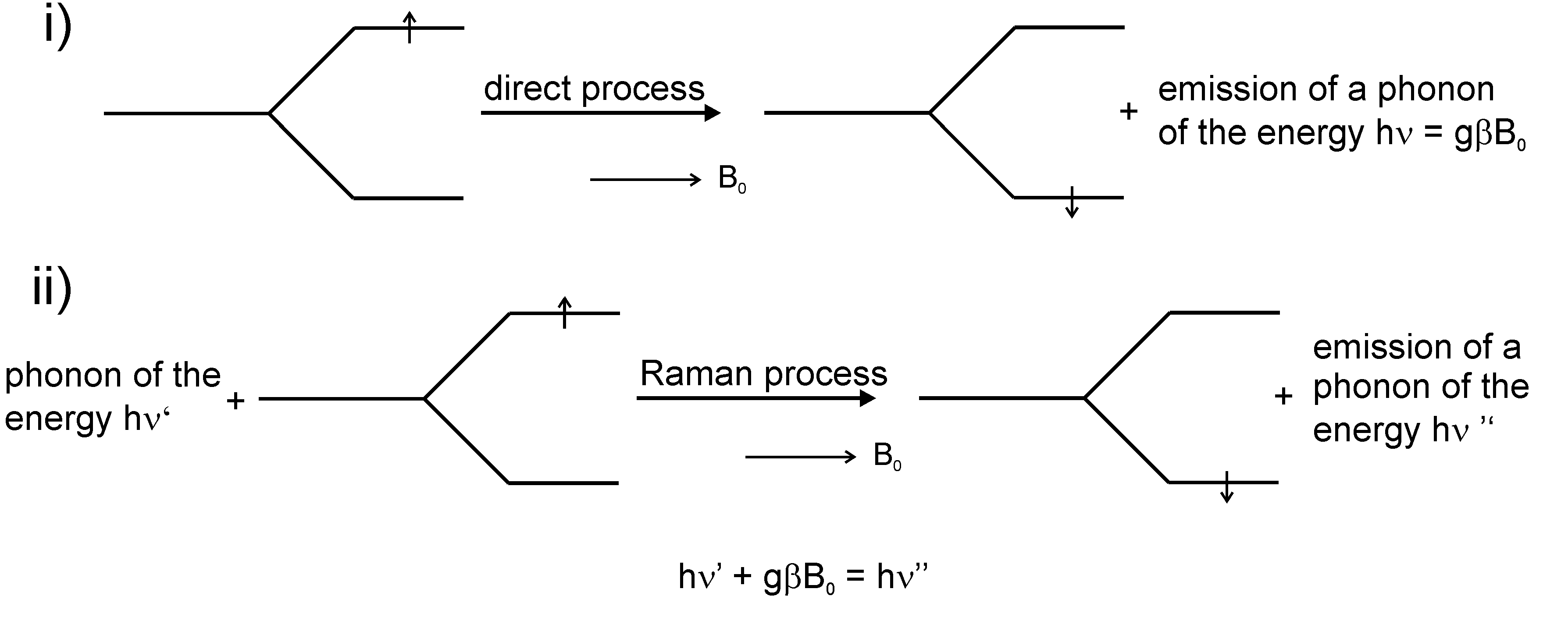
, (collecting magnetic dipolar and exchange spin-spin interactions) which is usually used for the parametrization of the ESR fine structure [Equation (5)], does not contain a mass depending term. Thus, for the description of the respective influence of isotopes the interaction with phonons have to be considered, e.g., in form of the spin-phonon operators [29,83] as it was shown by Shrivastava [84] for the system 52Cr, 53Cr in the system MgO:Cr3+.2.4. Spin Relaxation
 gB0. Process (ii) is characterized by the action of a phonon of the energy hν’ on a spin with mS = +1/2 and E = ½ gβB0 and in a non-elastic scattering process the spin state changes to mS = −1/2 and energy E = −½ gβB0 under emission of a phonon with the energy hν”. The following energy results: hν’ + gβB0 = hν” [4].
gB0. Process (ii) is characterized by the action of a phonon of the energy hν’ on a spin with mS = +1/2 and E = ½ gβB0 and in a non-elastic scattering process the spin state changes to mS = −1/2 and energy E = −½ gβB0 under emission of a phonon with the energy hν”. The following energy results: hν’ + gβB0 = hν” [4].
2.5. Spin Exchange
 .
.
 (τ)dτ))−(ψ- exp (−i
(τ)dτ))−(ψ- exp (−i  (τ)dτ)))̸
(τ)dτ)))̸ 

2.6. The Magnetic Isotope Effect
 . It decreases with increasing
. It decreases with increasing  . In this case the triplet states [3,27] T+ and T− of the singulet-triplet evolution are separated and only the S-T0 channel is active. At lower fields all three channels contribute.
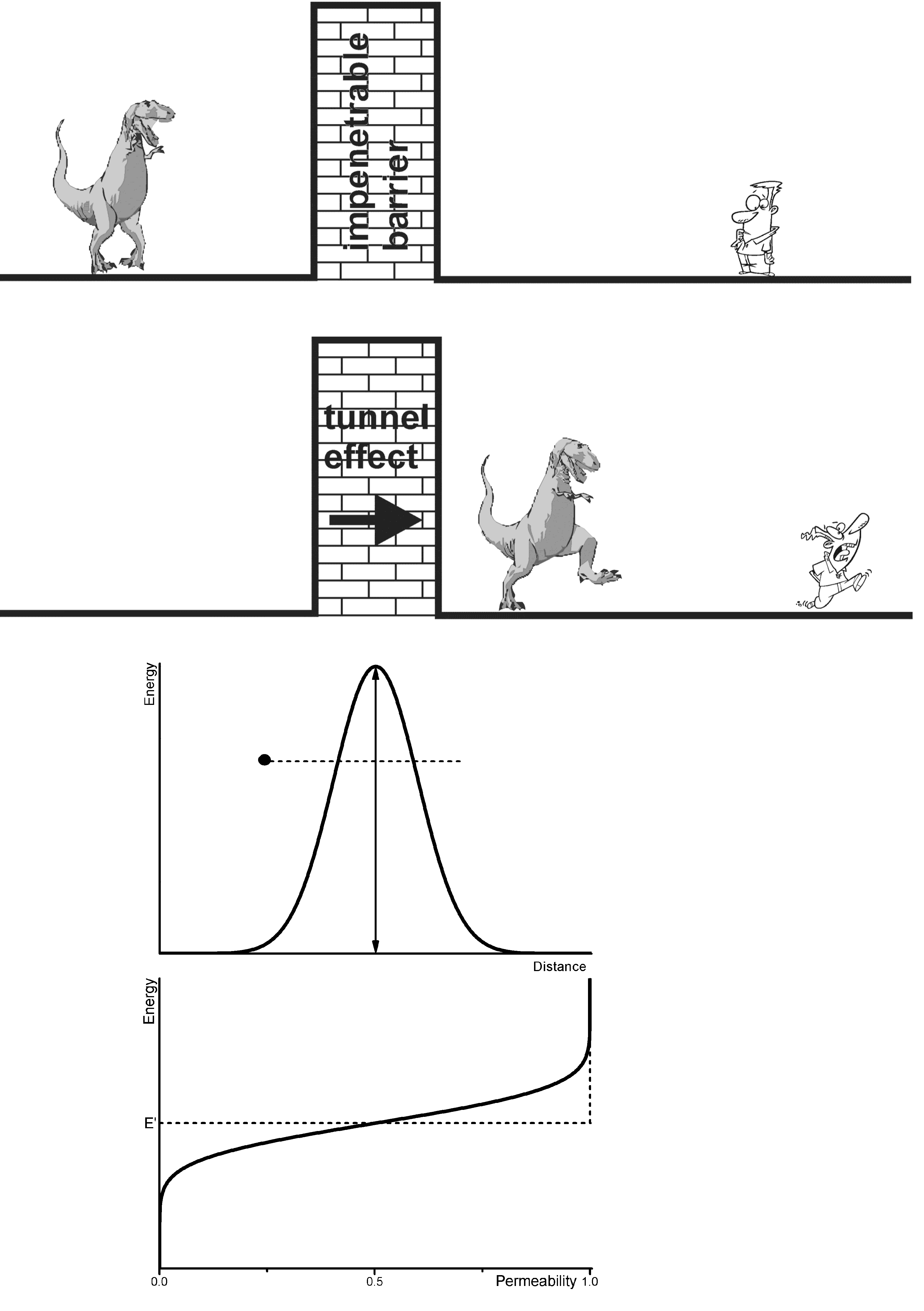
. In this case the triplet states [3,27] T+ and T− of the singulet-triplet evolution are separated and only the S-T0 channel is active. At lower fields all three channels contribute.2.7. Tunnel Effect Phenomenon and Its Relation to ESR and to Isotope Substitution
 exp (−γ r)
exp (−γ r)

2.8. The Jahn-Teller Effect (JTE)
2.9. Zero-Point Vibrations and Isotope Effects
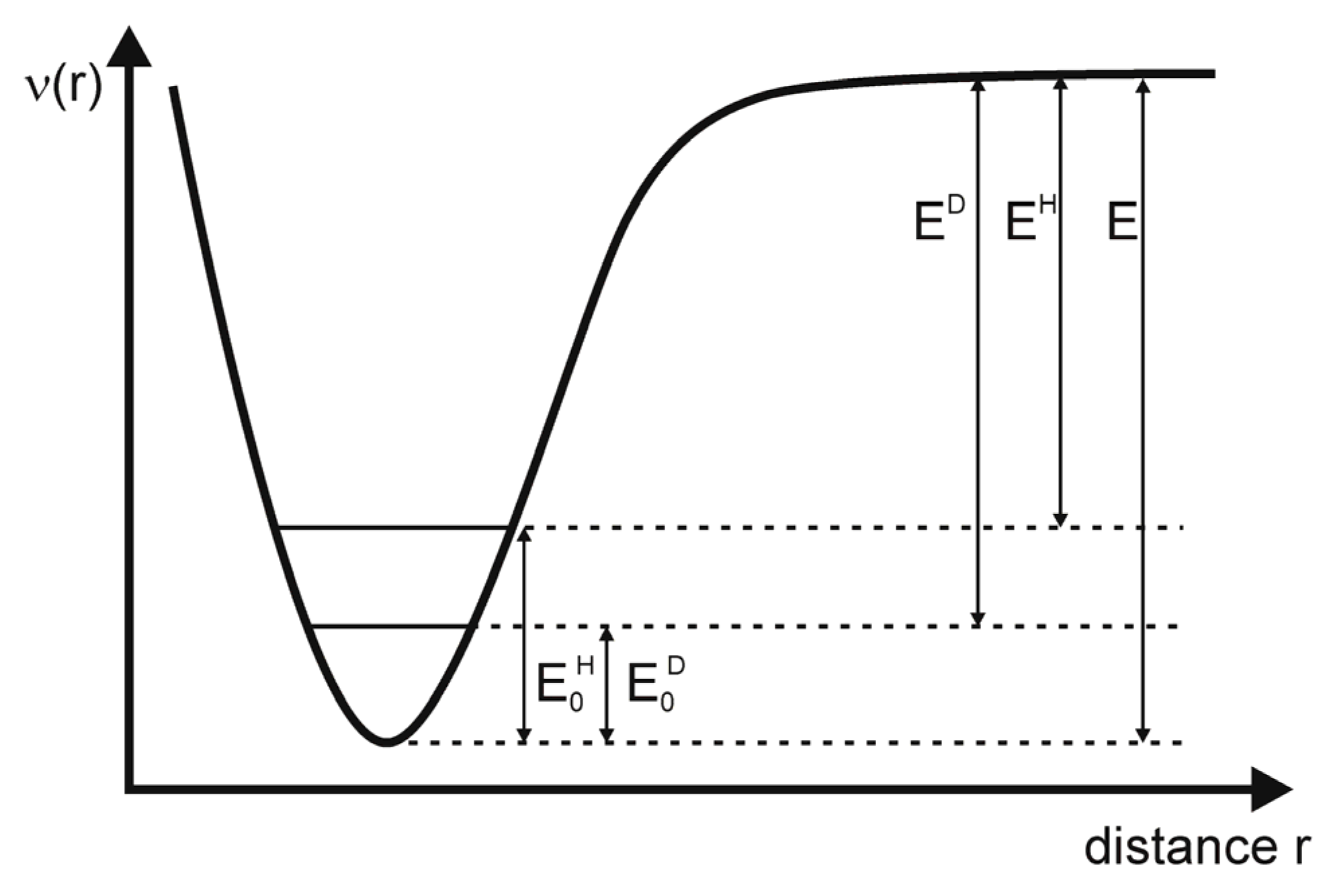
 ), but the reduced mass is smaller for the X-H than for the X-D fragment and consequently the oscillation frequencies νH > νD (in reality the ratio νH/νD is about 0.7) and the zero-point energies behave as E0H > E0D [33]. Thus, the dissociation energies are EH > ED (see Figure 5) and the dissociation of the hydrogen component will be faster (there is a smaller activation energy). This is, among others, manifested in the higher pH value of D2O in comparison to H2O at the same temperature.
), but the reduced mass is smaller for the X-H than for the X-D fragment and consequently the oscillation frequencies νH > νD (in reality the ratio νH/νD is about 0.7) and the zero-point energies behave as E0H > E0D [33]. Thus, the dissociation energies are EH > ED (see Figure 5) and the dissociation of the hydrogen component will be faster (there is a smaller activation energy). This is, among others, manifested in the higher pH value of D2O in comparison to H2O at the same temperature.
2.10. Parameters Determining the Habit of the ESR Spectra (ESR Response)
 , ti, Grad Bi,
, ti, Grad Bi,  , …)
, …)
 ); νRF: radio frequency; PM, PRF: microwave or radio frequency power;
); νRF: radio frequency; PM, PRF: microwave or radio frequency power;  : outer ‘static’ magnetic induction; ti: different time functions for the variation of the other parameters (cw and impulse techniques); Grad Bi: gradient of one component of
: outer ‘static’ magnetic induction; ti: different time functions for the variation of the other parameters (cw and impulse techniques); Grad Bi: gradient of one component of  , e.g., for recording spatially dissolved spectra (ESR tomography);
, e.g., for recording spatially dissolved spectra (ESR tomography);  : electric field e.g., for interaction with quadrupole moments.
: electric field e.g., for interaction with quadrupole moments. ,
,  , ci, …(ci: concentration of components which, e.g., determine the pH value or the concentration of educts or products of chemical reactions);
, ci, …(ci: concentration of components which, e.g., determine the pH value or the concentration of educts or products of chemical reactions);  is responsible for polarizations or chemical reactions (e.g., electrode reactions; see below); the state of magnetization of the sample may by varied by
is responsible for polarizations or chemical reactions (e.g., electrode reactions; see below); the state of magnetization of the sample may by varied by  of different origins.
of different origins. ,
,  ), orbital (
), orbital (  ), and quadrupole moments Q ††, as well as electric (
), and quadrupole moments Q ††, as well as electric (  ), and magnetic fields (
), and magnetic fields (  ) ‡‡ for symbolizing the spin couplings (with “fixed” atomic nuclei §§).
) ‡‡ for symbolizing the spin couplings (with “fixed” atomic nuclei §§).
 |  |  |  |  |  | |
|---|---|---|---|---|---|---|
 | I | II | II | IV | V | • |
 | VI | VII | VIII | • | • | |
 | IX | X | • | • | ||
 | • | • | • | |||
 | • | XI | ||||
 | *** |
 ; VI: spin-spin interaction HSS with coupling parameter
; VI: spin-spin interaction HSS with coupling parameter  (ESR fine structure) or J, respectively; VII: ESR hyperfine interactions, Fermi-contact interactions, dipolar hyperfine splitting; VIII: spin-orbit coupling with parameter λ; IX: nuclear spin-nuclear spin interaction Jij; X: nuclear spin-orbit interaction; XI: nuclear quadrupole-
(ESR fine structure) or J, respectively; VII: ESR hyperfine interactions, Fermi-contact interactions, dipolar hyperfine splitting; VIII: spin-orbit coupling with parameter λ; IX: nuclear spin-nuclear spin interaction Jij; X: nuclear spin-orbit interaction; XI: nuclear quadrupole-  interaction; *** these interactions will not be considered and explained.
interaction; *** these interactions will not be considered and explained.2.11. Models for the Parameterization of ESR Spectra; Phenomenological ESR Parameters
 or
or  ), line form (Gauss, Lorentz, and Voigt profiles) as well as line widths.
), line form (Gauss, Lorentz, and Voigt profiles) as well as line widths.3. Selected Examples
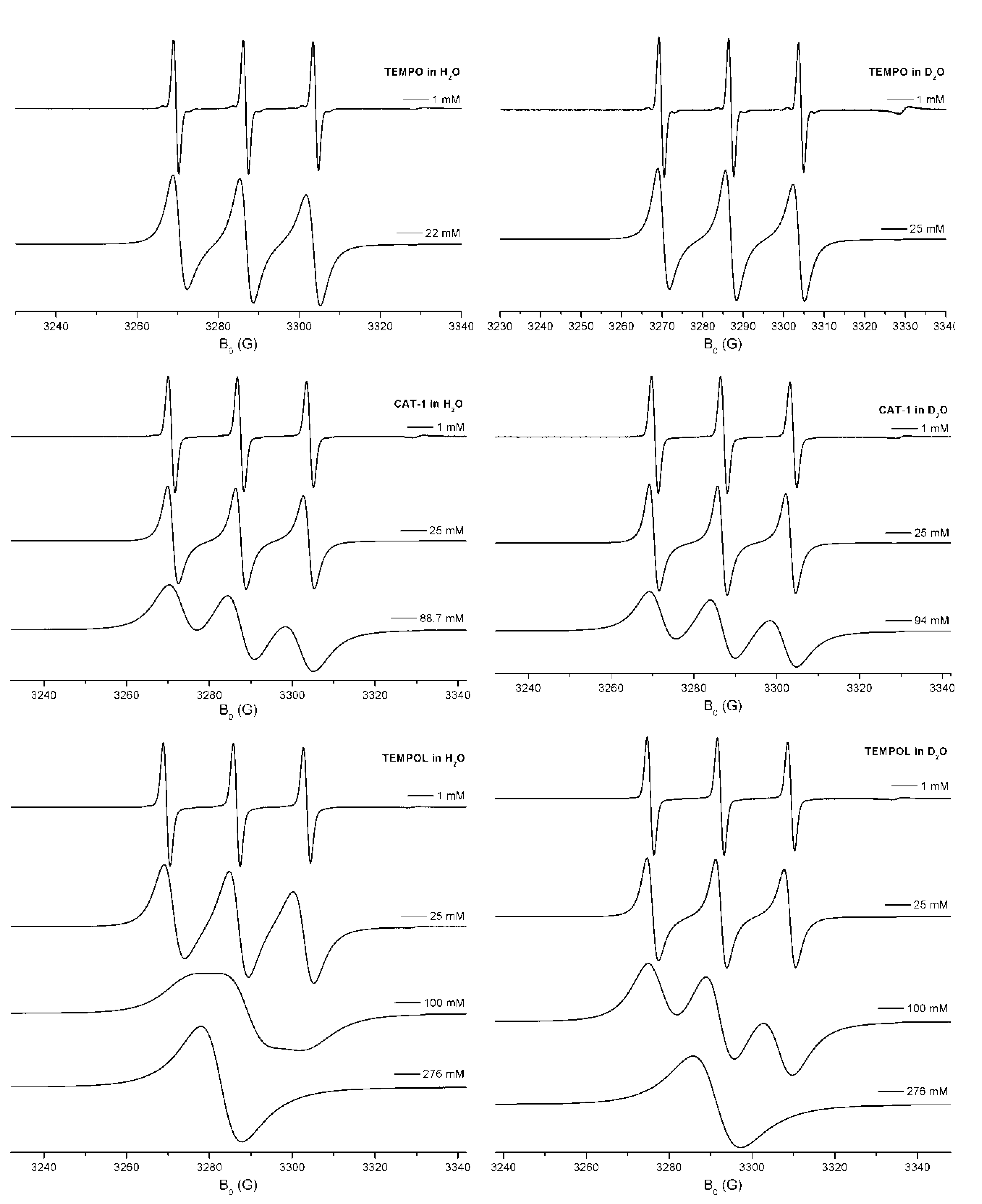
3.1. Determination of Concentrations of Paramagnetic Particles in a Sample by Means of ESR
3.2. Influence of the H-D Substitution on the g Factor
 the contributions of the orbital momentum has to be considered. The influence of the nuclear masses of the involved atoms is very small, thus there are only very few investigations with respect to the influence of isotopes in that field.
the contributions of the orbital momentum has to be considered. The influence of the nuclear masses of the involved atoms is very small, thus there are only very few investigations with respect to the influence of isotopes in that field.3.3. Selected Hyperfine Effects
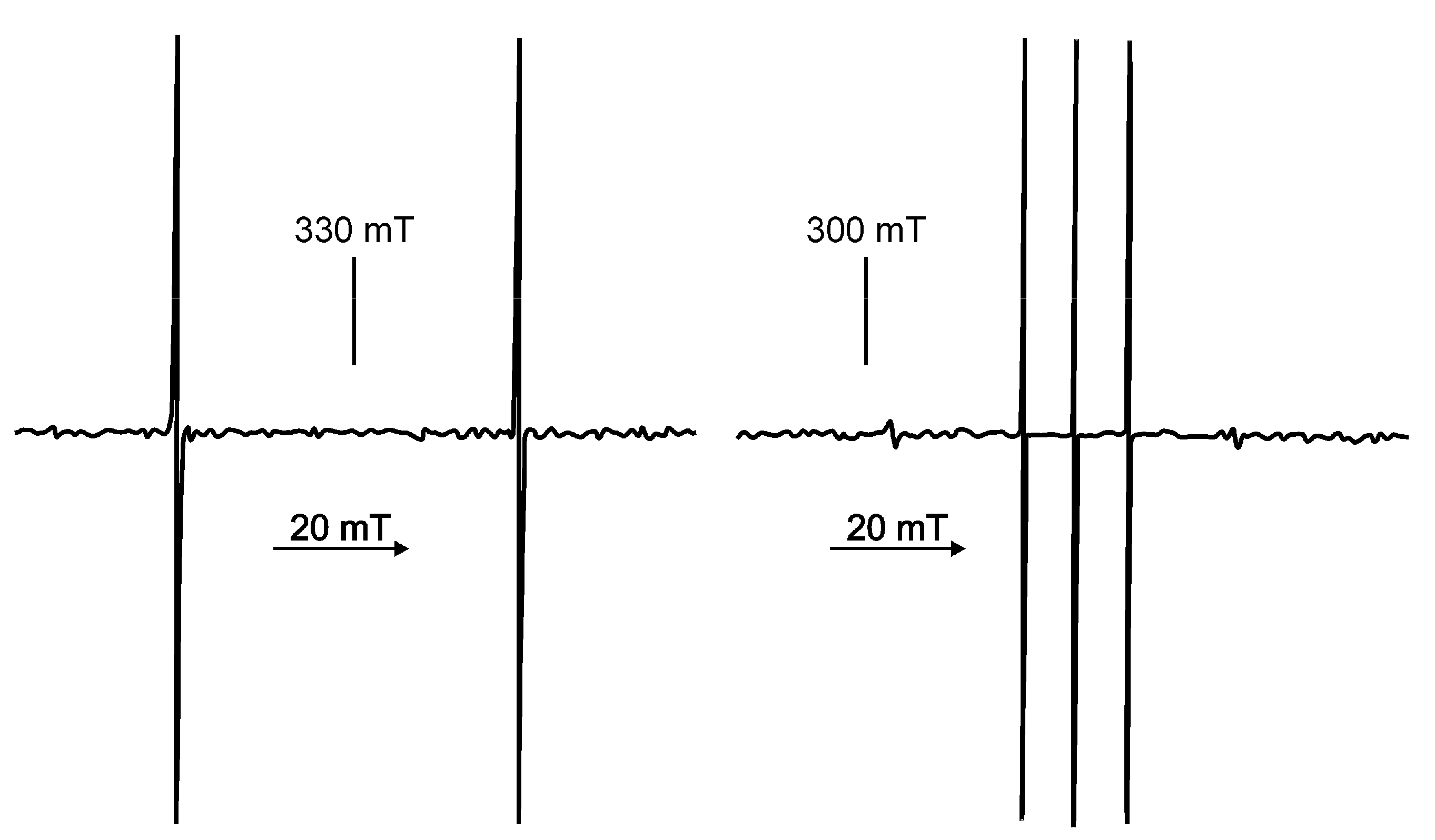
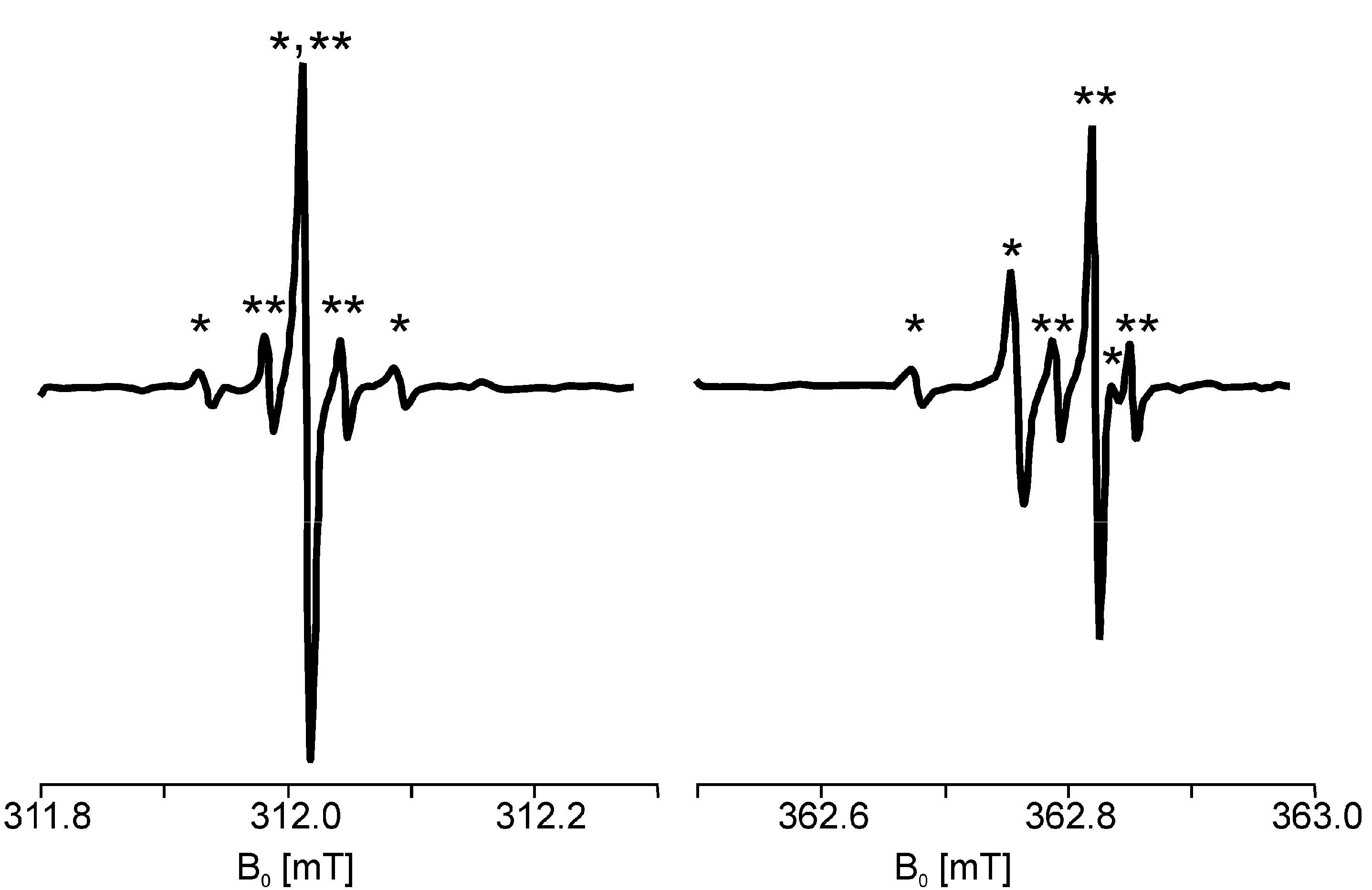
3.3.1. ESR Detection of Effect of Isotopes in Natural Abundance

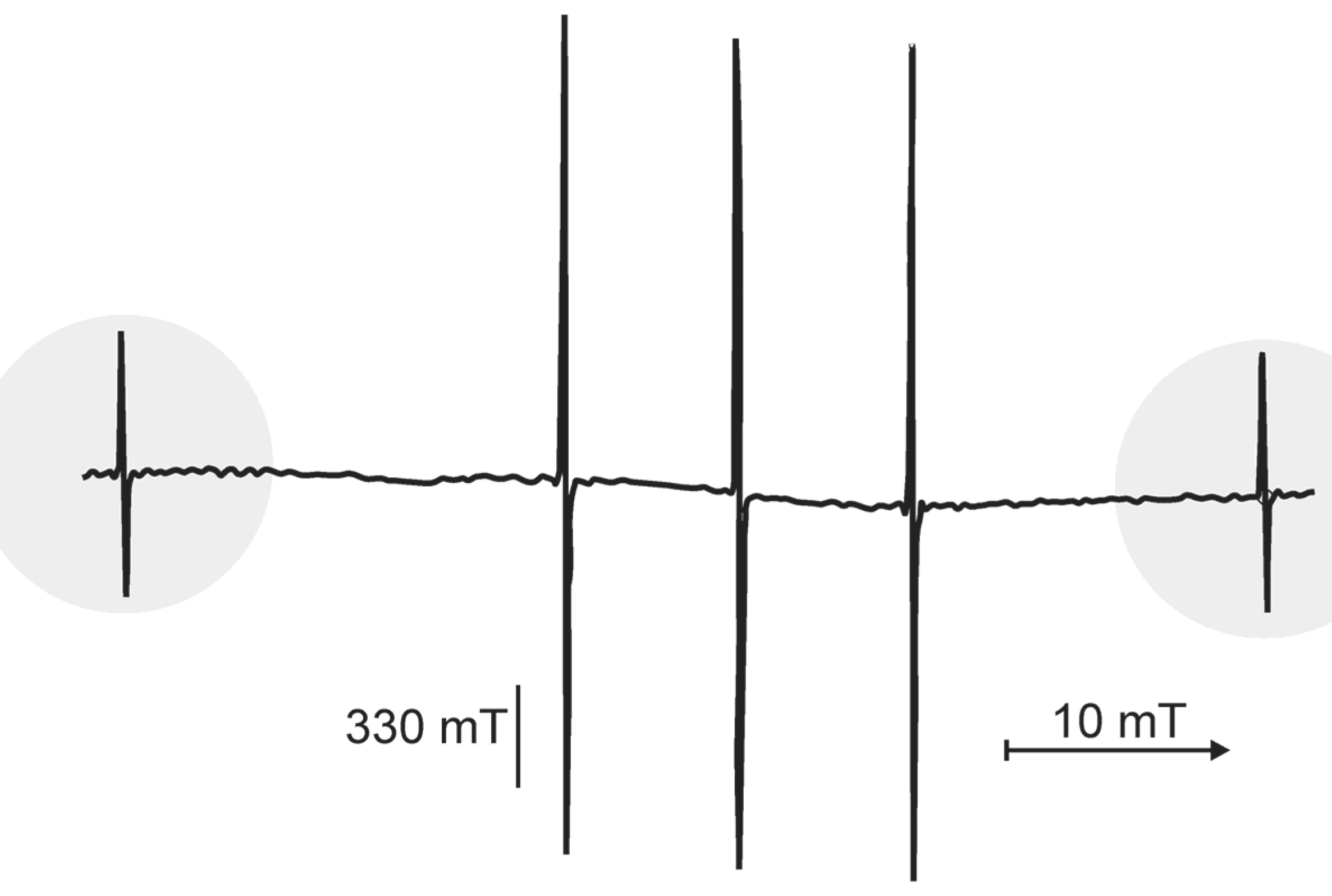
3.3.2. Evidence for Trapped Muonium, Hydrogen, and Deuterium Atoms at Ambient Temperature





3.4. Selected zfs Effects
3.5. Examples for the Jahn-Teller Effect and Vibronic Couplings
3.6. Examples for the Tunnel Effect
3.7. Influence of the Zero-Point Vibrations
3.8. Isotope Effects of the Matrix
3.8.1. Stationary Effects
3.8.1.1. H-D Exchange and Fermi Contact
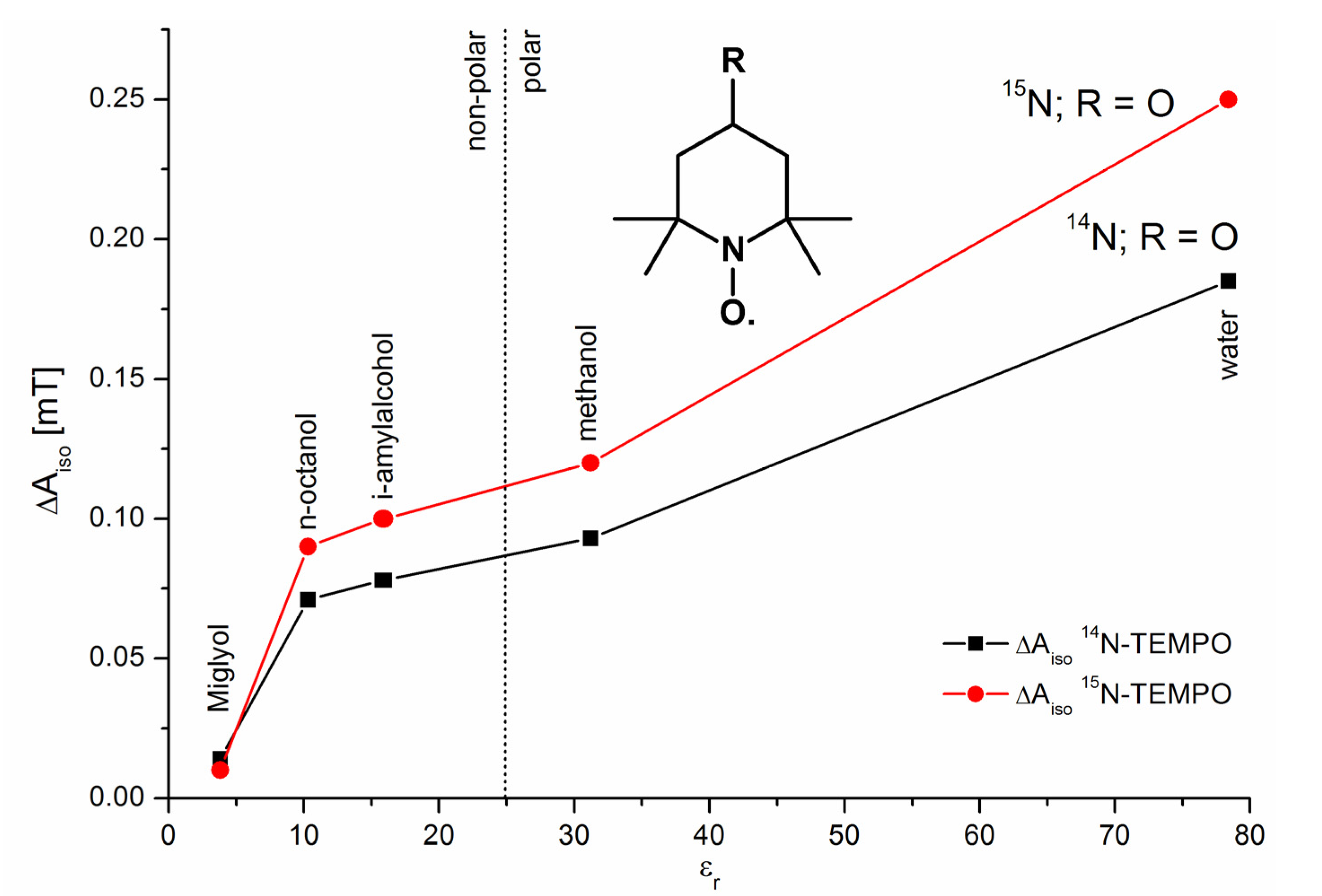
3.8.1.2. Spin Probes and Polarity of the Matrix

3.8.1.3. Spin trapping
3.8.2. Dynamic Effects
3.8.2.1. Spin Relaxation
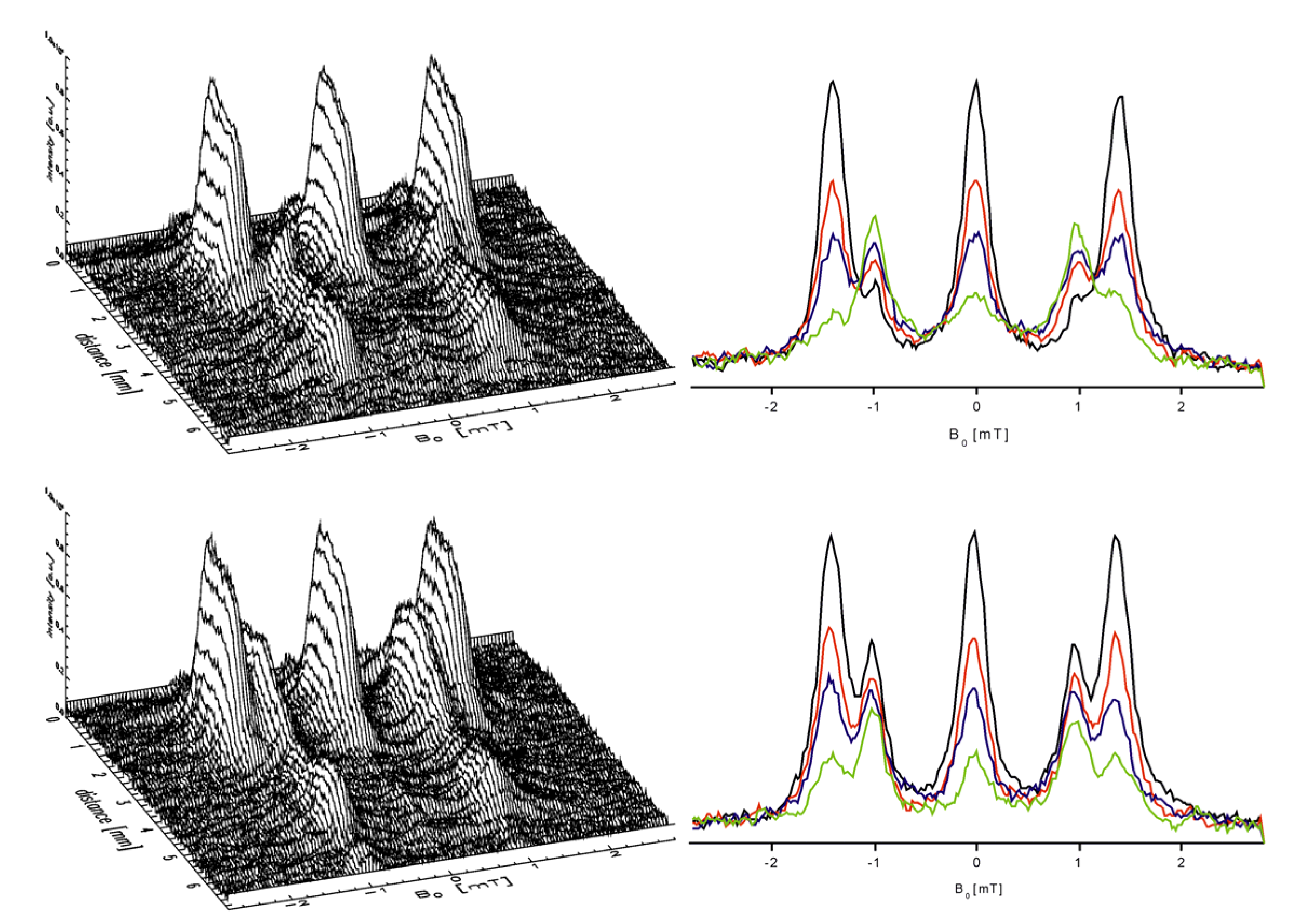
3.8.2.2. Diffusion of Spin Probes and ESR Tomography


3.8.2.3. Spin Exchange and Chemical Reactions
 , too. Examples for this are the changes of
, too. Examples for this are the changes of  in the course of the Maillard reactions [165], the analysis of water samples, in which
in the course of the Maillard reactions [165], the analysis of water samples, in which  provides environmental information about the sum of further dissolved substances and about activity and state of biosystems.
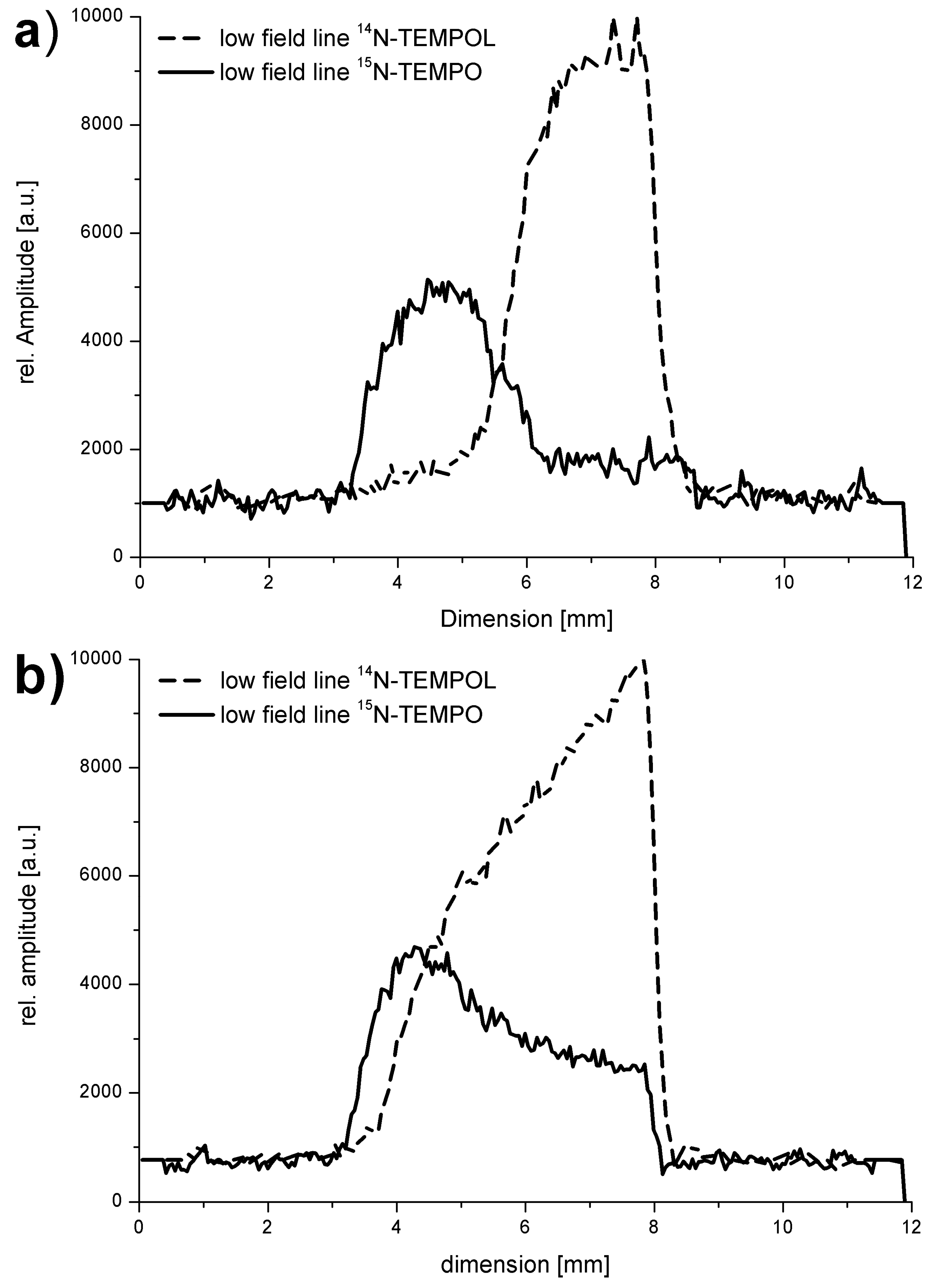
provides environmental information about the sum of further dissolved substances and about activity and state of biosystems. . The marking of the mainly used nitroxyl spin probes with the isotope 15N instead of 14N is advantageous due to the increase in signal amplitude because the complete signal intensity is distributed over two lines only. Additional signal splittings can be avoided by substitution of 1H by 2H, which leads to smaller line widths. Altogether at comparable sensitivity the spin probe concentration can be reduced. Additionally the reduction of the dipole-dipole interactions between the probes leads to an increase of sensitivity of the
. The marking of the mainly used nitroxyl spin probes with the isotope 15N instead of 14N is advantageous due to the increase in signal amplitude because the complete signal intensity is distributed over two lines only. Additional signal splittings can be avoided by substitution of 1H by 2H, which leads to smaller line widths. Altogether at comparable sensitivity the spin probe concentration can be reduced. Additionally the reduction of the dipole-dipole interactions between the probes leads to an increase of sensitivity of the  . Disadvantageous seems to be the case that only one parameter (K [165]) is used which usually even depends on the applied radical concentration. Moreover, the signal-to-noise ratio decreases because the spectra are recorded with small modulation amplitudes. E.g., the reduced information content results already from the fact that out of 18 hyperfine lines only two are used.
. Disadvantageous seems to be the case that only one parameter (K [165]) is used which usually even depends on the applied radical concentration. Moreover, the signal-to-noise ratio decreases because the spectra are recorded with small modulation amplitudes. E.g., the reduced information content results already from the fact that out of 18 hyperfine lines only two are used.
3.9. Indication of Some Sources Dealing with Non-cw Techniques and Modern Applications of ESR
4. Conclusions
Acknowledgments
Conflicts of Interest
References
- Isotope Effects in Chemistry and Biology; Kohen, A.; Limbach, H.-H. (Eds.) CRC Press — Taylor & Francis: Boca Raton, FL, USA, 2006.
- Bigeleisen, J. Gilbert N. Lewis and the beginnings of isotope chemistry. J. Chem. Educ. 1984, 61, 108–116. [Google Scholar] [CrossRef]
- Weil, J.A.; Bolton, J.R. Electron. Paramagnetic Resonance; John Wiley & Sons: Hoboken, NJ, USA, 2007. [Google Scholar]
- Pake, G.E. Paramagnetic Resonance; W.A. Benjamin, Inc: New York, NY, USA, 1969. [Google Scholar]
- Moss, R.A.; Platz, M.A.; Jones, Jr., M. Reactive Intermediate Chemistry; Wiley Interscience: Hoboken, NJ, USA, 2004. [Google Scholar]
- Spin Labeling—Theory and Applications; Berliner, L.J. (Ed.) Academic Press: New York, NY, USA, 1976.
- Spin Labeling. II—Theory and Applications; Berliner, L.J. (Ed.) Academic Press: New York, NY, USA, 1979.
- Brodsky, S.J.; Lebed, R.E. Production of the smallest QED atom: True muonium (μ+μ−). Phys. Rev. Lett. 2009, 102, 213401. [Google Scholar] [CrossRef]
- Griscom, D.L.; Friebele, E.J. Fundamental defect centers in glass: 29Si hyperfine structure of the nonbridging oxygen hole center and the peroxy radical in α-SiO2. Phys. Rev. B 1981, 24, 4896–4898. [Google Scholar] [CrossRef]
- Freed, J.H. Theory of slow tumbling esr spectra for nitroxides. In Spin Labeling—Theory and Applications; Berliner, L.J., Ed.; Academic Press: New York, NY, USA, 1976; pp. 53–132. [Google Scholar]
- Luz, Z.; Silver, B.L.; Eden, C. Sign of the 17O hyperfine coupling constant in fremy’s salt. J. Chem. Phys. 1966, 44, 4421–4426. [Google Scholar] [CrossRef]
- Kirmse, R.; Stach, J.; Abram, U.; Marov, I.N. Zu struktur, bindung und ligandenaustauschverhalten von nitrosyltechnetium(II)-verbindungen-eine EPR-untersuchung. Z. Anorg. Allg. Chem. 1984, 518, 210–226. [Google Scholar] [CrossRef]
- Reinel, M.; Höcher, T.; Abram, U.; Kirmse, R. ein beitrag zu rhenium(ii)-, osmium(ii)- und technetium(ii)-thionitrosylkomplexen vom typ [M(NS)Cl4py]: Darstellung, strukturen und EPR-Spektren. Z. Anorg. Allg. Chem. 2003, 629, 853–861. [Google Scholar] [CrossRef]
- Schweiger, A.; Jeschke, G. Principles of Pulse Electron Paramagnetic Resonance; Oxford University Press: Oxford, UK, 2001. [Google Scholar]
- Mehring, M.; Weberruß, V.A. Object-Oriented Magnetic Resonance; Academic Press: New York, NY, USA, 2001. [Google Scholar]
- Eaton, G.R.; Eaton, S.S.; Barr, D.P.; Weber, R.T. Quantitative EPR; Springer Wien: New York, NY, USA, 2010. [Google Scholar]
- McWeeny, R. Spins in Chemistry; Dover Publications Inc: Mineola, NY, USA, 1998. [Google Scholar]
- McWeeny, R. Methods of Molecular Quantum Mechanics; Academic Press London: New York, NY, USA, 1992. [Google Scholar]
- Abragam, A.; Bleaney, B. Electron. Paramagnetic Resonance of Transition Ions; Clarendon Press: Oxford, UK, 1970. [Google Scholar]
- Pake, G.E.; Townsend, J.; Weissmann, S.I. Hyperfine structure in the paramagnetic resonance of the ion (SO3)2NO−. Phys. Rev. 1952, 85, 682–683. [Google Scholar] [CrossRef]
- Waller, I. Über die magnetisierung von paramagnetischen kristallen in wechselfeldern. Z. Phys. 1932, 79, 370–388. [Google Scholar] [CrossRef]
- Abragam, A.; Pryce, M.H.L. The theory of the nuclear hyperfine structure of paramagnetic resonance spectra in the copper tutton salts. Proc. R. Soc. Lond. A 1951, 206, 164–172. [Google Scholar] [CrossRef]
- Bagguley, D.M.S.; Bleaney, B.; Griffiths, J.H.E.; Penrose, R.P.; Plumpton, B.I. Paramagnetic resonance in salts of the iron group—A preliminary survey: I. Theoretical discussion. Proc. Phys. Soc. 1948, 61, 542–550. [Google Scholar] [CrossRef]
- Bagguley, D.M.S.; Bleaney, B.; Griffiths, J.H.E.; Penrose, R.P.; Plumpton, B.I. Paramagnetic resonance in salts of the iron group—A preliminary survey: II. Experimental results. Proc. Phys. Soc. 1948, 61, 551–561. [Google Scholar] [CrossRef]
- Bersuker, I.B. The Jahn—Teller. Effect; Cambridge University Press: Cambridge, UK, 2006. [Google Scholar]
- Stößer, R.; Scholz, G.; Möckel, K.; Backhaus, E. Structural and dynamic aspects of the paramagnetic Cu(II)methylpyrazole complexes. J. Mol. Struct. 1994, 319, 203–210. [Google Scholar] [CrossRef]
- Stößer, R. Diradikale und Triplettzustände. Z. Chem. 1983, 23, 7–17. [Google Scholar]
- Uhlenbeck, G.E.; Goudsmit, S. Ersetzung der Hypothese vom unmechanischen zwang durch eine forderung bezüglich des inneren Verhaltens jedes einzelnen Elektrons. Naturwiss 1925, 13, 953–954. [Google Scholar] [CrossRef]
- Altschuler, S.A.A.; Kosyrew, B.M. Paramagnetische Elektronenresonanz; Teubner, B.G., Ed.; Verlagsgesellschaft: Leipzig, Germany, 1963. [Google Scholar]
- Salikhov, K.M.; Molin, Yu.N.; Sagdaev, R.Z.; Buchachencko, A.L. Spin Polarization and Magnetic Effects in Radical Reactions; Akademiai Kiado: Budapest, Hungary, 1984. [Google Scholar]
- Jahn, H.A.; Teller, E. Stability of polyatomic molecules in degenerate electronic states. I. Orbital Degeneracy. Proc. Royal Soc. Lond. A 1937, 161, 220–235. [Google Scholar]
- Zamaraev, K.I.; Chairutdinov, R.F.; Schdanov, W.P. Tunnelirowanie elektrona w chimii (russ.); Isdatelstwo Nauka: Nowosibirsk, Russia, 1985. [Google Scholar]
- Bell, R.P. The Tunnel Effect in Chemistry; Chapman and Hall: London, UK, 1980. [Google Scholar]
- Sagdeev, R.Z.; Molin, Yu. N.; Salikhov, K.M. Effect of magnetic field on radical reactions in solution. Soviet Sci. Rev. B 1979, 1, 1–67. [Google Scholar]
- Molin, Yu.N.; Salikhov, K.M.; Zamaraev, K.I. Spin Exchange. Principles and Applications; Springer Berlin: Berlin, Germany, 1980. [Google Scholar]
- Stößer, R.; Herrmann, W.; Feist, M. Austauschwechselwirkung Von der magnetischen Ordnung in Festkörpern zum Spinaustausch in Lösungen und Biosystemen. Humboldt.-Spektrum. 2011, 104–108. [Google Scholar]
- Herrmann, W.; Stößer, R.; Borchert, H.-H. ESR imaging investigations of two-phase systems. Magn. Reson. Chem. 2007, 45, 496–507. [Google Scholar] [CrossRef]
- Bleaney, B. Microwave spectroscopy in oxford: The first decade part I: Microwave gas spectroscopy. Contemp. Phys. 1984, 25, 315–329. [Google Scholar] [CrossRef]
- Abragam, A. The Principles of Nuclear Magnetic Resonance; Clarendon Press: Oxford, UK, 1961. [Google Scholar]
- Trigg, G.L. Landmark Experiments in Twentieth Century Physics; Dover Publications: Mineola, NY, USA, 1995. [Google Scholar]
- Goudsmit, S.; Back, E. Feinstrukturen und Termordnung des Wismutspektrums. Z. Phys. 1927, 43, 321–334. [Google Scholar] [CrossRef]
- Breit, G.; Doermann, F.W. The Magnetic Moment of the Li7 Nucleus. Phys. Rev. 1930, 36, 1262–1264. [Google Scholar] [CrossRef]
- Jackson, D.A. Hyperfine structure in the arc spectrum of caesium and nuclear rotation. Proc. Royal Soc. Lond. A 1928, 121, 432–447. [Google Scholar] [CrossRef]
- Jackson, D.A. The Magnetic moment of the nucleus of caesium. Proc. Royal Soc. Lond. A 1934, 147, 500–513. [Google Scholar] [CrossRef]
- Gasiorowicz, S. Quantenphysik; Oldenbourg Verlag: München, Germany, 2002. [Google Scholar]
- Schpolski, E.W. Atomphysik Teil I; Deutscher Verlag der Wissenschaften: Berlin, Germany, 1965. [Google Scholar]
- Schpolski, E.W. Atomphysik Teil II; Deutscher Verlag der Wissenschaften: Berlin, Germany, 1965. [Google Scholar]
- Urey, H.C.; Brickwedde, F.G.; Murphy, G.M. A Hydrogen Isotope of Mass 2. Phys. Rev. 1932, 39, 164–165. [Google Scholar] [CrossRef]
- Rabi, I.I.; Zacharias, J.R.; Millman, S.; Kusch, P. A new method of measuring nuclear magnetic moment. Phys. Rev. 1938, 53, 318. [Google Scholar] [CrossRef]
- Kopfermann, H. Kernmomente; Akademische Verlagsgesellschaft: Frankfurt, Germany, 1956. [Google Scholar]
- Pryce, M.H.L. Theory. Nature 1948, 162, 539–540. [Google Scholar] [CrossRef]
- Griffiths, D.J. Hyperfine splitting in the ground state of hydrogen. Am. J. Phys. 1982, 50, 698–703. [Google Scholar] [CrossRef]
- Bleaney, B. Hyperfine interactions-un peu d'histoire. Hyperfine Interact. 1990, 63, 3–12. [Google Scholar] [CrossRef]
- Bleaney, B. Hyperfine Structure in Paramagnetic Resonance. Physica 1951, 17, 175–190. [Google Scholar] [CrossRef]
- Owen, J.; Stevens, K.W.H. Paramagnetic resonance and covalent bonding. Nature 1953, 171, 836. [Google Scholar] [CrossRef]
- Bleaney, B.; Stevens, K.W.H. Paramagnetic resonance. Rep. Progr. Phys. 1953, 16, 108–159. [Google Scholar]
- Elliott, R.J.; Stevens, K.W.H. The Theory of magnetic resonance experiments on salts of the rare earths. Proc. R. Soc. London, Ser. A 1953, 218, 553–566. [Google Scholar] [CrossRef]
- Gorter, C.J. A new suggestion for aligning certain atomic nuclei. Physica 1948, 14, 504. [Google Scholar] [CrossRef]
- Penrose, R.P. Hyperfine structure in the solid state. Nature 1949, 163, 992. [Google Scholar] [CrossRef]
- Bleaney, B.; Penrose, R.P. Hyperfine structure of the paramagnetic resonance spectrum of divalent cobalt: Experimental. Nature 1949, 163, 985. [Google Scholar]
- Bleaney, B.; Bowers, K.D.; Pryce, M.H.L. Paramagnetic resonance in diluted copper salts. III. Theory, and evaluation of the nuclear electric quadrupole moments of 63Cu and 65Cu. Proc. R. Soc. Lond. A 1955, 228, 166–174. [Google Scholar] [CrossRef]
- Calculation of NMR and EPR Parameters; Kaupp, M.; Bühl, M.; Malkin, V.G. (Eds.) Wiley VCH Verlag: Weinheim, Germany, 2004.
- Bleaney, B.; Bowers, K.D. Nuclear Spin of 2453Chromium. Proc. Phys. Soc. A 1951, 64, 1135–1136. [Google Scholar] [CrossRef]
- Bleaney, B.; Scovil, H.E.D. Nuclear spins of neodymium 143 and 145. Proc. Phys. Soc. A 1950, 63, 1369. [Google Scholar] [CrossRef]
- Baker, J.M.; Bleaney, B. The nuclear spin and magnetic moment of terbium 159. Proc. Phys. Soc. A 1955, 68, 257. [Google Scholar] [CrossRef]
- Baker, J.M.; Bleaney, B. The nuclear magnetic moment of holmium 165. Proc. Phys. Soc. A 1955, 68, 1090–1091. [Google Scholar] [CrossRef]
- Baker, J.M.; Bleaney, B.; Llewellyn, P.M.; Shaw, P.F.D. Nuclear spins and magnetic moments of cobalt 56 and 57. Proc. Phys. Soc. A 1956, 69, 353–354. [Google Scholar] [CrossRef]
- Bleaney, B.; Hutchison, C.A.; Llewellyn, P.M.; Pope, D.F.D. Paramagnetic resonance absorption in 235UCl3, and the nuclear electric quadrupole moment of 235U. Proc. Phys. Soc. B 1956, 69, 1167–1169. [Google Scholar] [CrossRef]
- Bleaney, B.; Daniels, J.M.; Grace, M.A.; Halban, H.; Kurti, N.; Robinson, F.N.H.; Simon, F.E. Experiments on nuclear orientation at very low temperatures. I. Establishment of a method of nuclear alinement and its application to cobalt-60. Proc. R. Soc. London, Ser. A 1954, 221, 170–188. [Google Scholar] [CrossRef]
- Ursu, I. Rezonanta. Magnetica. Compusi. cu uraniu; Editura Academici Republicii Socialiste Romania: Bucharest, Romania, 1979. [Google Scholar]
- Bleaney, B.; Llewellyn, P.M.; Jones, D.A. Paramagnetic resonance of uranium Ions. Proc. Phys. Soc. B 1956, 69, 858–860. [Google Scholar] [CrossRef]
- Stevens, K.W.H. On the magnetic properties of covalent XY6 complexes. Proc. R. Soc. London, Ser. A 1953, 219, 542–554. [Google Scholar] [CrossRef]
- Griffiths, J.H.E.; Owen, J. Complex hyperfine structures in microwave spectra of covalent iridium compounds. Proc. R. Soc. London, Ser. A 1954, 226, 96–111. [Google Scholar] [CrossRef]
- Weinhold, F.; Landis, C.R. Discovering Chemistry with Natural Bond. Orbitals; Wiley: Hoboken, NJ, USA, 2012. [Google Scholar]
- Ranby, B.; Rabek, J.F. ESR Spectroscopy in Polymer Research; Springer: Berlin-Heidelberg, Germany, 1977. [Google Scholar]
- Rao, G.T. Simple Derivation of the electron-nucleus contact hyperfine interaction. Am. J. Phys. 1962, 30, 716–718. [Google Scholar]
- Ketterle, W. Hyperfine interaction. In Lecture Notes for Physics 8.421; M.I.T, Department of Physics: Cambridge, CA, USA, 2006. [Google Scholar]
- Bleaney, B. Hyperfine structure in paramagnetic resonance. Physica. 1951, 17, 175–190. [Google Scholar] [CrossRef]
- Sobel'man, I.I.; Vainshtein, L.A.; Yukov, E.A. Excitation of Atoms. and Broadening. of Spectral Lines; Springer: Berlin-Heidelberg, Germany, 1995. [Google Scholar]
- Guinea, F.; Calderon, M.J.; Brey, L. Spin Dependent Tunneling. Lect. Notes Phys. 2001, 569, 159–171. [Google Scholar] [CrossRef]
- Morton, J.R.; Preston, K.F. Isotope shifts in EPR spectra. J. Chem. Phys. 1981, 75, 1126–1128. [Google Scholar] [CrossRef]
- Stösser, R.; Scholz, G.; Buzaré, J.-Y.; Silly, G.; Nofz, M.; Schultze, D. A magnetic resonance investigation of the process of corundum formation starting from solgel precursors. J. Am. Ceram. Soc. 2005, 88, 2913–2922. [Google Scholar]
- Mattuck, R.D.; Strandberg, M.W.P. Spin-phonon interaction in paramagnetic crystals. Phys. Rev. 1960, 119, 1204–1217. [Google Scholar] [CrossRef]
- Shrivastava, K.N. Isotope effect in electron paramagnetic resonance. Phys. Lett. A 1973, 42, 385–386. [Google Scholar] [CrossRef]
- Guseinov, D. V.; Ezhevskii, A. A. Spin relaxation of electrons localized on shallow and deep donor centers in silicon with different isotopic composition. Magn. Reson. Sol. 2006, 8, 48–52. [Google Scholar]
- Dilger, H.; Roduner, E.; Scheuermann, R.; Major, J.; Schefzik, M.; Stößer, R.; Päch, M.; Fleming, D.G. Mass and temperature effects on the hyperfine coupling of atomic hydrogen isotopes in cages. Physica B 2000, 289 & 290, 482–486. [Google Scholar]
- Eckert, H.; Roduner, E.; Limbach, H.-H. Kern- und Elektronenspins als strukturelle, Dynamische und kinetische Sonden. Bunsen.-Magazin. 2002, 4, 22–29. [Google Scholar]
- Trakhtenberg, L.I.; Klochikhin, V.L.; Pshezhetsky, S.Y. Theory of tunnel transitions of atoms in solids. Chem. Phys. 1992, 69, 121–134. [Google Scholar]
- Le Roy, R.J.; Murai, H.; Williams, F. Tunneling model for hydrogen abstraction reactions in low-temperature solids. Application to reactions in alcohol glasses and acetonitrile crystals. J. Am. Chem. Soc. 1980, 102, 2325–2334. [Google Scholar] [CrossRef]
- Wang, J.-T.; Williams, F. Hydrogen atom abstraction by methyl radicals in gamma-irradiated crystalline methyl isocyanide at 77–125 K. J. Am. Chem. Soc. 1972, 94, 2930–2934. [Google Scholar] [CrossRef]
- Sprague, E.D. Deuterium isotope effect on hydrogen atom abstraction by methyl radicals in acetonitrile at 77 K. Evidence for tunnelling. J. Phys. Chem. 1977, 81, 516–520. [Google Scholar] [CrossRef]
- Toriyama, K.; Iwasaki, M. Tunneling effect on hydrogen abstraction by hydrogen atoms and their trapping in some hydrocarbons irradiated at low temperatures. J. Phys. Chem. 1978, 82, 2056–2061. [Google Scholar] [CrossRef]
- Tachiya, M.; Mozumder, A. Decay of trapped electrons by tunnelling to scavenger molecules in low-temperature glasses. Chem. Phys. Lett. 1974, 28, 87–89. [Google Scholar] [CrossRef]
- Landau, L. D.; Lifshitz, E. M. Course of Theoretical Physics (Volume 3) — Quantum Mechanics. Non-relativistic Theory; Pergamon Press: Oxford, UK, 1991. [Google Scholar]
- Stryukov, V.B.; Stunzhas, P.A.; Kirillov, S.T. 14N and 15N isotopes in the study of rapid rotational diffusion of nitroxide radicals. Chem. Phys. Lett. 1975, 25, 453–456. [Google Scholar]
- Stößer, R.; Herrmann, W.; Pahlke, A. Spin Exchange of Nitroxyl Radicals in H2O and D2O. J. Phys. Chem. A 2012, 116, 952–961. [Google Scholar] [CrossRef]
- Levitt, M.H. Spin Dynamics: Basics of Nuclear Magnetic Resonance; John Wiley & Sons: Hoboken, NJ, USA, 2002. [Google Scholar]
- Bloch, F. Nuclear induction. Phys. Rev. 1946, 70, 460–474. [Google Scholar] [CrossRef]
- Böttger, H. Aufspaltung der ESR-linie durch spin-phonon-wechselwirkung an ionen mit spin 1. Phys. Stat. Sol. B 1967, 24, 65–75. [Google Scholar] [CrossRef]
- Jansson, M. Quantum chemical calculations on ESR, Core excitations and isotope effects in molecular systems. PhD Theses, Uppsala University, Uppsala, 2004. [Google Scholar]
- Itagaki, Y.; Wang, P.; Isamoto, N.; Shiotani, M.; Hasegawa, A.; Jansson, M.; Lunell, S. Static and dynamic structures of halogenated dimethyl ether radical cations: An EPR and MO study. Phys. Chem. Chem. Phys. 2002, 4, 2524–2529. [Google Scholar] [CrossRef]
- Shiotami, M.; Isamoto, N.; Hayashi, M.; Fängström, T.; Lunell, S. Deuterium isotope effects on rotation of methyl hydrogens. A study of the dimethyl ether radical cation by ESR spectroscopy and ab initio and density functional theory. J. Am. Chem. Soc. 2000, 122, 12281–12288. [Google Scholar] [CrossRef]
- Poole, C.P. Electron Spin Resonance: A Comprehensive Treatise on Experimental Techniques; Courier Dover Publications: Chelmsford, MA, USA, 1983. [Google Scholar]
- Hochkirch, U.; Herrmann, W.; Stößer, R.; Moll, K.-P.; Gallego Llerena, J.; Linscheid, M.; Borchert, H.-H. Determination of spin concentrations in ESR tomography as applied for the spatial distribution of spin labels in human skin. Appl. Magn. Reson. 2008, 35, 173–184. [Google Scholar]
- Yordanov, N.D.; Mladenova, B.; Petkov, P. Studies on the uncertainties in quantitative EPR estimations due to the construction of the cavities used. Anal. Chim. Acta 2002, 453, 155–162. [Google Scholar] [CrossRef]
- Mazur, M. A dozen useful tips on how to minimise the influence of sources of error in quantitative electron paramagnetic resonance (EPR) spectroscopy—A review. Anal. Chim. Acta 2006, 561, 1–15. [Google Scholar] [CrossRef]
- Kurylo, M.J.; Hollinden, G.A.; Timmons, R.B. ESR Study of the kinetic isotope effect in the reaction of H and D atoms with CH4. J. Chem. Phys. 1970, 52, 1773–1782. [Google Scholar] [CrossRef]
- Jungmann, K.-P. Spectroscopy of the muonium atom. Lect. Notes Phys. 2001, 570, 81–102. [Google Scholar] [CrossRef]
- Roduner, E. Muonium - An Ultra-Light Isotope of Hydrogen. In Isotope Effects in Chemistry and Biology; Kohen, A., Limbach, H.-H., Eds.; Taylor & Francis: Boca Raton, FL, USA, 2006; pp. 433–450. [Google Scholar]
- Walther, F.G.; Phillips, W.D.; Kleppner, D. Effect of nuclear mass on the bound-electron g factor. Phys. Rev. Lett. 1972, 28, 1159–1161. [Google Scholar] [CrossRef]
- Tiedeman, J.S.; Robinson, H.G. Determination of gJ(1H, 12S1/2)/gs(e): Test of mass-independent corrections. Phys. Rev. Lett. 1977, 39, 602–604. [Google Scholar] [CrossRef]
- Kinoshita, T. Present Status of g-2 of electron and muon. Lect. Notes Phys. 2001, 570, 157–175. [Google Scholar] [CrossRef]
- Mohr, P.J.; Taylor, B.N. Fundamental constants and the hydrogen atom. Lect. Notes Phys. 2001, 570, 145–156. [Google Scholar] [CrossRef]
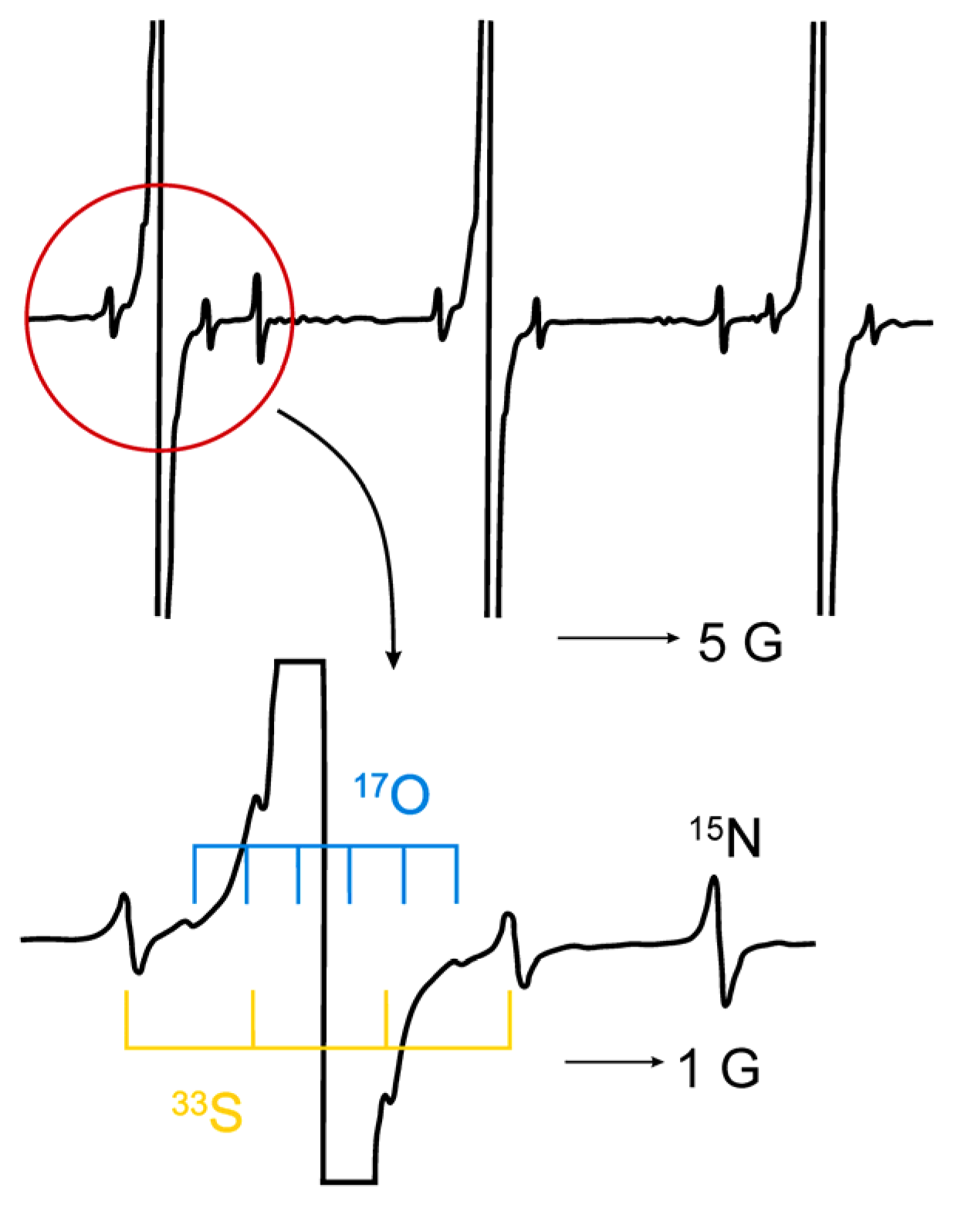
- Okazaki, M.; Toriyama, K. Ultra high-resolution ESR using a reverse micelle 33S, 17O, and 15N satellite lines of fremy’s salt at natural isotope abundance. J. Magn. Reson. 1988, 79, 158–162. [Google Scholar]
- Windle, J.J.; Wiersema, A.K. Electron paramagnetic resonance of naturally abundant N15 and S33 in peroxylamine disulfonate ion [NO(SO3)2]+. J. Chem. Phys. 1962, 39, 1139–1140. [Google Scholar] [CrossRef]
- Goldman, S.A.; Bruno, G.V.; Freed, J.H. ESR studies of anisotropic rotational reorientation and slow tumbling in liquid and frozen media. II. Saturation and nonsecular effects. J. Chem. Phys. 1973, 59, 3071–3091. [Google Scholar] [CrossRef]
- Tsujii, K.; Sunamoto, J.; Fendler, J.H. Microscopic viscosity of the interior water pool in dodecylammonium propionate reversed micelles. Bull. Chem. Soc. Jpn. 1983, 56, 2889–2893. [Google Scholar] [CrossRef]
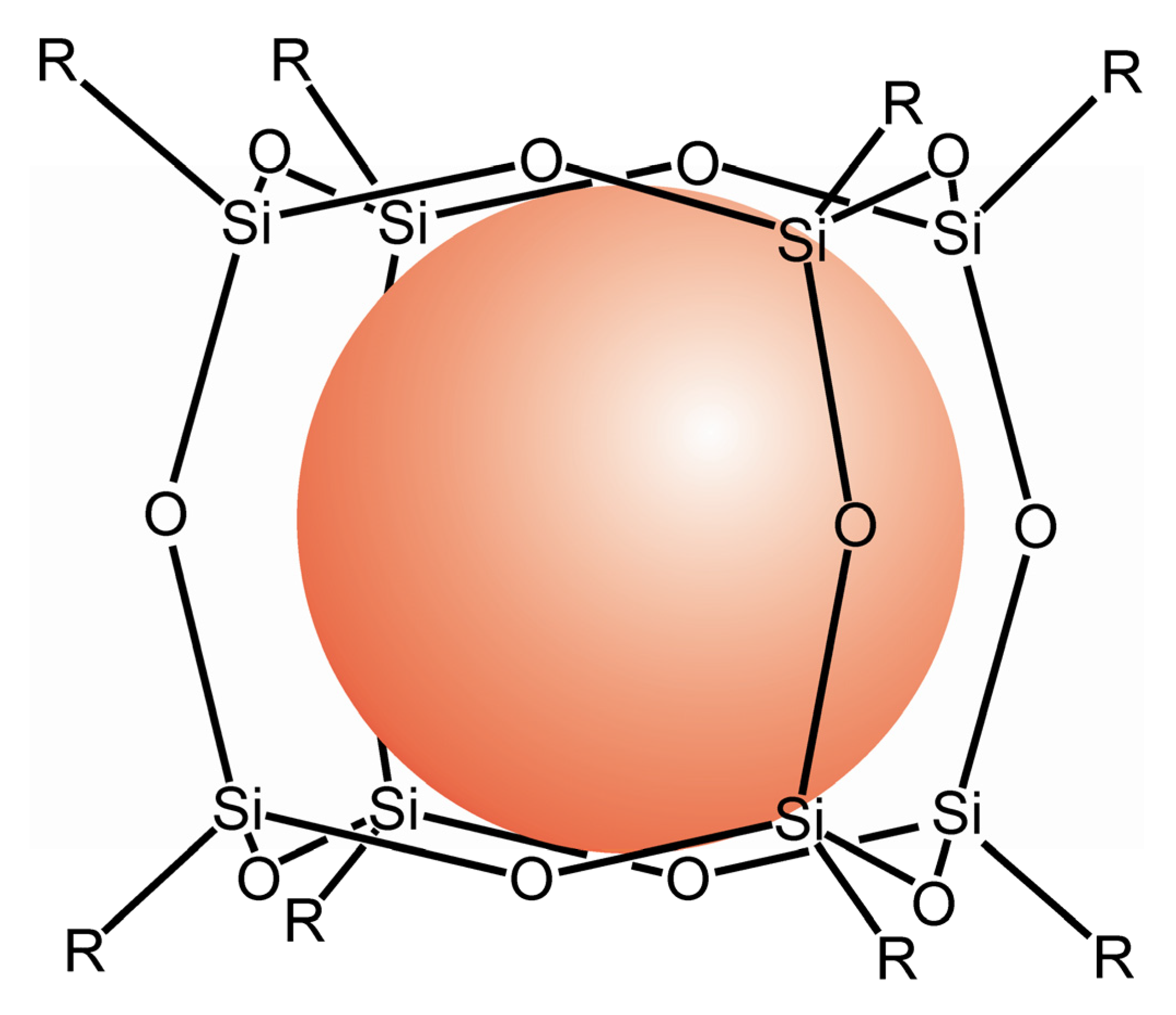
- Päch, M.; Macrae, R.M.; Carmichael, I. Hydrogen and deuterium atoms in octasilsesquioxanes: Experimental and computational studies. J. Am. Chem. Soc. 2006, 128, 6111–6125. [Google Scholar] [CrossRef]
- Weiden, N.; Päch, M.; Dinse, K.P. Pulsed EPR and ENDOR investigation of hydrogen atoms in silsesquioxane cages. Appl. Magn. Reson. 2001, 21, 507–516. [Google Scholar] [CrossRef]
- Päch, M.; Stößer, R. Octasilsesquioxanes as Traps for Atomic Hydrogen at Room Temperature. In Organosilicon Chemistry IV: From Molecules to Materials; Auner, N., Weis, J., Eds.; Wiley: Hoboken, NJ, USA, 2000; pp. 521–525. [Google Scholar]
- Sasamori, R.; Okaue, Y.; Isobe, T.; Matsuda, Y. Stabilization of atomic hydrogen in both solution and crystal at room temperature. Science 1994, 265, 1691–1693. [Google Scholar]
- Päch, M.; Stösser, R. Scavenger assisted trapping of atomic hydrogen in Si8O12-Cages. J. Phys. Chem. A 1997, 101, 8360–8365. [Google Scholar] [CrossRef]
- Roduner, E.; Louwrier, P.W.F.; Brinkman, G.A.; Garner, D.M.; Reid, I.D.; Arseneau, D.J.; Senba, M.; Fleming, D.G. Quantum phenomena and solvent effects on addition of hydrogen isotopes to benzene and to dimethylbutadiene. Ber. Bunsenges. Phys. Chem. 1990, 94, 1224–1230. [Google Scholar]
- Roduner, E.; Percival, P.W.; Han, P.; Bartels, D.M. Isotope and temperature effects on the hyperfine interaction of atomic hydrogen in liquid water and in ice. J. Chem. Phys. 1995, 102, 5989–5997. [Google Scholar] [CrossRef]
- Scholz, G.; Stösser, R. Atomic hydrogen as spin probe in thermally and mechanically activated materials. Phys. Chem. Chem. Phys. 2002, 4, 5448–5457. [Google Scholar] [CrossRef]
- Holuj, F.; Kwan, C.T. Isotope effects in the ESR of Mn2+ in Ca(OH)2 and Ca(OD)2. Phys. Rev. B 1974, 9, 3673–3677. [Google Scholar] [CrossRef]
- Marshall, S.A.; Hodges, J.A.; Serway, R.A. Isotopic shift in the electron-spin-resonance absorption spectrum of Cr3+ in magnesium oxide. Phys. Rev. 1964, 136, A1024–A1029. [Google Scholar] [CrossRef]
- Höhne, M.; Stasiw, M.; Watterich, A. ESR of Ni2+ in AgCl and isotope effect in crystal field. Phys. Stat. Sol. B 1969, 34, 319–327. [Google Scholar] [CrossRef]
- Höhne, M.; Stasiw, M. ESR Investigation of Ni+ in illuminated AgCl and AgBr. Phys. Stat. Sol. B 1969, 33, 405–409. [Google Scholar] [CrossRef]
- Mchedlidze, T.; Suezawa, M. Features of isotopic shift in the fine structure term of ESR spectra from iron–vacancy pair in silicon. Physica B 2003, 340–342, 556–560. [Google Scholar] [CrossRef]
- Tomioka, H.; Okada, H.; Watanabe, T.; Banno, K; Komatsu, K.; Hirai, K. Polymethylated and poly(tert)butylated diphenylcarbenes. Generation, reactions, kinetics, and deuterium isotope effects of sterically congested triplet carbenes. J. Am. Chem. Soc. 1997, 119, 1582–1593. [Google Scholar] [CrossRef]
- Iwasaki, M.; Toriyama, K.; Nunome, K. Electron spin resonance study of electronic and geometrical structures of C2H6+ and other simple alkane cations at 4.2 K: Possible evidence for Jahn-Teller distortion. J. Am. Chem. Soc. 1981, 103, 3591–3592. [Google Scholar] [CrossRef]
- Symons, M.C.R. Radical cations in condensed phases. Chem. Soc. Rev. 1984, 13, 393–439. [Google Scholar] [CrossRef]
- Toriyama, K.; Okazaki, M.; Jansson, M.; Lund, A.; Lunell, S. Isotope effect on the J–T distortion of partially deuteriated benzene cation radicals: an experimental EPR and theoretical DFT study. Phys. Chem. Chem. Phys. 2004, 6, 1658–1665. [Google Scholar] [CrossRef]
- Lawler, R.G.; Bolton, J.R.; Fraenkel, G.K.; Brown, T.H. Orbital Degeneracy and the electron spin resonance spectrum of the benzene-1-d negative Ion. J. Am. Chem. Soc. 1964, 86, 520–521. [Google Scholar] [CrossRef]
- Wang, P.; Shiotani, M.; Lunell, S. Structure and dynamics of radical cations of selectively deuteriated cyclohexanes: an ESR and ab initio study. Chem. Phys. Lett. 1984, 292, 110–114. [Google Scholar] [CrossRef]
- Iwasaki, M.; Toriyama, K.; Nunome, K. ESR observation of isotopic preference in static distortions of Jahn-Teller active radical cations: Partially deuterated ethane radical cations in SF6 at 4 K. Chem. Phys. Lett. 1984, 111, 309–314. [Google Scholar] [CrossRef]
- Nicovich, J.M.; Ravishankara, A.R. Reaction of hydrogen atom with benzene. Kinetics and mechanism. J. Phys. Chem. 1984, 88, 2534–2541. [Google Scholar] [CrossRef]
- Zamaraev, K.I.; Khairutdinov, R.F. The role of long range tunneling in electron transfer processes in condensed media. Chem. Phys. 1974, 4, 181–195. [Google Scholar] [CrossRef]
- Williams, F.; Sprague, E.D. Evidence for hydrogen atom abstraction by methyl radicals in the solid state at 77°K. J. Am. Chem. Soc. 1971, 93, 787–788. [Google Scholar] [CrossRef]
- Le Roy, R.J.; Sprague, E.D.; Williams, F. Quantum mechanical tunneling in hydrogen atom abstraction for solid acetonitrile at 77–87°K. J. Phys. Chem. 1972, 76, 546–551. [Google Scholar] [CrossRef]
- Sprague, E.D.; Schulte-Frohlinde, D. Electron Spin Resonance Investigation of the Disappearance of Trapped Hydrogen Atoms in γ-Irradiated Sulfuric Acid Glasses. J. Phys. Chem. 1973, 77, 1222–1225. [Google Scholar] [CrossRef]
- Campion, A.; Williams, F. Hydrogen atom abstraction by methyl radicals in methanol glasses at 67–77°K. J. Am. Chem. Soc. 1972, 94, 7633–7637. [Google Scholar] [CrossRef]
- Brunton, G.; Griller, D.; Barclay, L.R.C.; Ingold, K.U. Kinetic applications of electron paramagnetic resonance spectroscopy. 26. Quantum-mechanical tunneling in the isomerization of sterically hindered aryl radicals. J. Am. Chem. Soc. 1976, 98, 6803–6811. [Google Scholar] [CrossRef]
- Jakimtschenko, O.E.; Lebedev, Y.S. Radikalnye pary w oblutschenom dimetiloxim: Isotopnye I tunnelnnye effekty w prewroschtschenijach par. Int. J. Radiat. Phys. Chem. 1971, 3, 17–44. [Google Scholar] [CrossRef]
- Kurita, Y. Electron Spin Resonance Study of radical pairs trapped in irradiated single crystals of dimethylglyoxime at liquid-nitrogen temperature. J. Chem. Phys. 1964, 41, 3926–3927. [Google Scholar] [CrossRef]
- Pfister, G.; Känzig, W. Isotopeneffekt in der paraelastischen relaxation. Phys. Kondens. Mat. 1969, 10, 231–264. [Google Scholar]
- Spaeth, J.M. Influence of zero-point vibration on the superhyperfine interactions of hydrogen and deuterium centres in KCl. Phys. Stat. Sol. B 1969, 34, 171–181. [Google Scholar] [CrossRef]
- Badalyan, A.G.; Baranov, P.G. EPR of hydrogen and deuterium atoms in alkali halide crystals. Phys. Stat. Sol. B 1980, 97, 95–99. [Google Scholar] [CrossRef]
- Komaguchi, K.; Marutani, T.; Shiotani, M.; Hasegawa, A. Isotope effects in partially deuterated tetramethylsilane cations studied by EPR spectroscopy. Phys. Chem. Chem. Phys. 2001, 3, 3536–3540. [Google Scholar] [CrossRef]
- Gendell, J.; Miller, W.R., Jr.; Fraenkel, G.K. Electron spin resonance studies of hydroxysemiquinone radicals. Hydrogen-deuterium isotope effects in intramolecular hydrogen bonds. J. Am. Chem. Soc. 1969, 91, 4369–4380. [Google Scholar] [CrossRef]
- Alberti, A.; Guerra, M.; Pedulli, G.F. Effect of temperature and isotopic substitution on the NH proton hyperfine splitting in 1-hydropyridinyl radicals. J. Magn. Reson. 1979, 34, 233–236. [Google Scholar]
- Ewert, U.; Stößer, R.; Tomaschewski, G.; Freyer, W. Pyrazolonderivate IX: EPR-spektroskopische untersuchungen an kupfer(II)-komplexen mit liganden vom 4-Amino-methylen-Δ2-pyrazolin-(5)-Typ. Monatshefte. für Chemie. 1982, 113, 973–982. [Google Scholar] [CrossRef]
- Evans, J.C.; Morgan, P.H.; Renaud, R.H. Simulation of electron spin resonance spectra by fast fourier transform: A Novel method of calculating spectra to include isotopic substitution, superhyperfine coupling, instrument time constant and modulation broadening in fluid and crystalline media. Anal. Chim. Acta 1978, 103, 175–187. [Google Scholar] [CrossRef]
- Magin, R.L.; Morse, II, P.D. Rapid measurement of drug release from temperature-sensitive liposomes by electron paramagnetic resonance and radioisotope techniques. Biochim. Biophys. Acta 1983, 760, 357–362. [Google Scholar] [CrossRef]
- Pfirrmann, S.; Limberg, C.; Herwig, C.; Knispel, C.; Braun, B.; Bill, E.; Stösser, R. A Reduced β-diketiminato-ligated Ni3H4 unit catalyzing H/D exchange. J. Am. Chem. Soc. 2010, 132, 13684–13691. [Google Scholar] [CrossRef]
- Reichardt, C. Polarity of ionic liquids determined empirically by means of solvatochromic pyridiniumN-phenolate betaine dyes. Green Chem. 2005, 7, 339–351. [Google Scholar] [CrossRef]
- Jeschke, G.; Zimmermann, H.; Godt, A. Isotope selection in distance measurements between nitroxides. J. Magn. Reson. 2006, 180, 137–146. [Google Scholar] [CrossRef]
- Dikalov, S.; Jiang, J.; Mason, R.P. Characterization of the high-resolution ESR spectra of superoxide radical adducts of 5-(diethoxyphosphoryl)-5-methyl-1-pyrroline N-oxide (DEPMPO) and 5,5-dimethyl-1-pyrroline N-oxide (DMPO). Analysis of conformational exchange. Free Rad. Res. 2005, 39, 825–836. [Google Scholar] [CrossRef]
- Zhang, H.; Joseph, J.; Vasquez-Vivar, J.; Karoui, H.; Nsanzumuhire, C.; Martásek, P.; Tordo, P.; Kalyanaraman, B. Detection of superoxide anion using an isotopically labeled nitrone spin trap: potential biological applications. FEBS Lett. 2000, 473, 58–62. [Google Scholar] [CrossRef]
- Du, J.L.; Eaton, G.R.; Eaton, S.S. Electron-spin-lattice relaxation in natural abundance and isotopically enriched oxo-chromium(V)bis (2-hydroxy-2-ethylbutyrate). J. Magn. Reson. A 1995, 115, 236–240. [Google Scholar] [CrossRef]
- Biller, J.R.; Meyer, V.; Elajaili, H.; Rosen, G.M.; Kao, J.P.Y.; Eaton, S.S.; Eaton, G.R. Relaxation times and line widths of isotopically-substituted nitroxides in aqueous solution at X-band. J. Magn. Reson. 2011, 212, 370–377. [Google Scholar] [CrossRef]
- Stoesser, R.; Herrmann, W.; Zehl, A.; Laschewsky, A.; Strehmel, V. Microviscisity and micropolarity effects of imidazolium based ionic liquids investigated by spin probes their diffusion and spin exchange. Z. Phys. Chem. 2006, 220, 1309–1342. [Google Scholar] [CrossRef]
- Bartels, D.M.; Han, P.; Percival, P.W. Diffusion and CIDEP of H and D atoms in solid H2O, D2O and isotopic mixtures. Chem. Phys. 1992, 164, 421–437. [Google Scholar] [CrossRef]
- Stoesser, R.; Klein, J.; Peschke, S.; Zehl, A.; Cämmerer, B.; Kroh, L.W. On the time behaviour of the concentration of pyrazinium radical cations in the early stage of the Maillard reaction. Spectrochim. Acta A 2007, 67, 1161–1168. [Google Scholar] [CrossRef]
- Khan, N.; Blinco, J.P.; Bottle, S.E.; Hosokawa, K.; Swartz, H.M.; Micallef, A.S. The evaluation of new and isotopically labeled isoindoline nitroxides and an azaphenalene nitroxide for EPR oximetry. J. Magn. Reson. 2011, 211, 170–177. [Google Scholar] [CrossRef] [Green Version]
- Halpern, H.J.; Peril, M.; Nguyen, T.-D.; Spencer, D.P.; Teicher, B.A.; Lin, Y.J.; Bowman, M.K. Selective isotopic labeling of a nitroxide spin label to enhance sensitivity for T2 oxymetry. J. Magn. Reson. 1990, 90, 40–51. [Google Scholar]
- Pahlke, A. Physikalische und chemische eigenschaften von leichtem und schwerem wasser-untersucht am Beispiel des Spinaustausches von stabilen Radikalen. B.Sc. Theses, Humboldt University, Berlin, Germany, 2010. [Google Scholar]
- Yankwich, P.E. Isotope effects in chemical reactions. Ann. Rev. Nucl. Sci. 1953, 3, 235–248. [Google Scholar] [CrossRef]
- Wolfsberg, M. Isotope effects on reaction rates and the reaction coordinate. J. Chem. Phys. 1960, 33, 21–22. [Google Scholar] [CrossRef]
- Sartori, E.; Khudyakov, I.V.; Lei, X.; Turro, N.J. A Time-resolved electron paramagnetic resonance investigation of the spin exchange and chemical interactions of reactive free radicals with isotopically symmetric (14N−X−14N) and isotopically asymmetric (14N−X−15N) nitroxyl biradicals. J. Am. Chem. Soc. 2007, 129, 7785–7792. [Google Scholar] [CrossRef]
- Burkhard, G.; Loss, D. Quantum Computing with Solid State Systems. In Lectures on Quantum Information; Bruß, D., Leuchs, G., Eds.; Wiley: Hoboken, NJ, USA, 2007; pp. 451–480. [Google Scholar]
- Cerletti, V.; Coish, W.A.; Gywat, O.; Loss, D. Recipes for spin-based quantum computing. Nanotechnology 2005, 16, R27–R49. [Google Scholar] [CrossRef]
- Hanson, R.; Witkamp, B.; Vandersypen, L.M. K.; van Beveren, L.W. H.; Elzerman, J.M.; Kouwenhoven, L.P. Zeeman energy and spin relaxation in a one-electron quantum dot. Phys. Rev. Lett. 2003, 91, 196802–196805. [Google Scholar] [CrossRef]
- Engel, H.-A.; Loss, D. Detection of single spin decoherence in a quantum dot via charge currents. Phys. Rev. Lett. 2001, 86, 4648–4651. [Google Scholar] [CrossRef]
- Loss, D.; DiVincenzo, D.P. Quantum computation with quantum dots. Phys. Rev. A 1998, 57, 120–126. [Google Scholar] [CrossRef]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Stößer, R.; Herrmann, W. Isotope Effects in ESR Spectroscopy. Molecules 2013, 18, 6679-6722. https://doi.org/10.3390/molecules18066679
Stößer R, Herrmann W. Isotope Effects in ESR Spectroscopy. Molecules. 2013; 18(6):6679-6722. https://doi.org/10.3390/molecules18066679
Chicago/Turabian StyleStößer, Reinhard, and Werner Herrmann. 2013. "Isotope Effects in ESR Spectroscopy" Molecules 18, no. 6: 6679-6722. https://doi.org/10.3390/molecules18066679