Synthesis of RP 48497, an Impurity of Eszopiclone

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
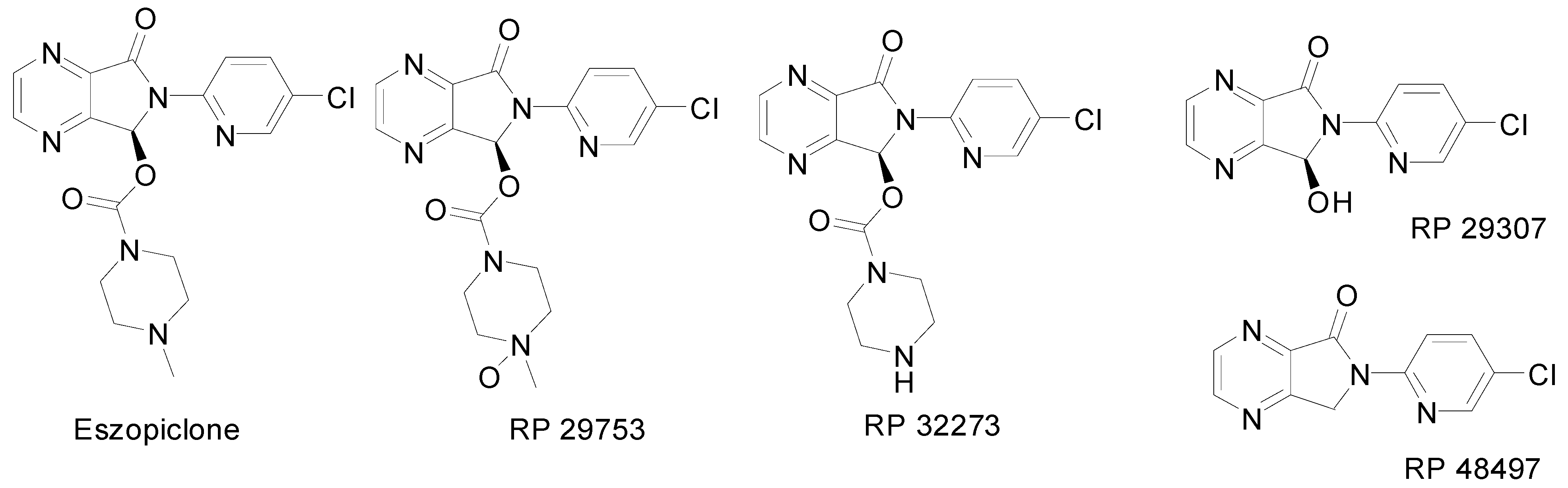
:Introduction


Results and Discussion
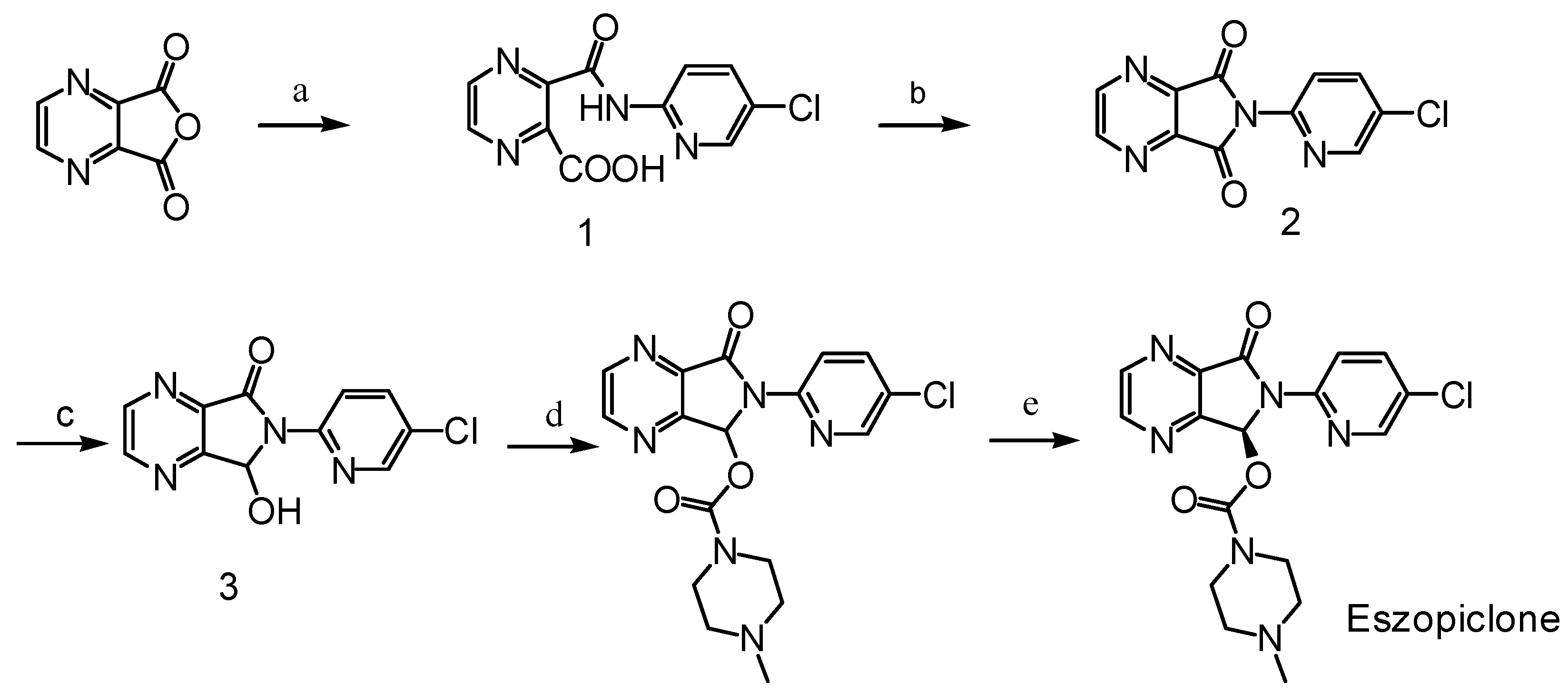
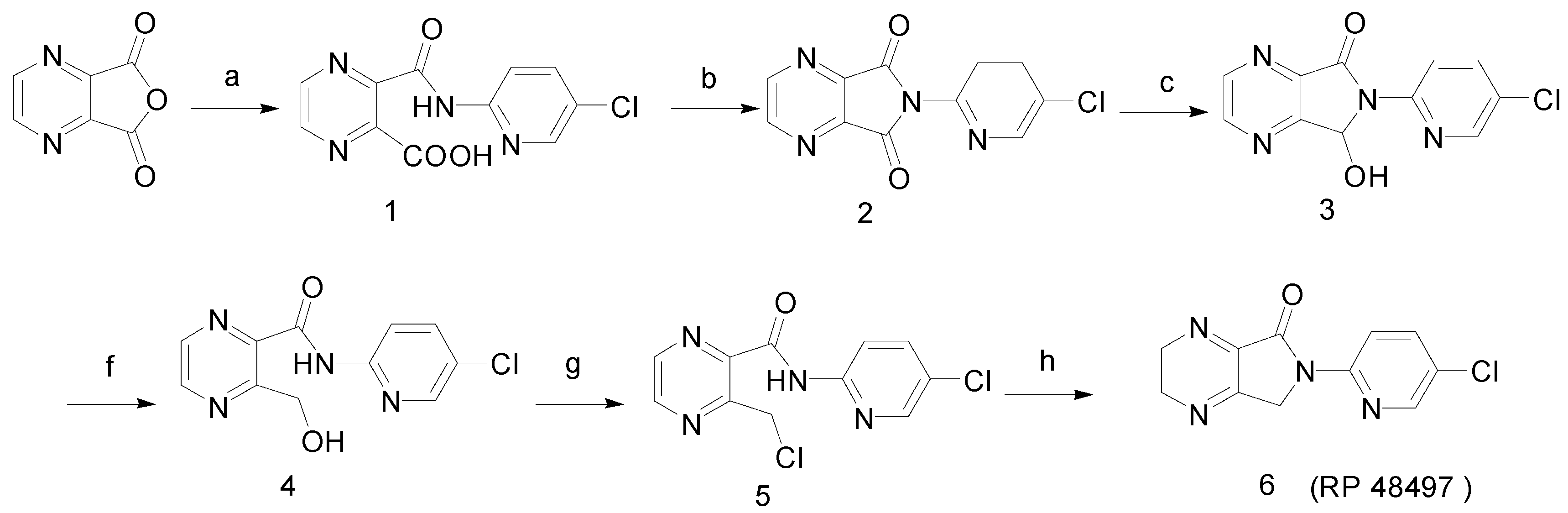
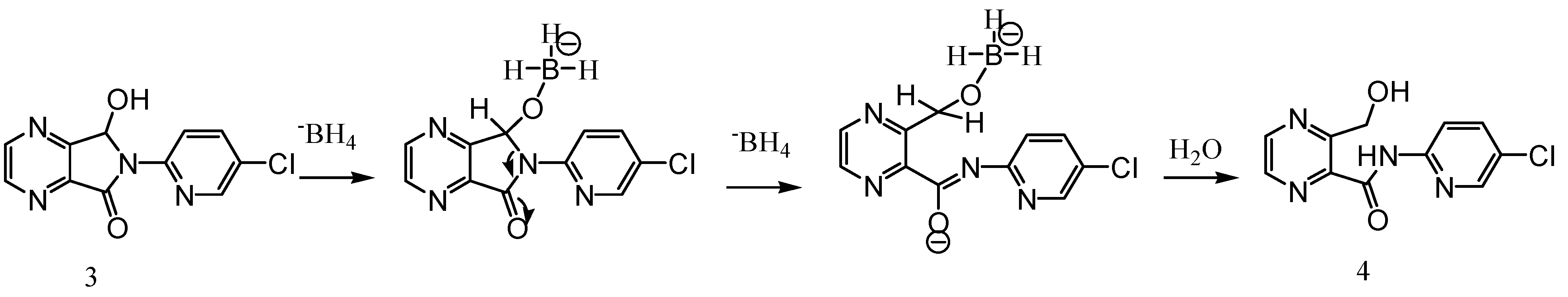
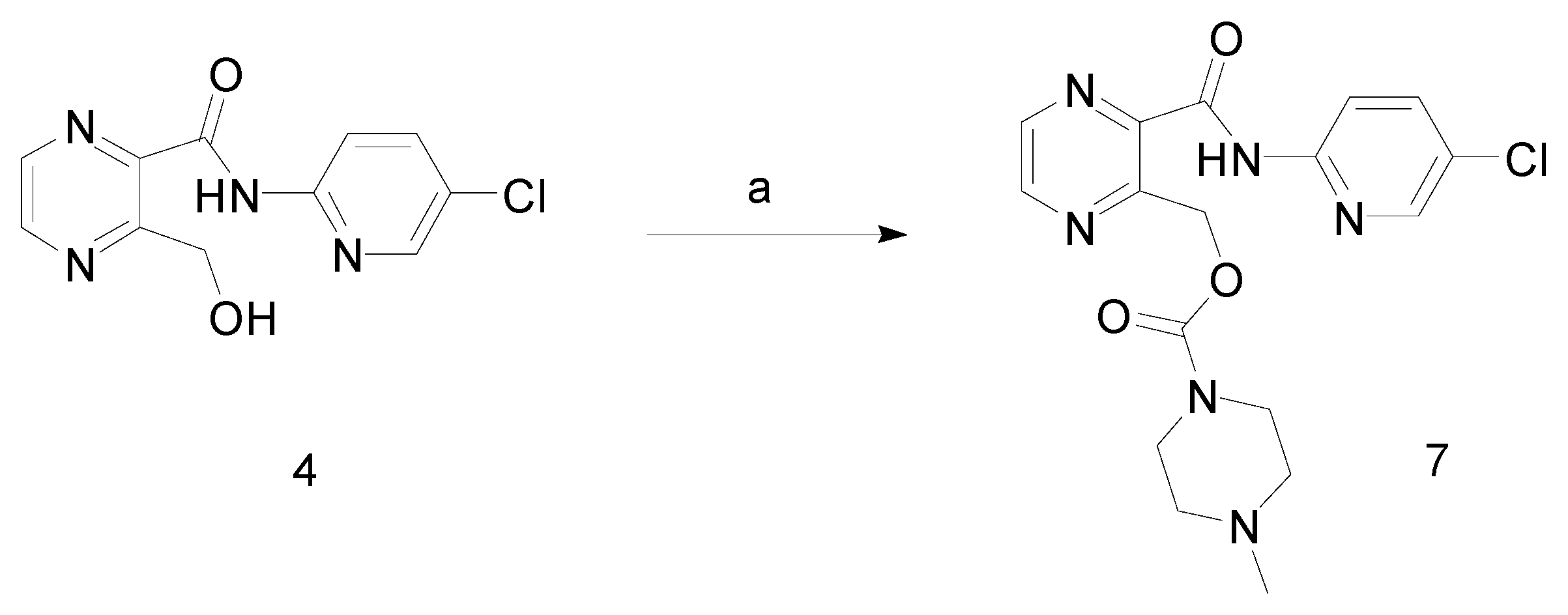
Synthesis



Experimental
General
3-Hydroxymethyl-pyrazine-2-carboxylic acid (5-chloro-pyridin-2-yl)-amide (4)
3-Chloromethylpyrazine-2-carboxylic acid (5-chloro-pyridin-2-yl)-amide (5)
6-(5-Chloropyridin-2-yl)-6,7-dihydropyrrolo[3,4-b]pyrazin-5-one (RP 48497, 6)
References
- Piperak, S; Parissi-Poulou, M. Enantiomeric separation of zopiclone, its metabolites and products of degradation on a cyclodextrin bonded phase. J. Chromatogr. A. 1996, 729, 19–28. [Google Scholar] [CrossRef]
- Cotrel, C.; Jeanmart, C.; Messer, M.N. Pyrrolo[3,4-b]pyrazine derivatives. U.S. Pat. 3,862,149, 1975. [Google Scholar]
- Cotrel, C.; Jeanmart, C.; Messer, M.N.; Crisan, C. Heterocyclic compounds. U.S. Pat. 4,220,646, 1980. [Google Scholar]
- Chen, H.C.; Liu, Z.Z.; Zhang, C.X. Synthesis of sedative hypnotic Zopiclone. J. Zheng Zhou Univ. (zirankexueban) 1993, 12, 73–76. [Google Scholar]
- Zhang, Y.F.; Zuo, D.S.; Tian, K.P.; Shen, J.M. Synthesis of the new sedative hypnotic Zopiclone. Chin. J. Med. Chem. 1994, 4, 62–64. [Google Scholar]
- Horii, Z.; Iwata, C.; Tamura, Y. Reduction of phthalimides with sodium borohydride. J. Org. Chem. 1961, 26, 2273–2276. [Google Scholar] [CrossRef]
- Sample Availability: Samples are available from the authors.
© 2008 by the authors. Licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Sharov, Y.; Zhang, L.; Du, G.-J.; Ren, J.; Cheng, M.-S. Synthesis of RP 48497, an Impurity of Eszopiclone. Molecules 2008, 13, 1817-1821. https://doi.org/10.3390/molecules13081817
Sharov Y, Zhang L, Du G-J, Ren J, Cheng M-S. Synthesis of RP 48497, an Impurity of Eszopiclone. Molecules. 2008; 13(8):1817-1821. https://doi.org/10.3390/molecules13081817
Chicago/Turabian StyleSharov, Yu, Lei Zhang, Gui-Jie Du, Jian Ren, and Mao-Sheng Cheng. 2008. "Synthesis of RP 48497, an Impurity of Eszopiclone" Molecules 13, no. 8: 1817-1821. https://doi.org/10.3390/molecules13081817