Novel Pyrazolo[3,4-d]pyrimidine Derivatives as Potential Antitumor Agents: Exploratory Synthesis, Preliminary Structure-Activity Relationships, and in Vitro Biological Evaluation

Abstract
:1. Introduction


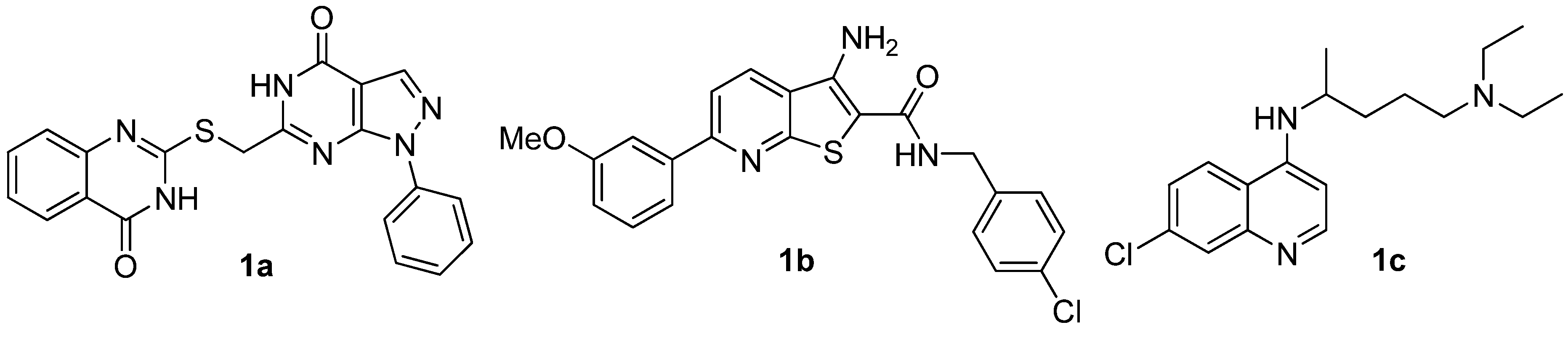{kind=link}
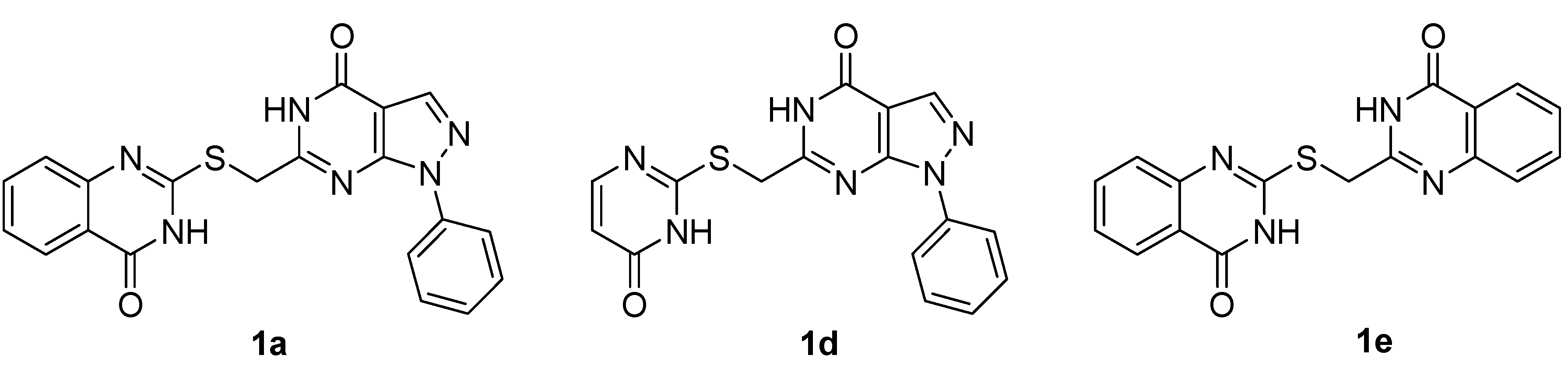{kind=link}
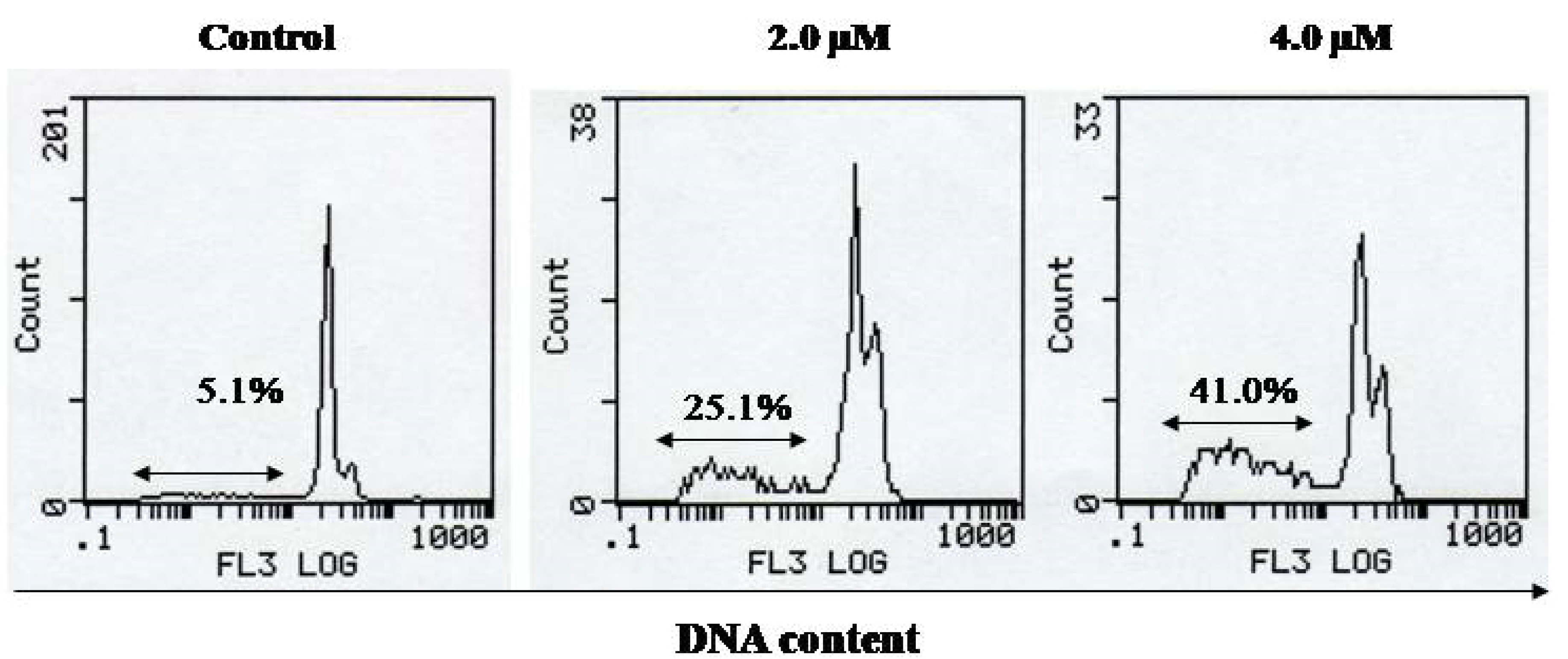{kind=link}
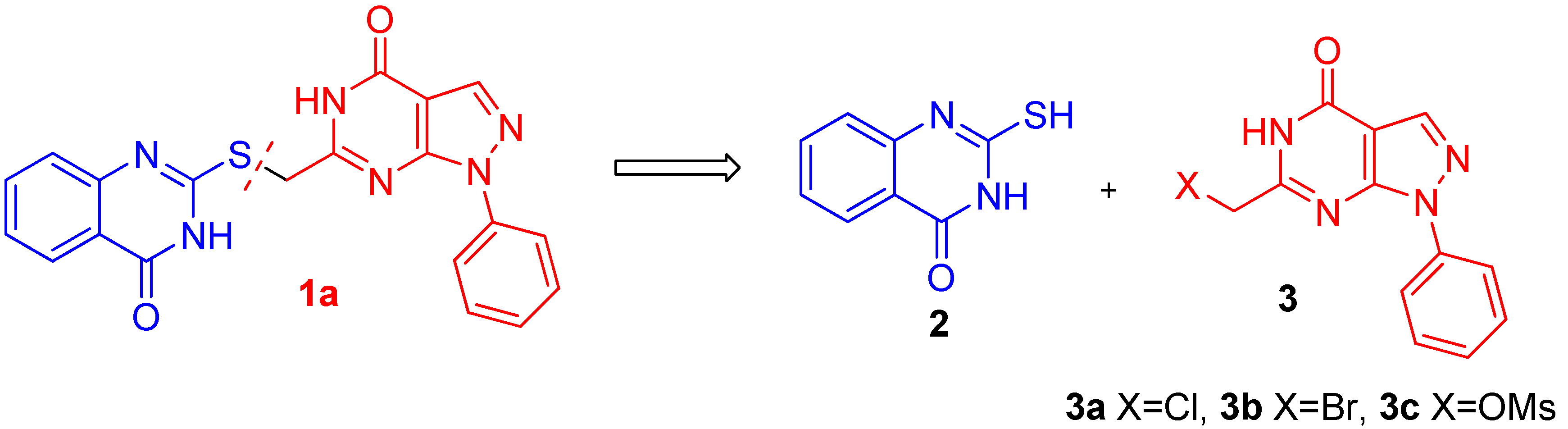{kind=link}
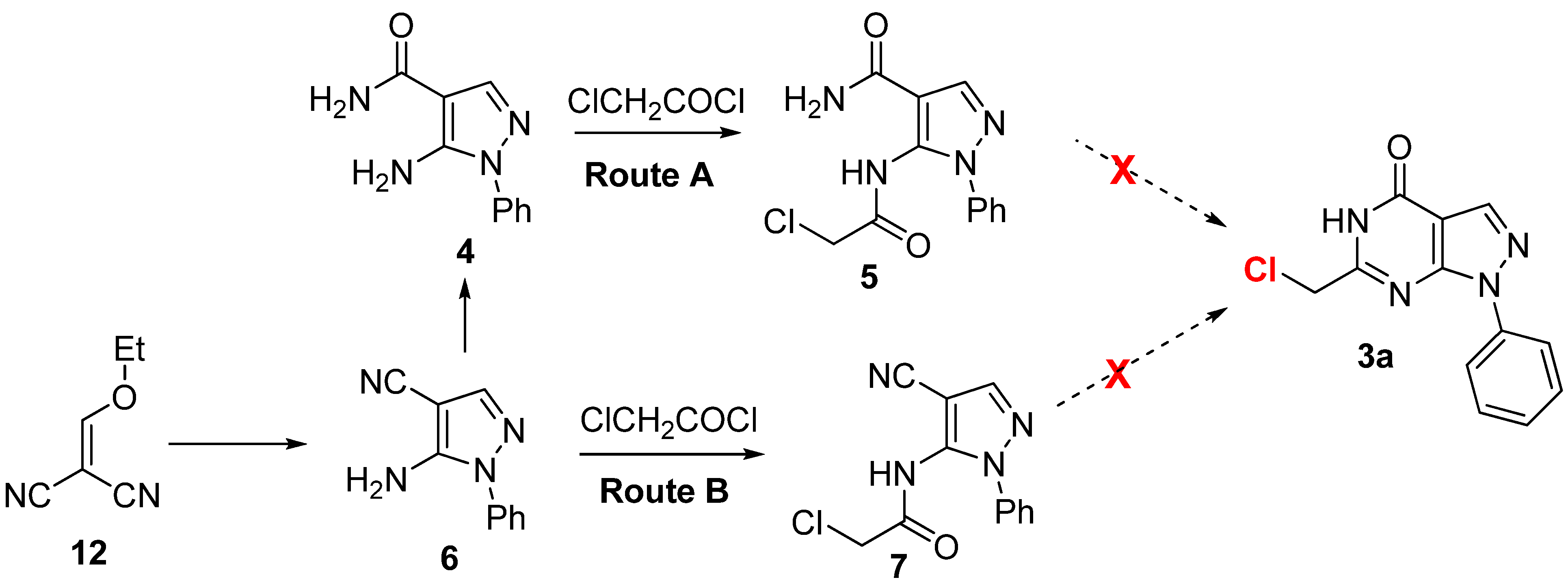{kind=link}
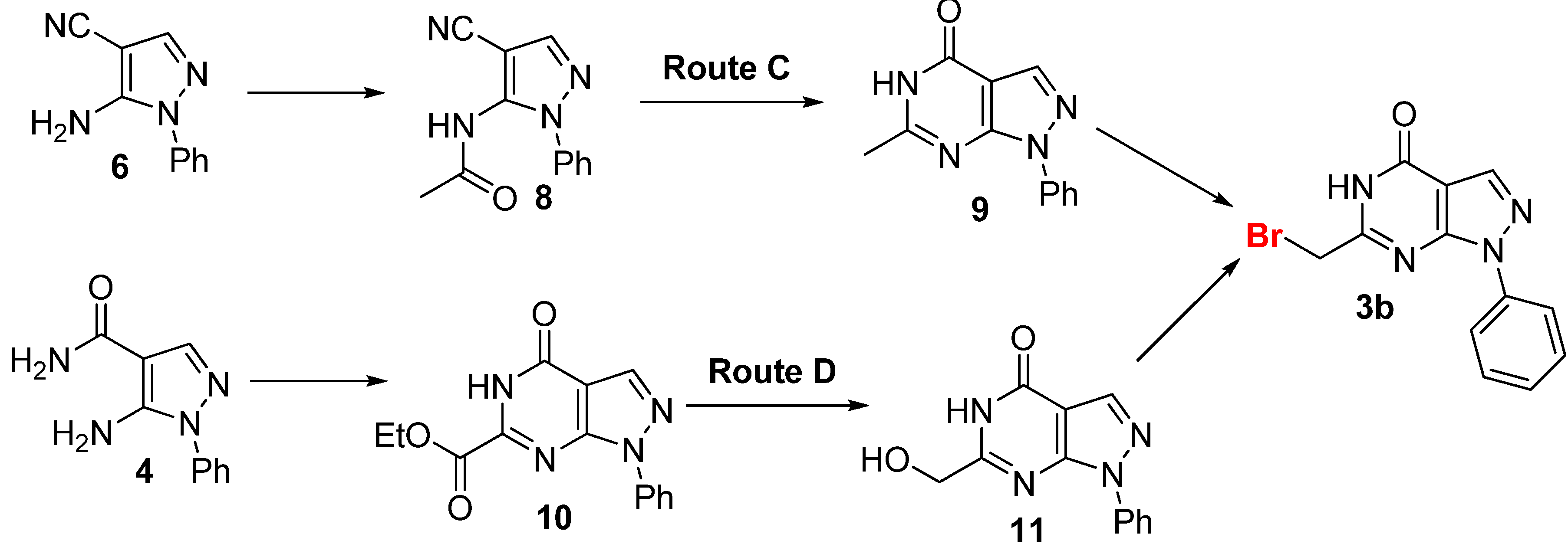{kind=link}
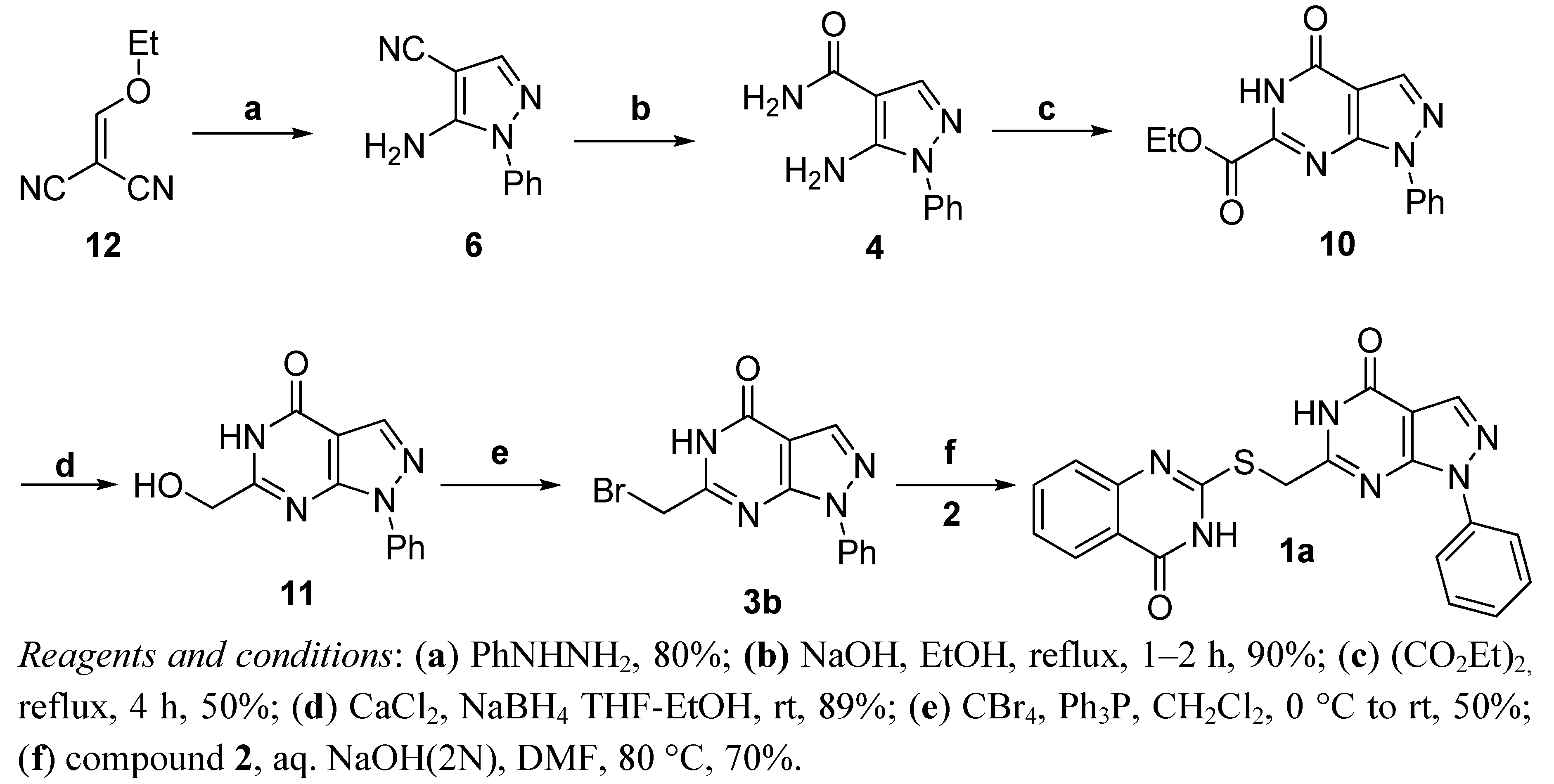{kind=link}
| Compound | IC50 (µM) a | |||
|---|---|---|---|---|
| HepG2 | MCF-7 | A549 | PC-3 | |
| 1a | 13.9 | 42.3 | 2.24 | 26.6 |
| 1d | 25.2 | 1.74 | 5.20 | >100 |
| 1e | >100 | >100 | 47.0 | >100 |
| Doxorubicin | 0.54 | 0.75 | 9.20 | 0.60 |
2. Results and Discussion
2.1. Chemistry




2.2. Anticancer Activity


3. Experimental
General
4. Conclusions
Acknowledgements
References and Notes
- Jemal, A.; Siegel, R.; Xu, J.; Ward, E. Cancer Statistics 2010. CA Cancer J. Clin. 2010, 60, 277–300. [Google Scholar] [CrossRef]
- Simonetti, R.G.; Liberati, A.; Angiolini, C.; Pagliaro, L. Treatment of hepatocellular carcinoma: A systematic review of randomized controlled trials. Ann. Oncol. 1997, 8, 117–136. [Google Scholar] [CrossRef]
- Zheng, Y.Z.; Zhao, Y.L.; Deng, X.Q.; Yang, S.Y.; Mao, Y.Q.; Li, Z.G.; Jiang, P.D.; Zhao, X.; Wei, Y.Q. Chloroquine inhibits colon cancer cell growth in vitro and tumor growth in vivo via induction of apoptosis. Cancer Invest. 2009, 27, 286–292. [Google Scholar] [CrossRef]
- Xie, H.Z.; Li, L.L.; Ren, J.X.; Zou, J.; Yang, L.; Wei, Y.Q.; Yang, S.Y. Pharmacophore modeling study based on known spleen tyrosine kinase inhibitors together with virtual screening for identifying novel inhibitors. Bioorg. Med. Chem. Lett. 2009, 19, 1944–1949. [Google Scholar] [CrossRef]
- Wang, H.Y.; Cao, Z.X.; Li, L.L.; Jiang, P.D.; Zhao, Y.L.; Luo, S.D.; Yang, L.; Wei, Y.Q.; Yang, S.Y. Pharmacophore modeling and virtual screening for designing potential PLK1 inhibitors. Bioorg. Med. Chem. Lett. 2008, 18, 4972–4977. [Google Scholar]
- Luo, Y.F.; Xu, Y.B.; Chen, L.J.; Hu, J.; Peng, C.; Xie, D.C.; Shi, J.Y.; Huang, W.C.; Xu, G.B.; Peng, M.; Han, J.; Li, R.; Yang, S.Y.; Wei, Y.Q. Semi-synthesis and anti-proliferative activity evaluation of novel analogues of Honokiol. Bioorg. Med. Chem. Lett. 2009, 19, 4702–4705. [Google Scholar]
- Wei, Y.Q.; Wang, Q.R.; Zhao, X.; Yang, L.; Tian, L.; Lu, Y.; Kang, B.; Lu, C.J.; Huang, M.J.; Lou, Y.Y.; et al. Immunotherapy of tumors with xenogeneic endothelial cells as a vaccine. Nat. Med. 2000, 6, 1160–1166. [Google Scholar] [CrossRef]
- Yang, L.; Zhao, Y.L.; Yang, S.Y.; Yu, L.T.; Wei, Y.Q. CN Patent 200810304724.X, 28 September 2008.
- Deng, X.Q.; Wang, H.Y.; Zhao, Y.L.; Xiang, M.L.; Jiang, P.D.; Cao, Z.X.; Zheng, Y.Z.; Luo, S.D.; Yu, L.T.; Wei, Y.Q.; et al. Pharmacophore modelling and virtual screening for identification of new Aurora-A kinase inhibitors. Chem. Biol. Drug Des. 2008, 71, 533–539. [Google Scholar] [CrossRef]
- Jiang, P.D.; Zhao, Y.L.; Shi, W.; Deng, X.Q.; Xie, G.; Mao, Y.Q.; Li, Z.G.; Zheng, Y.Z.; Yang, S.Y.; Wei, Y.Q. Cell growth inhibition, G2/M cell cycle arrest, and apoptosis induced by chloroquine in human breast cancer cell line Bcap-37. Cell. Physiol. Biochem. 2008, 22, 431–440. [Google Scholar] [CrossRef]
- Toque, H.A.F.; Priviero, F.B.M.; Texeira, C.E.; Perissutti, E.; Fiorino, F.; Severino, B.; Frecentese, F.; Lorenzetti, R.; Baracat, J.S.; Santagada, V.; et al. Synthesis and Pharmacological Evaluations of Sildenafil Analogues for Treatment of Erectile Dysfunction. J. Med. Chem 2008, 51, 2807–2815. [Google Scholar]
- Gillespie, R.J.; Cliffe, I.A.; Dawson, C.E.; Dourish, C.T.; Gaur, S.; Jordan, A.M.; Knight, A.R.; Lerpiniere, J.; Misra, A.; Pratt, R.M.; et al. Antagonists of the human adenosine A2A receptor. Part 3: Design and synthesis of pyrazolo[3,4-d]pyrimidines, pyrrolo[2,3-d]pyrimidines and 6-arylpurines. Bioorg. Med. Chem. Lett. 2008, 18, 2924–2929. [Google Scholar]
- Amjad, A.; Gayle, E.T.; Ken, E.; Georgianna, H.; Ronald, P.; Lynn, L.S.; Katherine, Y. Novel Pyrazolo[3,4-d]pyrimidine-Based Inhibitors of Staphlococcus aureus DNA Polymerase III: Design, Synthesis, and Biological Evaluation. J. Med. Chem. 2003, 46, 1824–1830. [Google Scholar] [CrossRef]
- Wang, H.M.; Eiji, S.; Tang, Y.P.; Min, C.; Maureen, K.; Zuo, W.Q.; Daphne, A.R.; Peter, J.A.; Zhang, C.; Hiromi, M.; et al. Inducible protein knockout reveals temporal requirement of CaMKII reactivation for memory consolidation in the brain. Proc. Natl. Acad. Sci. USA 2003, 100, 4287–4292. [Google Scholar]
- Kim, D.C.; Lee, Y.R.; Yang, B.S.; Shin, K.J.; Kim, D.J.; Chung, B.Y.; Yoo, K.H. Synthesis and biological evaluations of pyrazolo[3,4-d]pyrimidines as cyclin-dependent kinase 2 inhibitors. Eur. J. Med. Chem. 2003, 38, 525–532. [Google Scholar] [CrossRef]
- Carraro, F.; Naldini, A.; Pucci, A. Pyrazolo[3,4-d]pyrimidines as Potent Antiproliferative and Proapoptotic Agents toward A431 and 8701-BC Cells in Culture via Inhibition of c-Src Phosphorylation. J. Med. Chem. 2006, 49, 1549–1561. [Google Scholar] [CrossRef]
- Schenone, S.; Bruno, O.; Bondavalli, F.; Ranise, A.; Mosti, L.; Menozzi, G.; Fossa, P.; Donnini, S.; Santoro, A.; et al. Antiproliferative activity of new 1-aryl-4-amino-1H-pyrazolo[3,4-d]pyrimidine derivatives toward the human epidermoid carcinoma A431 cell line. Eur. J. Med. Chem. 2004, 39, 939–946. [Google Scholar] [CrossRef]
- The sample of 1a for screening was purchased from Specs Company. Although 1a is a known compound and commercially available on the milligram scale, there is no report concerning its synthesis and activities except our manuscript. Because there is no synthetic literature of it, we had to synthesize compound 1a and its analogs for further research.
- Bol'but, A.V.; Vovk, M.V. Condensed pyrimidine systems 5: 6-methyl-functionalized in pyrazolo[3,4-d]pyrimidin-4(5H)-ones. Zh. Org. Farm. Khim. 2006, 4, 57–61. [Google Scholar]
- Harden, F.A.; Quinn, R.J.; Scammells, P.J. Synthesis and Adenosine Receptor Affinity of a Series of Pyrazolo[3,4-d]pyrimidine Analogues of 1-Methylisoguanosine. J. Med. Chem. 1991, 34, 2892–2898. [Google Scholar] [CrossRef]
- Bavetsias, V. A Facile Route to Quinazolin-4(3H)-ones Functionalised at the 2-Position. Syn. Commun. 1998, 28, 4547–4559. [Google Scholar] [CrossRef]
- Cheng, C.C.; Robins, R.K. Potential Purine Antagonists VII. Synthesis of 6-Alkylpyrazolo-[3,4-d] pyrimidine. J. Org. Chem. 1958, 23, 191–200. [Google Scholar] [CrossRef]
- Hermecz, I.; Kökösi, J.; Podányi, B.; Szász, G. Syntheses of indolyl-(3H)-quinazolinones. Heterocycles 1994, 37, 903–914. [Google Scholar] [CrossRef]
- Masayuki, I.; Yukuo, E. Syntheses of 2-hydroxymethyl-4(3H)-quinazolinone and its analogs. Heterocycles 1981, 16, 31–34. [Google Scholar] [CrossRef]
- Hu, T.S.; Yu, Q.; Wu, Y.L.; Wu, Y. Enantioselective syntheses of monotetrahydrofuran Annonaceous acetogenins tonkinecin and annonacin starting from carbohydrates. J. Org. Chem. 2001, 66, 853–861. [Google Scholar] [CrossRef]
- Faul, M.M.; Winneroski, L.L.; Krumrich, C.A. Synthesis of Rebeccamycin and 11-Dechlororebeccamycin. J. Org. Chem. 1999, 64, 2465–2470. [Google Scholar] [CrossRef]
- Baker, B.R.; Almaula, P.I. Nonclassical Antimetabolites. X.1, 2 A Facile Synthesis of 4-Quinazolone-2-carboxylic Acid and the Structure of Bogert's Ammonium Salt. J. Org. Chem. 1962, 27, 4672–4764. [Google Scholar] [CrossRef]
- Brown, H.C.; Narasimhan, S.; Choi, Y.M. Selective Reductions. 30. Effect of Cation and Solvent on the Reactivity of Saline Borohydrides for Reduction of Carboxylic Esters. Improved Procedures for the Conversion of Esters to Alcohols by Metal Borohydrides. J. Org. Chem. 1982, 47, 4702–4708. [Google Scholar] [CrossRef]
- Compound 1d and 1e were also prepared with the same synthetic sequence of 1a as shown in Scheme 4. The synthetic details is shown in supplementary data file.
- Briefly, cells (2,000/well) were seeded in 96-well plates and cultured for 24 hours, followed by treatment with the compounds for 48 h. Ten microliters of 10 mg/mL MTT was added per well and incubated for another 2.5 h at 37 °C. Then the supernatant fluid was removed and 150 μL/well DMSO was added for 15–20 minutes. The absorbance (OD) of each well was measured at 570 nm, using a SpectraMAX M5 microplate spectrophotometer (Molecular Devices).
- Peng, F.; Wei, Y.Q.; Tian, L.; Yang, L.; Zhao, X.; Lu, Y.; Mao, Y.Q.; Kan, B.; Lei, S.; Wang, G.S.; et al. Induction of apoptosis by norcantharidin in human colorectal carcinoma cell lines: Involvement of the CD95 receptor/ligand. J. Cancer Res. Clin. Oncol. 2002, 128, 223–230. [Google Scholar] [CrossRef]
- Wei, Y.Q.; Zhao, X.; Kariya, Y.; Fukata, H.; Teshigawara, K.; Uchida, A. Induction of apoptosis by quercetin: Involvement of heat shock protein. Cancer Res. 1994, 54, 4952–4957. [Google Scholar]
- Sample Availability: Samples of the compounds 1a, 1d and 1e are available from the authors.
© 2011 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
He, H.-Y.; Zhao, J.-N.; Jia, R.; Zhao, Y.-L.; Yang, S.-Y.; Yu, L.-T.; Yang, L. Novel Pyrazolo[3,4-d]pyrimidine Derivatives as Potential Antitumor Agents: Exploratory Synthesis, Preliminary Structure-Activity Relationships, and in Vitro Biological Evaluation. Molecules 2011, 16, 10685-10694. https://doi.org/10.3390/molecules161210685
He H-Y, Zhao J-N, Jia R, Zhao Y-L, Yang S-Y, Yu L-T, Yang L. Novel Pyrazolo[3,4-d]pyrimidine Derivatives as Potential Antitumor Agents: Exploratory Synthesis, Preliminary Structure-Activity Relationships, and in Vitro Biological Evaluation. Molecules. 2011; 16(12):10685-10694. https://doi.org/10.3390/molecules161210685
Chicago/Turabian StyleHe, Hai-Yun, Jin-Ni Zhao, Ruo Jia, Ying-Lan Zhao, Sheng-Yong Yang, Luo-Ting Yu, and Li Yang. 2011. "Novel Pyrazolo[3,4-d]pyrimidine Derivatives as Potential Antitumor Agents: Exploratory Synthesis, Preliminary Structure-Activity Relationships, and in Vitro Biological Evaluation" Molecules 16, no. 12: 10685-10694. https://doi.org/10.3390/molecules161210685