Library Preparation
The overall strategic design of the library was influenced by three main factors: cost, quality and time. The goal at the outset was to achieve the highest quality library possible at a moderate cost and within a reasonable time with the size of the natural products library not exceeding more than 10% of the entire screening library. Financial considerations not only include the total cost of library preparation from organism isolation to fermentation to fractionation and plating but also the cost to screen each sample. At a cost of 25 to 50 cents per well per assay, the cost can rapidly escalate. In this instance, screening a library of one million wells would cost between $250K and $500K per assay. Additionally, features that enable reduction in project cycle time post-HTS were considered in the design of the library. Simplified mixtures should help to reduce the time from hit identification to compound characterization as should the immediate availability of stored crude extract.
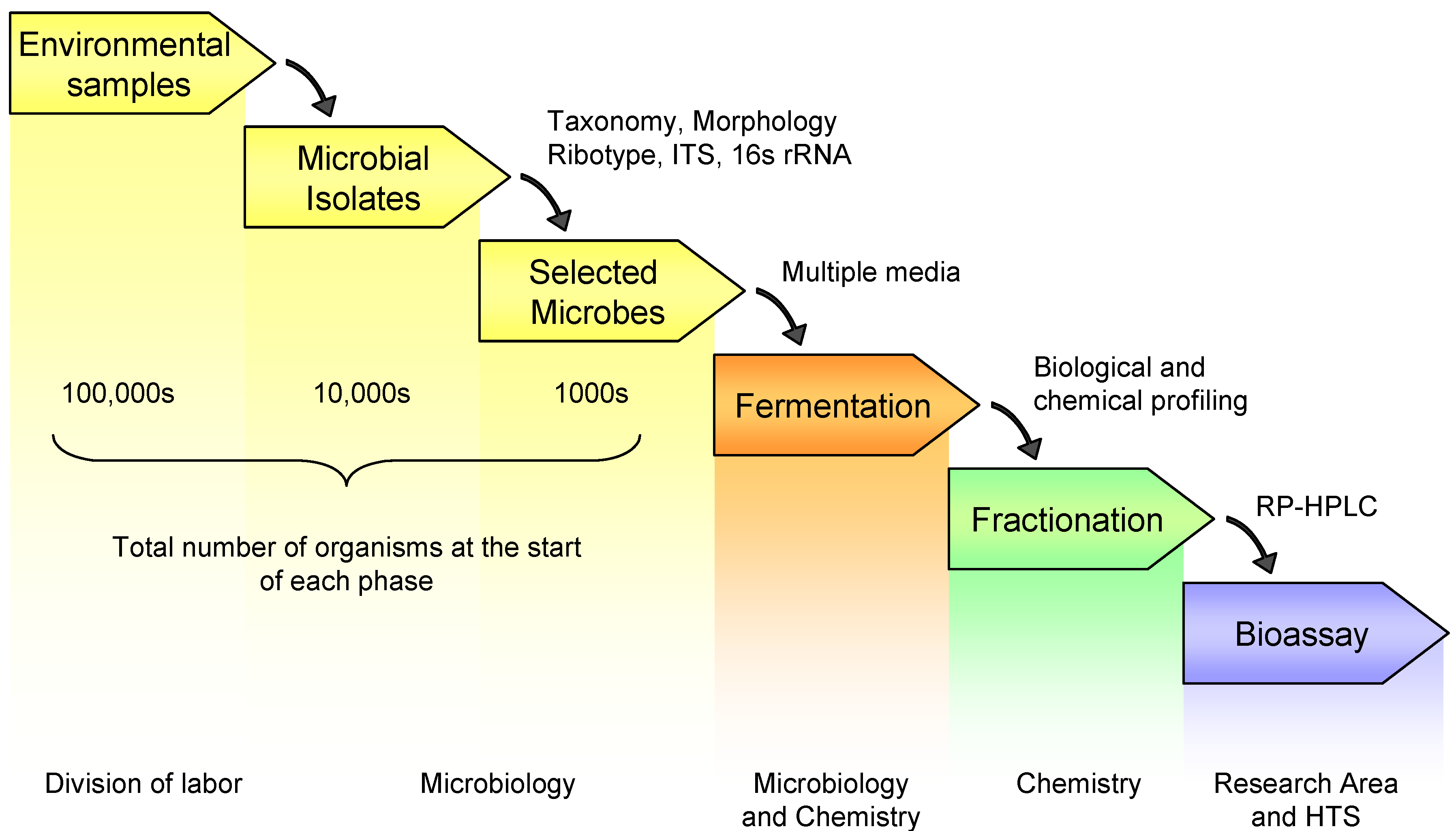
Figure 1 outlines the workflow for the production and screening of a pre-fractionated library and the division of labor. This process represents a joint effort between microbiology, chemistry and HTS. The initial steps in library generation result in the selection of a set of microorganisms by microbiologists. Environmental samples are collected for isolation of microbes that are then selected based on various criteria for fermentation. These first three steps are not unique to the preparation of a pre-fractionated library. However, because of the increased workload in downstream processing and the need to generate high quality within a fixed library size, more rigorous selection criteria need to be applied in the selection of microbes. More time and resources will be needed upfront for the pre-screening of strains using molecular techniques as well as classical taxonomy to remove redundancy and select for novel organisms. The selected microorganisms are then fermented using at least two different fermentation conditions to increase the potential for producing chemical diversity. The liquid fermentations are pooled prior to solid-phase extraction. By pooling the fermentations of each individual organism prior to extraction, the overall cost and time for extraction, profiling and fractionation are reduced by fifty percent. The resulting crude extracts are subjected to both biological and chemical profiling. Chemical profiles of the extracts are generated by LCMS-DAD-ELSD and analyzed for the presence of secondary metabolites. This chemical screening aids in the dereplication of known compounds and thus can be used to reduce redundancy in the overall library. Biological profiling in this instance involves the testing of the extracts in antimicrobial assays; the presence of antimicrobial activity is one indication that an organism is producing secondary metabolites. Biological screening can be more sensitive than chemical profiling and is useful for detecting minor compounds present in quantities below the limit of detection of the LCMS-DAD-ELSD. Select extracts are then fractionated and the fractionated samples plated and sent to the research areas and/or the HTS group for testing. Based on analysis of over 1000 combined extracts, it was determined that approximately 65% of the organisms fermented using our standard fermentation conditions produced secondary metabolites and were suitable for fractionation. Because only limited chemical and biological data is acquired on each sample, the organic extracts that are not fractionated are incorporated into the library as unfractionated crude extracts.
Figure 1.
General schematic for library generation and screening.
Figure 1.
General schematic for library generation and screening.
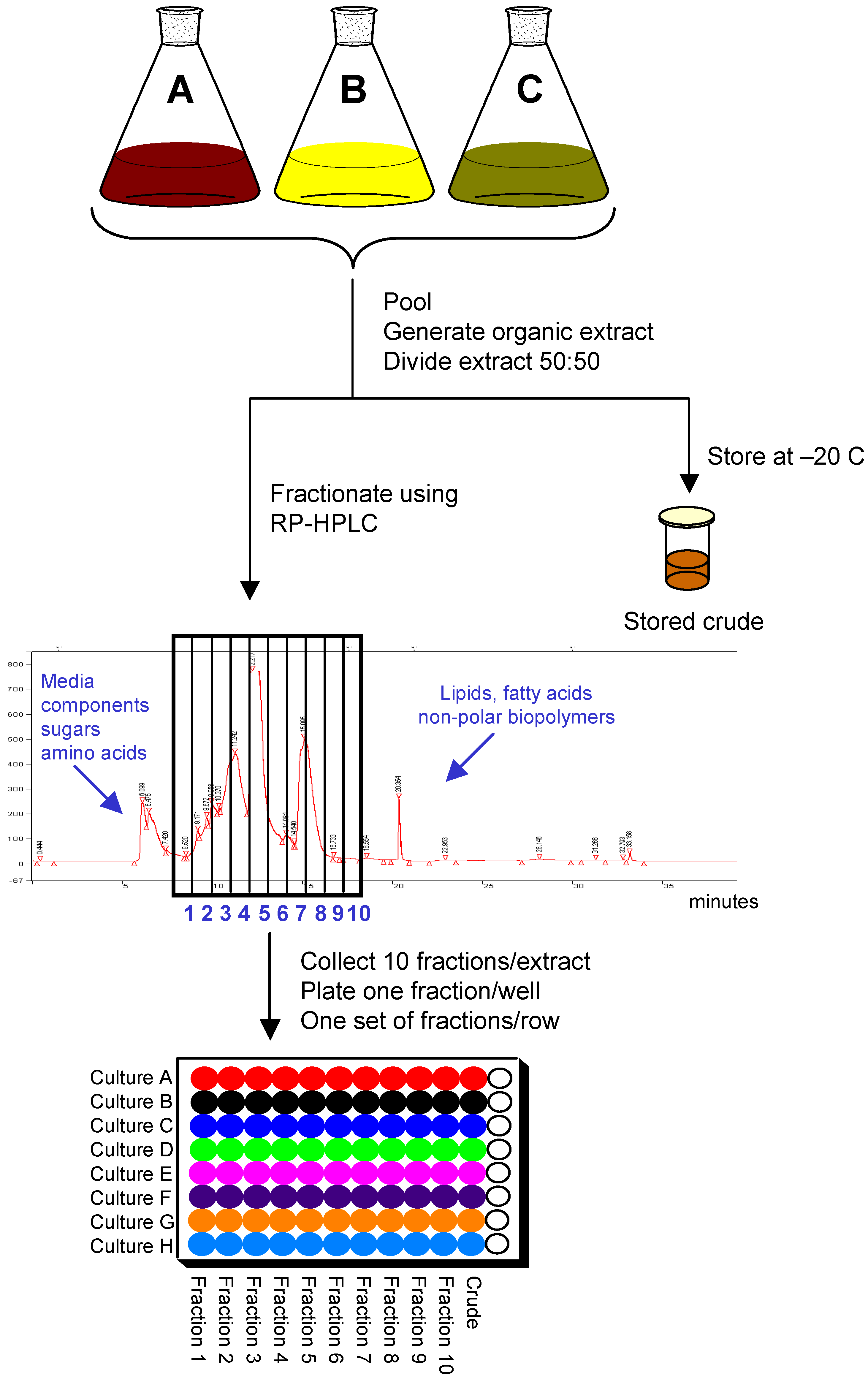
Extracts meeting the criteria for fractionation are concentrated and divided into two equal portions. One portion, the stored crude, is concentrated to dryness and stored at –20 ºC as reference material for processing should that sample show activity later when tested. The remaining half is used for fractionation and plating. Basic separation can be achieved with a number of different techniques such as solid-phase extraction, liquid-liquid partitioning, and column chromatography. Preparative reversed-phase HPLC was chosen as the method for fractionation because it is robust and reproducible, can be easily automated, requires minimal sample handling, and provides good resolution and recovery.
Figure 2.
Detailed overview of the production process for the pre-fractionated natural products library.
Figure 2.
Detailed overview of the production process for the pre-fractionated natural products library.
In order to balance the degree of resolution with the biological and chemical diversity while working within the library size dedicated for natural products, a total of ten fractions were collected per extract.
Figure 2 provides an overview of the fractionation scheme. To prepare for fractionation, the second half of the sample is resolubilized and one-tenth of that solution is removed for plating as the crude in column 11 of the 96-well plate used by the HTS group. Thus the total “saved” crude material plated for HTS is equivalent to 1/20
th of the original fermentation volume. The remaining nine-tenths of that solution is subjected to RP-HPLC using a Luna C-18 column to generate 10 fractions collected in 1-minute increments. Because the intent is to collect the fractions most likely to contain compounds with drug-like properties, the early eluting material consisting of media components and highly polar compounds is not collected nor is the late-eluting lipophilic portion. The total run time for each sample including a wash cycle of the column is 35 minutes. In an average 8-hour workday a total of thirteen runs can be completed. With a fully automated system, higher capacity can be achieved. The rate-limiting step in the fractionation is the drying of the fractions. Each SpeedVac system can handle two sets of fractions in a 24-hour period.
Dried fractions are resuspended in DMSO as a 100X concentrate. The 1/10 “saved” crude is resuspended in an equal volume of DMSO; thus the crude is roughly 10 times less concentrated than the fractions. Samples are then dispensed into 96-well plates using a Tecan robotic system. The resulting 96-well plate consists of samples from eight different cultures with the column number corresponding to the fraction number, column 11 containing crude extract, and column 12 empty and available for controls.
Library Analysis
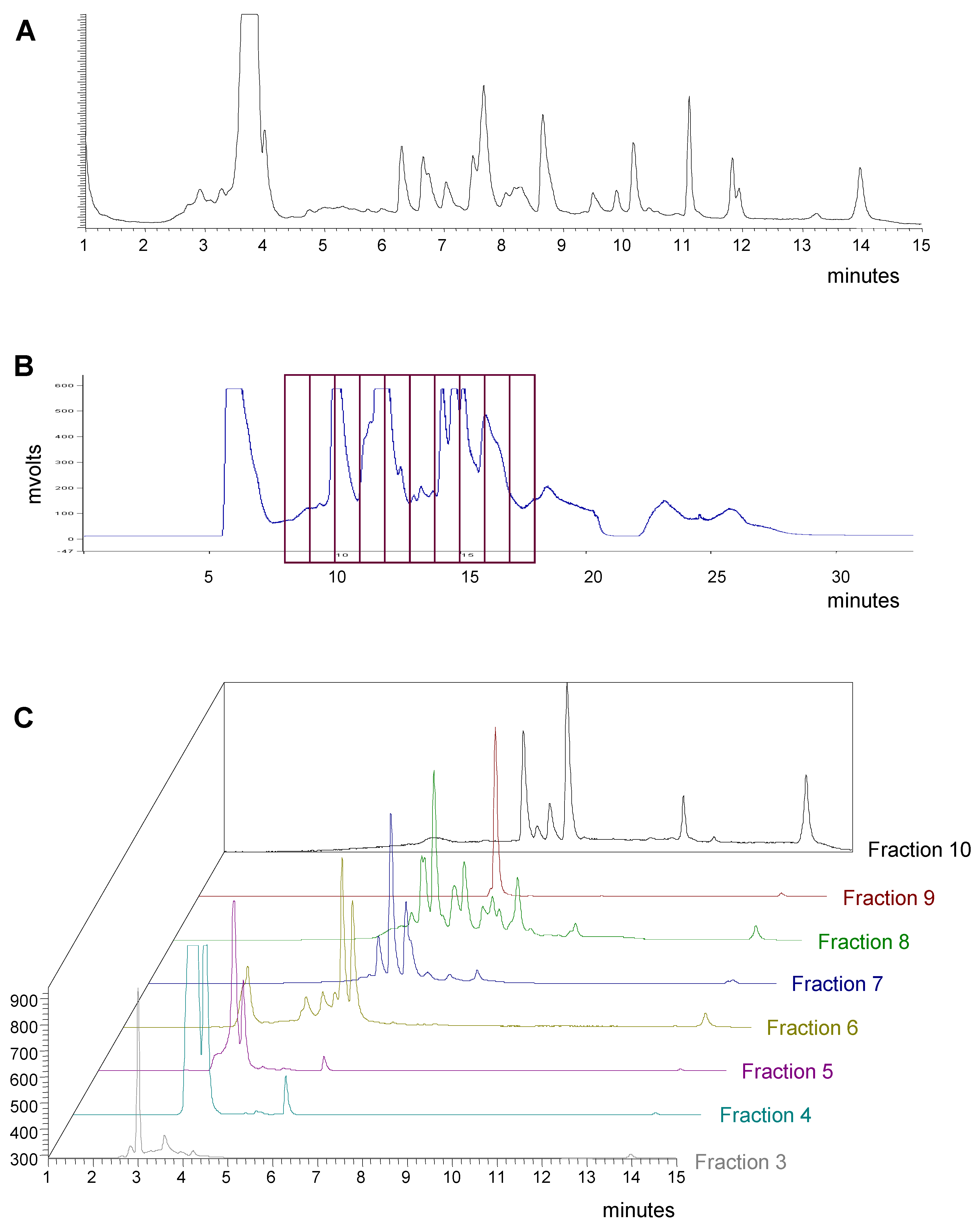
Based on simple visual examination of chromatograms, the pre-fractionation method shows several qualitative benefits. Early-eluting media and polar components are removed from the samples. The overall complexity of the samples to be tested in HTS is reduced with fewer compounds per well. Suitable resolution was achieved and the major components that may interfere or mask the activity of minor components are separated into individual fractions. These benefits are illustrated in the example shown in
Figure 3. Crude extract 70360-F11 contains more than 20 compounds as shown in the analytical HPLC-ELSD chromatogram (
Figure 3A). Based on the biological and chemical profiling results, this extract was chosen for preparative fractionation. The fractions were collected in 1-minute intervals beginning at 8 minutes as shown on the HPLC-UV chromatogram (
Figure 3B). The most polar components eluting between 0 and 7 minutes were discarded. The analytical HPLC-ELSD chromatograms of fractions 3 through 10 are shown in
Figure 3C. Each fraction shows reduced complexity compared to the original crude extract with fraction 9 appearing to contain a single component. In addition, the majority of the sample by weight is separated into fraction 4 as determined by light scattering detection.
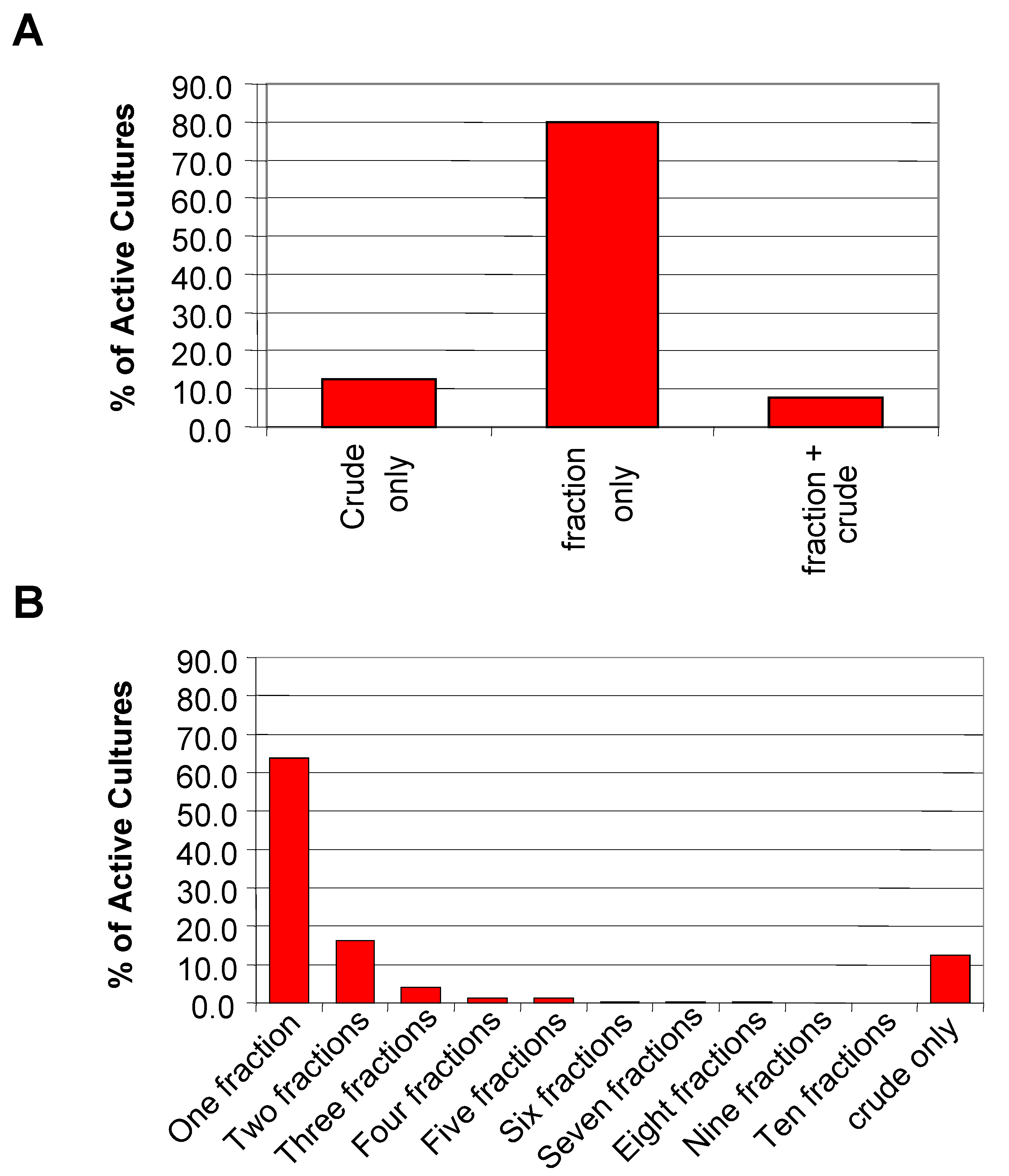
High-throughput screening data was used to do a more rigorous quantitative analysis of the pre-fractionation method. A total of 2750 primary hits from nine HTS assays were analyzed. The 2750 hits represent fractions and crude extracts from 1882 unique organisms. In 79.9% of the 1882 active cultures, the activity was found only in the fractions (
Figure 4A). These samples would have been missed if only the crude extracts had been screened. It is unclear from the analysis if this finding is a result of the increase in concentration of the fractions compared to the crude or the removal of interfering compounds. However, it should be noted that the increase in concentration was only achievable because the physical nature of the samples (i.e. viscosity) was improved by pre-fractionation.
Figure 3.
HPLC chromatograms of fungal extract 70360-F11. A) Analytical HPLC-ELSD chromatogram of the crude methanol extract 70360-F11. B) Preparative LC chromatogram of the crude methanol extract 70360-F11 (UV detection at 254 nm) with the fraction collection outlined. C) HPLC-ELSD chromatograms of fractions 3 through 10 from the preparative LC run of 70360-F11 shown in B.
Figure 3.
HPLC chromatograms of fungal extract 70360-F11. A) Analytical HPLC-ELSD chromatogram of the crude methanol extract 70360-F11. B) Preparative LC chromatogram of the crude methanol extract 70360-F11 (UV detection at 254 nm) with the fraction collection outlined. C) HPLC-ELSD chromatograms of fractions 3 through 10 from the preparative LC run of 70360-F11 shown in B.
For 12.5% of the active cultures, the activity was detected only in the crude fraction reinforcing the decision to plate the crude sample alongside its fractions. This result suggests that the active compound eluted outside the time range for collection, was unstable under the conditions used for fractionation, or was retained on the column. For more than 60% of the active cultures, the activity was concentrated into a single fraction with an additional 16.2% of the active cultures registering activity in only two of the ten fractions (
Figure 4B). Less than 1% of the active cultures showed activity spread over six or more fractions. Thus sufficient resolution was achieved with the pre-fractionation RP-HPLC method and collection parameters used.
Figure 4.
Distribution of the hits from nine HTS screens. A) General categorization of the HTS hits based on activity localization for 1882 active cultures. B) Detailed view of the activity distribution of the hits for the 1882 active cultures.
Figure 4.
Distribution of the hits from nine HTS screens. A) General categorization of the HTS hits based on activity localization for 1882 active cultures. B) Detailed view of the activity distribution of the hits for the 1882 active cultures.
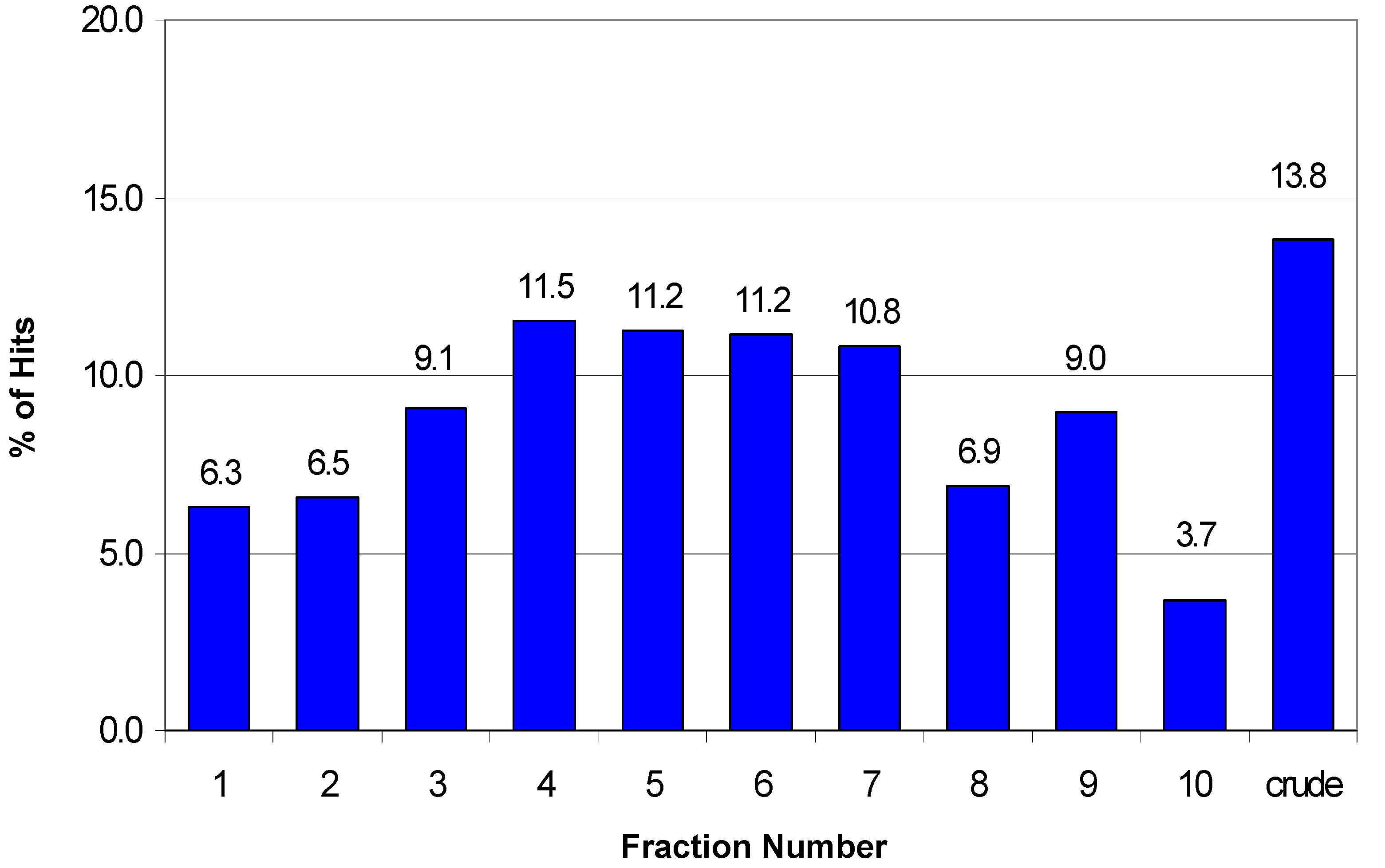
Figure 5 depicts the distribution of hits across the fractions for the 2750 hits identified in nine HTS assays. The pattern of activity across fractions approximates a Gaussian curve centered around fractions 5 and 6 with slightly fewer active samples found in the peripheral fractions. However, this distribution is clearly assay-dependent as can be seen when the activity pattern for each individual assay is analyzed as shown in
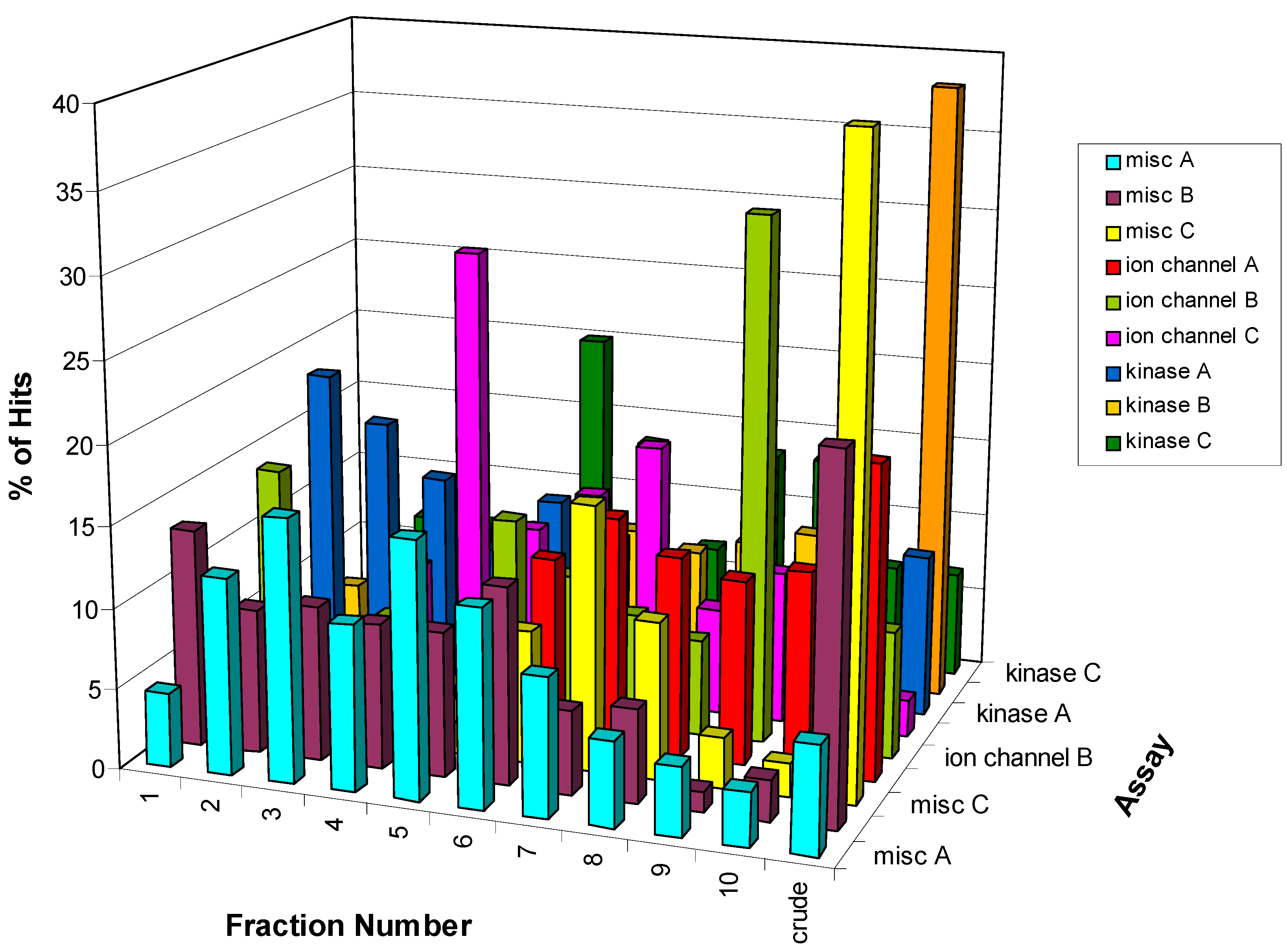
Figure 6. For three of the nine assays the crude fraction represents the highest percentage (>20%) of the hits in those specific assays (misc B, misc C, and kinase B). However, the greatest number of hits is found in a single fraction for some of the other assays. For example, 33% of the hits from the ion channel B assay are found in fraction 9 while fraction 4 represents 28% of the hits in the ion channel C assay. In some instances, the HTS hits are distributed over all 10 fractions plus the crude with no one fraction corresponding to >20% of the hits. The distribution of hits over all ten fractions suggests that any reduction in the collection interval would result in missed actives. Furthermore, these data could help in the analysis of the hits and the prioritization of the hits for follow-up as described below.
Figure 5.
Distribution of the hits across fractions for nine HTS assays (total number of hits = 2750).
Figure 5.
Distribution of the hits across fractions for nine HTS assays (total number of hits = 2750).
Figure 6.
Distribution of the 2750 HTS hits from 9 screens showing the assay-dependency.
Figure 6.
Distribution of the 2750 HTS hits from 9 screens showing the assay-dependency.
One of the major challenges in screening natural product crude samples is the prioritization of hits for follow-up. Even with screening a pre-fractionated library, the resolution of hits can be resource-intensive and thus focusing on the samples with the highest probability for success is preferential. Dereplication, the process of identifying known compounds, is possible with both crude and pre-fractionated extracts but there is no guarantee that the compound identified is responsible for the observed biological activity without additional experimental work. Differentiating between the hits that contain compounds of interest and those that contain ubiquitous or chemically unattractive compounds is not easily accomplished without proceeding through at least one round of bioassay-guided fractionation. A total of eleven data points are generated for each culture in the pre-fractionated library in comparison with a single data point per culture in the crude extract library. This additional information creates the opportunity for more effective data mining which can facilitate the selection of the best candidates for follow-up chemical processing and analysis. Comparison of the activity profiles of each culture can be informative. For example, the bioactivity profiles of 28 active actinomycete cultures tested in a kinase screen are shown in
Figure 7A. The potency of each sample is represented both by the size and the color of the circle (larger circle = greater activity; red = most active). Based on information captured in our in-house database, several of the samples (fractions and crudes) as indicated with green arrows have previously been identified as containing staurosporine (
1), a well-known kinase inhibitor [
13]. The seven cultures known to contain staurosporine show similar activity profiles suggesting that staurosporine is responsible for the observed activity and is found in fractions 5 and 6. A similar pattern of activity is seen for 16 additional extracts (samples AA, F, L-P and R-Z). Analysis of the LCMS-DAD-ELSD chromatograms collected during the chemical profiling prior to fractionation indicated that at least 13 of the sixteen extracts with activity between fractions 4 and 6 contain staurosporine. Although fraction 4 is the active fraction of sample X, this sample does not appear to contain staurosporine by LCMS-DAD-ELSD analysis. This pattern recognition allowed for the rapid identification of the known compound staurosporine and the deprioritization of 20 of the initial active cultures. The observed shift in activity from fractions 5 and 6 to fractions 4 and 5 corresponds to a modification in the preparative HPLC fractionation method and collection parameters.
The visual inspection of bioactivity profiles is not suitable for the massive data sets produced by high-throughput screening. Pattern recognition tools including self-organizing maps and principal components analysis are available for triaging cumbersome data sets. More than 15,000 data points were used to generate activity profiles for each fractionated extract by plotting the fraction number versus the activity for 1436 actinomycete extracts. The activity profiles were then clustered using a self-organizing map algorithm in Spotfire
® and binned as shown in
Figure 7B. Many of the extracts showed no activity and resulted in flat bioactivity profiles. Of the 25 extracts represented in bin 12, nineteen are known to contain staurosporine. The additional six cultures in bin 12 have a high probability of containing staurosporine. One additional staurosporine producer is located in bin 8. The bioactivity profiles can also be used in principal components analysis. In
Figure 7C, the samples known to produce staurosporine are shown in pink and are separated from the majority of the other extracts shown in light blue. Several extracts (circled in green) are in close proximity or overlap with the staurosporine-containing cluster and thus should be evaluated further for the presence of staurosporine.
Figure 7.
Examples of pattern recognition for actinomycete cultures tested in a kinase screen. A) Activity profiles for twenty-eight active actinomycete cultures. Color and size of the circle correlate with activity. Green arrows indicate samples known to contain staurosporine. B) Self-organizing map of the activity profiles for 1436 actinomycete cultures in a kinase screen. C) Principal components analysis of the activity profiles for 1436 actinomycete cultures. Samples in pink are known to contain staurosporine.
Figure 7.
Examples of pattern recognition for actinomycete cultures tested in a kinase screen. A) Activity profiles for twenty-eight active actinomycete cultures. Color and size of the circle correlate with activity. Green arrows indicate samples known to contain staurosporine. B) Self-organizing map of the activity profiles for 1436 actinomycete cultures in a kinase screen. C) Principal components analysis of the activity profiles for 1436 actinomycete cultures. Samples in pink are known to contain staurosporine.
Another implication of the high-throughput drug discovery pathway is the shortened project cycle times. Screening of pre-fractionated natural product libraries reduces the number of bioassay-guided fractionation cycles needed to isolate and identify the active component(s) and thus the initial hits for a project are more rapidly resolved. As shown previously in
Figure 3, the overall complexity of the pre-fractionated samples tested in HTS is greatly reduced which facilitates the isolation and identification process. In some instances, the samples generated by the pre-fractionation method contain one major component and no further isolation work may be needed as shown in the LCMS-ELSD chromatograms for two hits in a kinase assay (
Figure 8A and
Figure 8B).
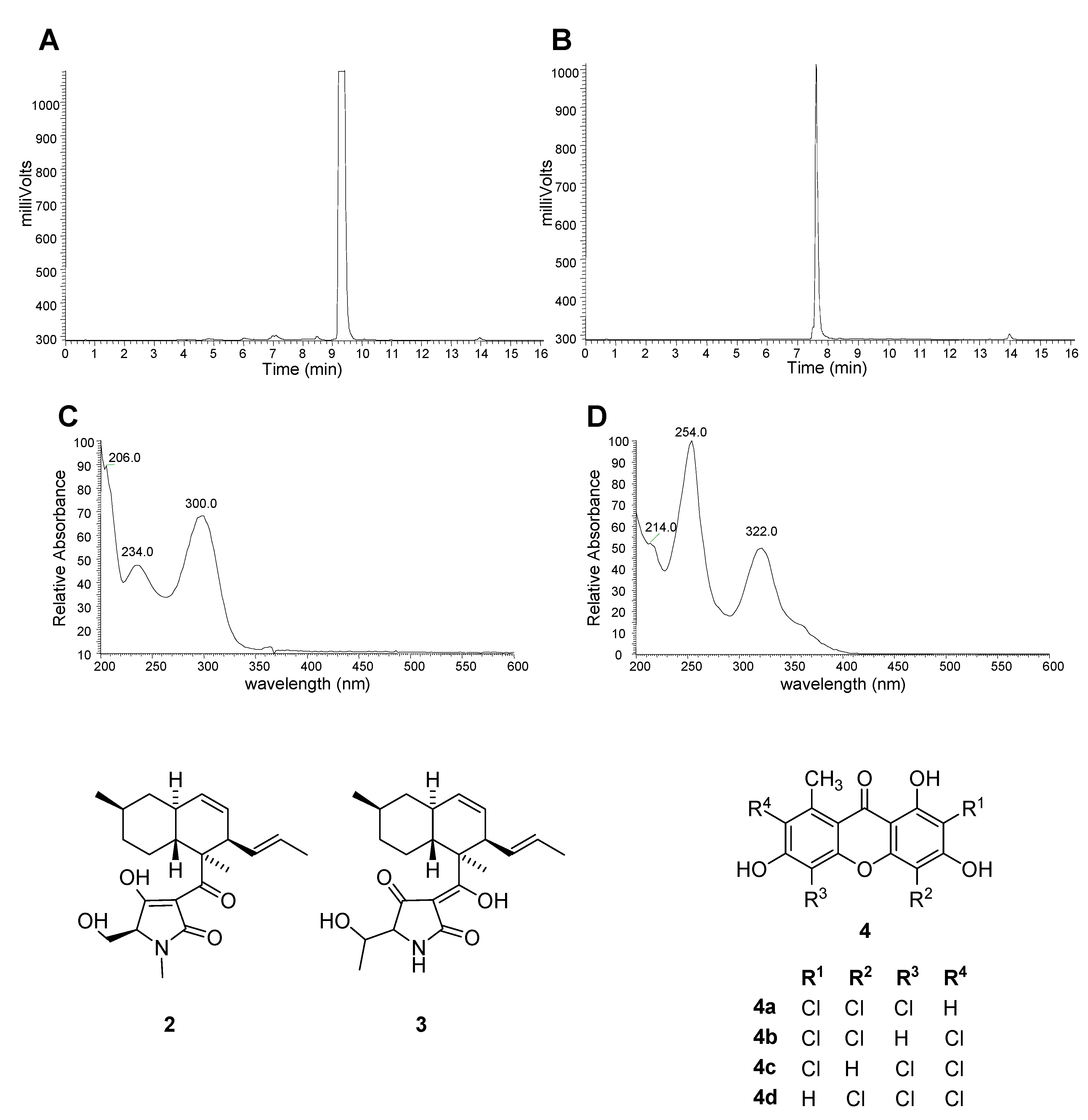
Figure 8.
Analytical data for select samples containing one major component. A) HPLC-ELSD chromatogram of 70254-F08. B) HPLC-ELSD chromatogram of 70360-F09. C) UV spectrum of 70254-F08. D) UV spectrum of 70360-F09.
Figure 8.
Analytical data for select samples containing one major component. A) HPLC-ELSD chromatogram of 70254-F08. B) HPLC-ELSD chromatogram of 70360-F09. C) UV spectrum of 70254-F08. D) UV spectrum of 70360-F09.
High-resolution mass spectral (HRMS) data and UV spectra can be readily obtained for these samples and used for dereplicating the sample. A molecular formula of C
22H
31NO
4 was determined using HRMS (
m/z 374.23236 [M + H]
+) for sample 70254-F08, a fraction from fermentation of a Fusarium sp. Combining the molecular formula with the UV spectrum (
Figure 8C), sample 70254-F08 was identified as a tenuazonic acid derivative. The UV spectrum and molecular formula are consistent with both equisetin (
2), a known antimicrobial metabolite produced by
Fusarium equiseti [
14], and the closely related paecilosetin (
3) produced by
Paecilomyces farinosus [
15]. Similarly, the major component of 70360-F09 was determined to be a member of the norlichexanthone (
4) family based on molecular formula (C
14H
7Cl
3O
5;
m/z 358.92850 [M – H]
-) and comparison of the UV spectrum (
Figure 8D) with literature values for that of arthothelin (
4a) [
16,
17]. The dereplication and identification process can be greatly facilitated by the availability of a comprehensive UV spectral library.
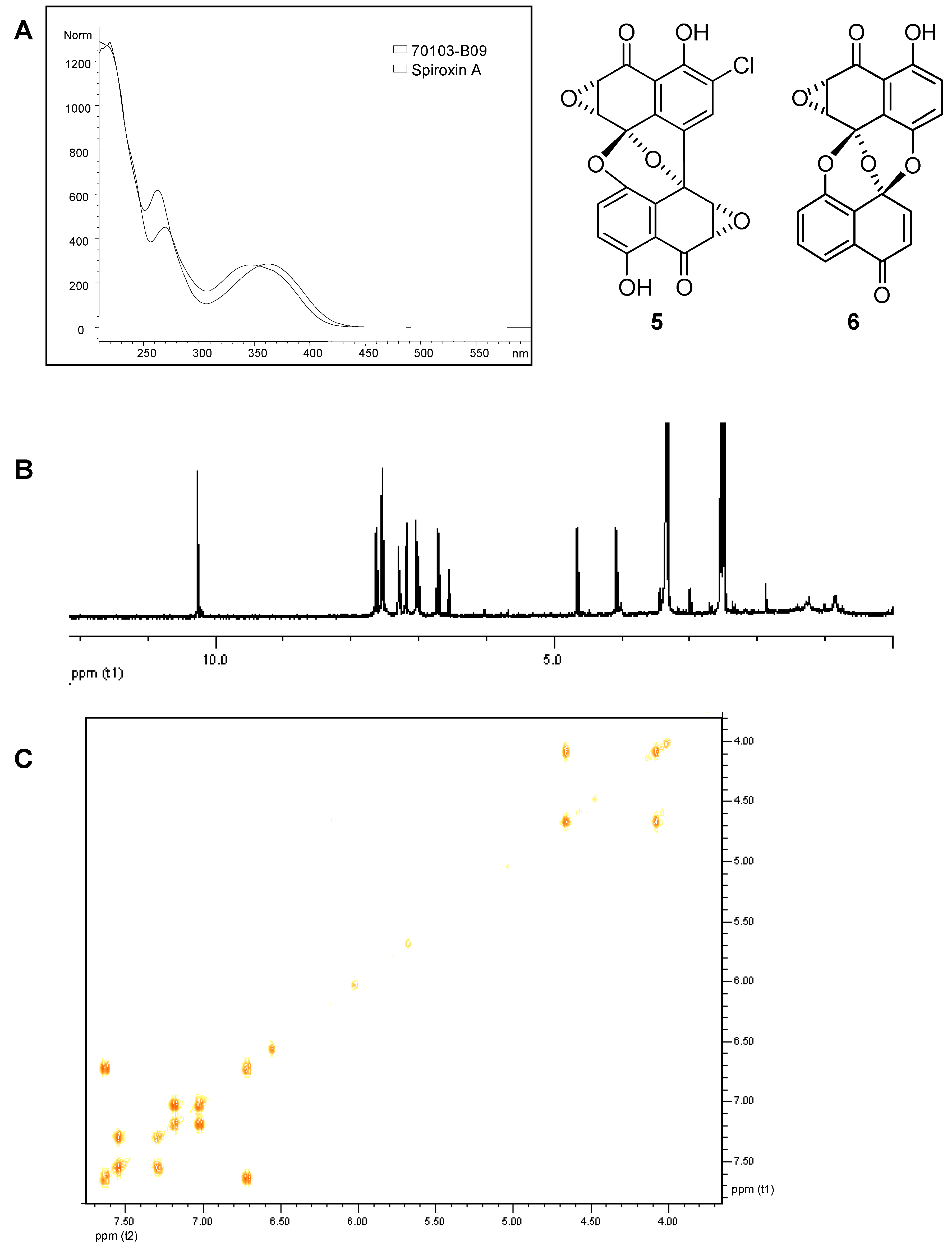
Full structural characterization may be possible if enough sample is available. Sample 70103-B09 was an initial hit from high-throughput screening against a kinase target. Searching our in-house library of UV spectra, the UV spectrum of 70103-B09 showed similarity to that of spiroxin A (
5) (
Figure 9A) but it was not an exact match [
18]. High-resolution mass spectral analysis indicated a molecular formula of C
20H
12O
7 for the major component in sample 70103-B09 whereas spiroxin A has a molecular formula of C
20H
9ClO
8. A 100 μL aliquot of the 100X solution of sample 70103-B09 was concentrated to dryness and resuspended in 200 μL of DMSO-
d6 (concentration of major component is unknown). The
1H-NMR and COSY spectra of this fraction acquired using a Bruker 3 mm probe on a Bruker Avance 400 MHz spectrometer are shown in
Figure 9B and
Figure 9C, respectively. Based on a combination of the HRMS, the UV spectrum and the NMR spectral data, the major component in this fraction is consistent with preussomerin G (
6). Preussomerin G was first isolated as an inhibitor of Ras farnesyl-protein transferase; this observed activity may be due to its ability to act as a Michael acceptor for strong nucleophiles [
19]. Although preussomerin G is a known compound, this example illustrates that full structure elucidation may be possible for novel compounds if enough material is available to obtain all homo- and heteronuclear correlations by high-resolution NMR.
In instances when the active component cannot be readily dereplicated or fully characterized from the available DMSO stock solution, the stored crude can be used for follow-up of the HTS hit. Because project cycle times are shortened in the high-throughput paradigm and the refermentation phase is typically one of the lengthiest steps in the process from identifying an active extract to resolving the active constituent, the decision was made to create a stored crude during the library generation phase. This aspect of the library design is not specific to pre-fractionated libraries but can be applied to the production of any library type including crude extract libraries. Stored crude material offers two main benefits. First, in cases where the stored crude contains enough material for isolation and identification, the overall process is reduced by at least 4 to 6 weeks corresponding to the amount of time needed to revive the organism from the cryovial, ferment the culture, and then process the fermentation to generate a crude organic extract. Second, the stored crude is identical to the original crude sample tested in HTS and thus eliminates the need for confirmation of the bioactivity. Because refermentations involve living organisms, the activity is not always reproducible and a freshly prepared extract must be assayed to confirm that the original observed activity was reproduced during the refermentation. The average turnaround time is 1 to 2 weeks for confirmation of bioactivity. Therefore, the availability of stored crude material can reduce the overall time from hit identification to resolution by up to 8 weeks. The processing of the frozen stored crude can follow two distinct paths. The standard path forward involves bioassay-guided fractionation where the samples are tested for activity after each round of separation. Alternatively, in select cases, the active constituent can be pinpointed from the sample tested by HTS or by preliminary work with an aliquot of the DMSO stock solution but the amount of material obtained is insufficient for full structure elucidation. In these instances, the path forward employs target-guided isolation from the stored crude using UV or MS data in combination with retention time. This path reduces the time to resolution by eliminating the need for all bioassay support during the isolation process.
Figure 9.
Analytical data for 70103-B09 used for identification of the major component. A) UV spectrum of 70103-B09 overlaid with the UV spectrum of spiroxin A (5). B) 1H- NMR spectrum of 70103-B09 in DMSO-d6. C) 1H-1H COSY spectrum of 70103-B09 in DMSO-d6.
Figure 9.
Analytical data for 70103-B09 used for identification of the major component. A) UV spectrum of 70103-B09 overlaid with the UV spectrum of spiroxin A (5). B) 1H- NMR spectrum of 70103-B09 in DMSO-d6. C) 1H-1H COSY spectrum of 70103-B09 in DMSO-d6.
The pre-fractionated natural products library offers many strategic advantages over a library of crude extracts as described above. However, associated with these advantages are increased costs. Some costs can be easily measured while others are less tangible. First, because the samples are 10-fold more concentrated than the previously prepared crude extract library and a stored crude is generated, 20 times more organic extract needed to be produced. This increase in extract amount is directly proportional to an increase in raw materials such as the media for fermentation and the solvents for extraction and processing. Other additional costs include the initial investment in enabling technologies dedicated to library preparation including the various instrumentation needed for molecular analysis of the microorganisms, for profiling of the extracts, and for HPLC separation of the extracts. A regular supply of reagents associated with each technique also increased the overall production cost. One extra FTE per year and additional expertise was needed to support the library production. More difficult to ascertain is the cost associated with biological and chemical diversity. The overall throughput for generation of the pre-fractionated library was approximately 4-fold less than that of generating a library of crude extracts. This reduction in throughput translates to a moderate decrease in overall diversity. However, the pre-fractionation protocol results in a higher quality library with samples that are more suitable for the high-throughput drug discovery paradigm currently popular with the pharmaceutical industry. Because the ultimate goal was to increase the probability of success, the quality of the library and the resulting HTS hits is more important than the quantity of hits.











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}