Cyclization-activated Prodrugs


Abstract
:1. Introduction

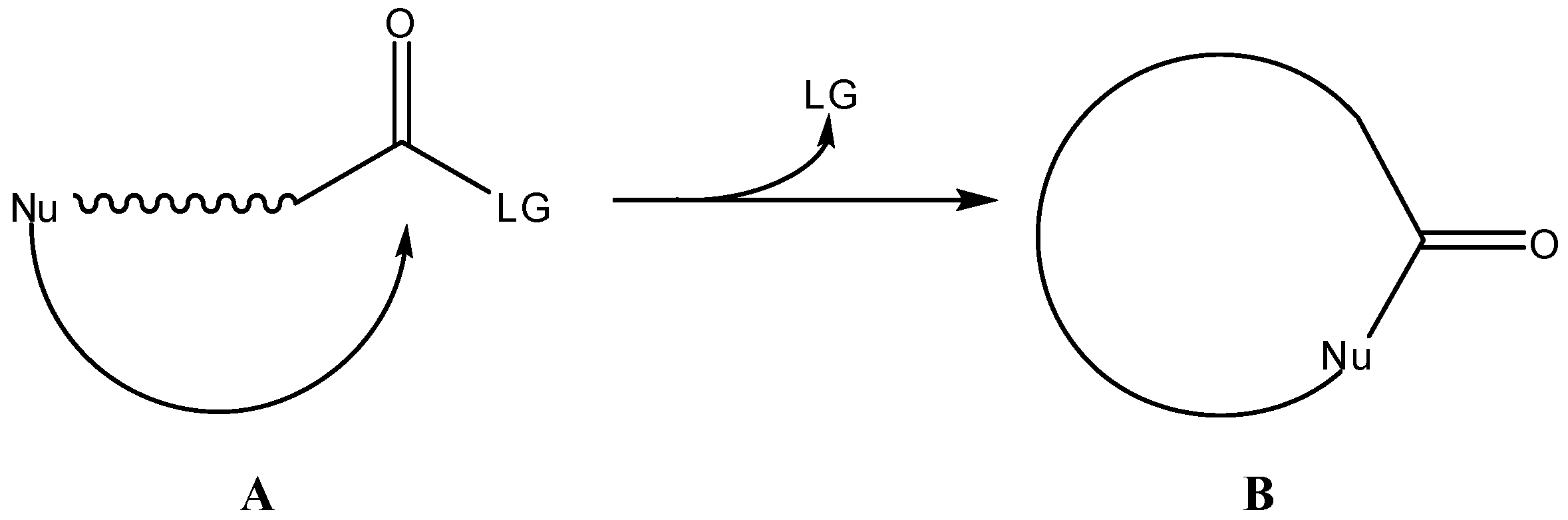
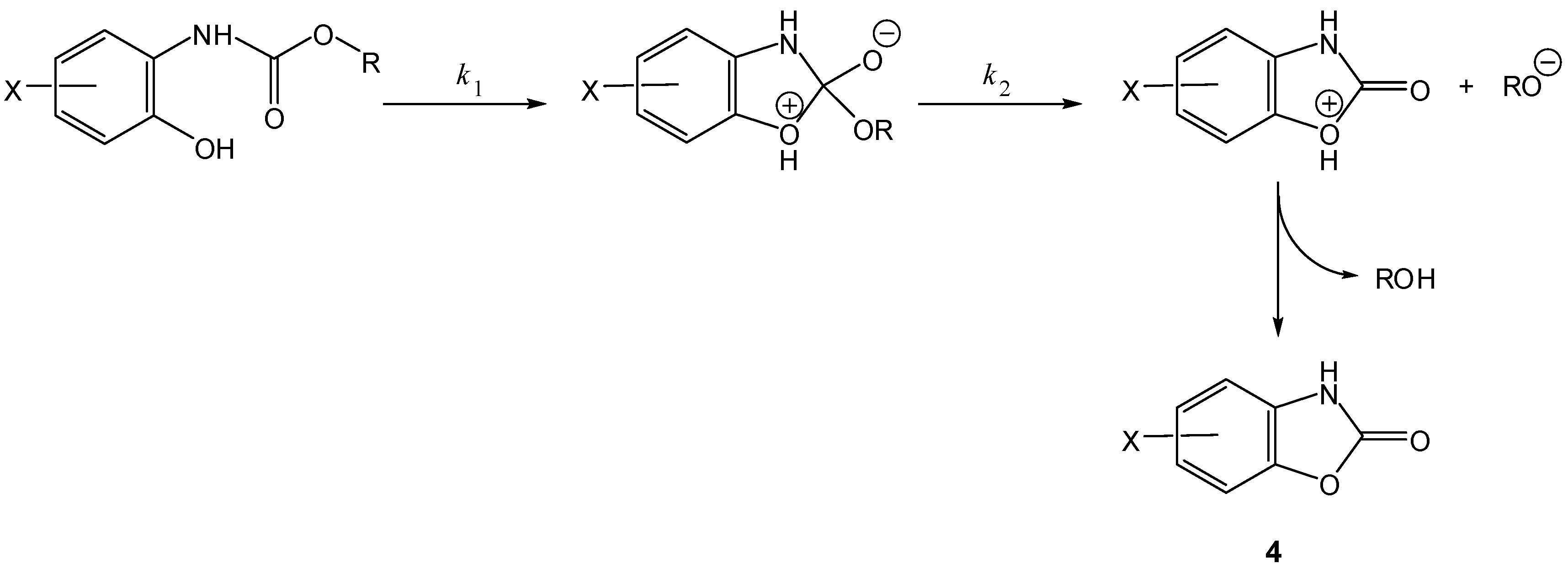
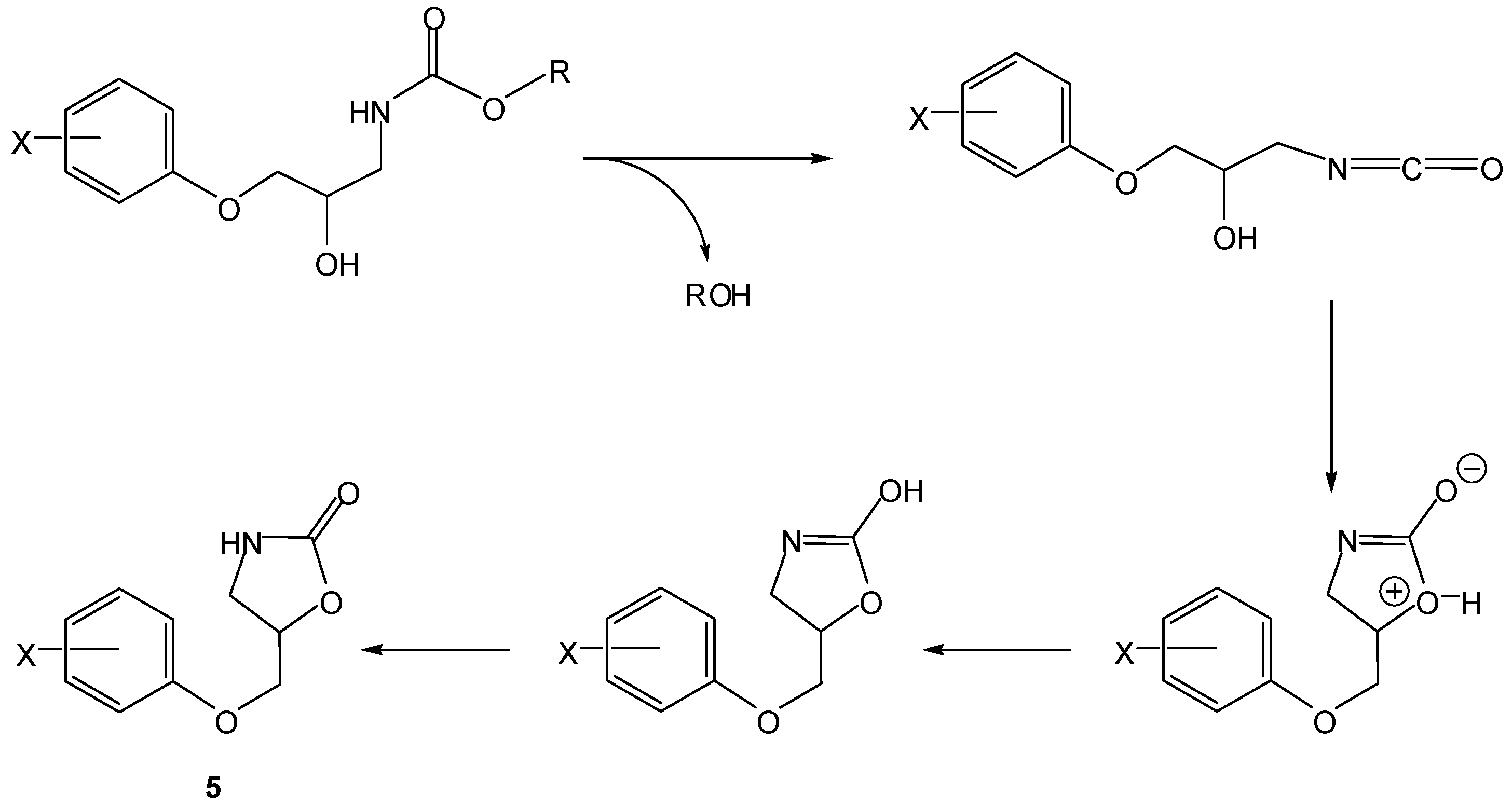
2. Active Drug as the Cyclic Product of Intramolecular Activation



3. Active Drug as the Leaving Group in the Cyclization-Elimination Reaction

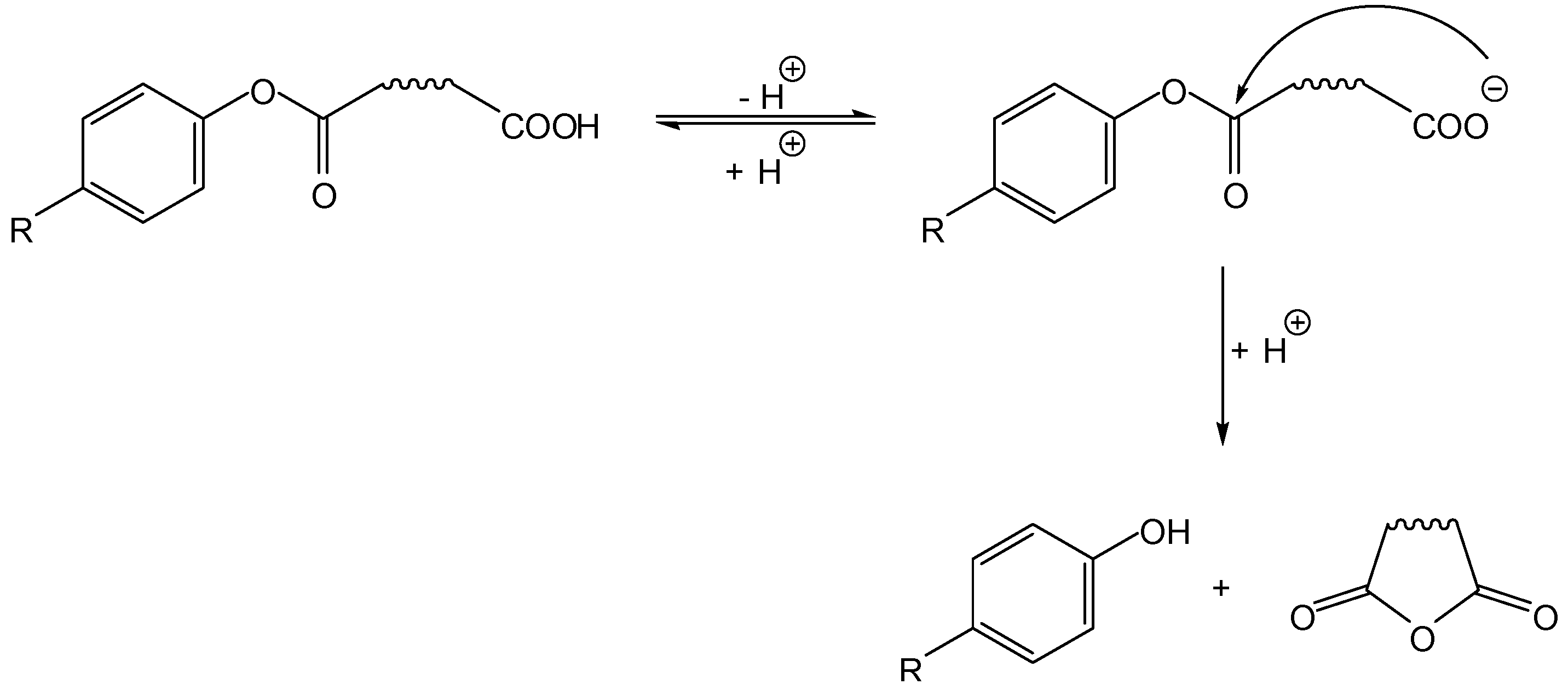
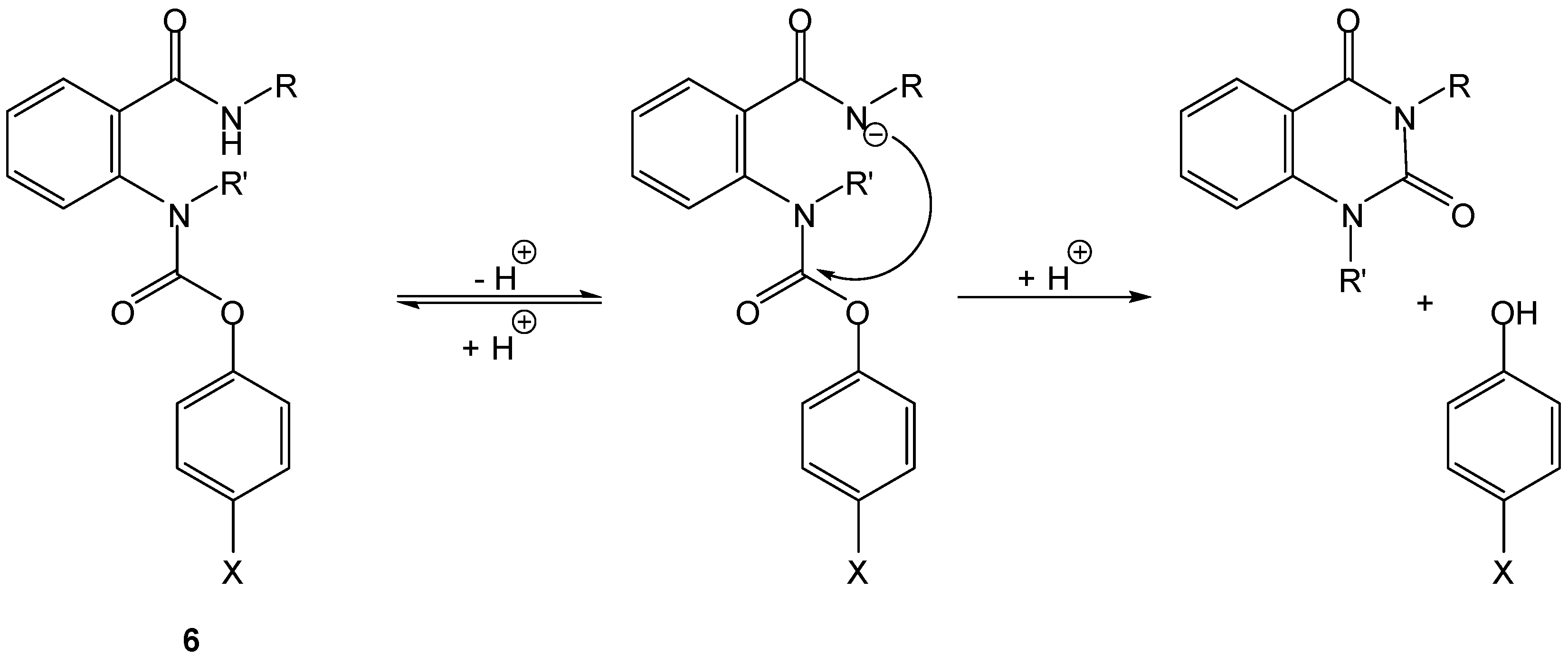
3.1. Cyclization-elimination involving an amido group


3.2. Cyclization-elimination involving an amino group
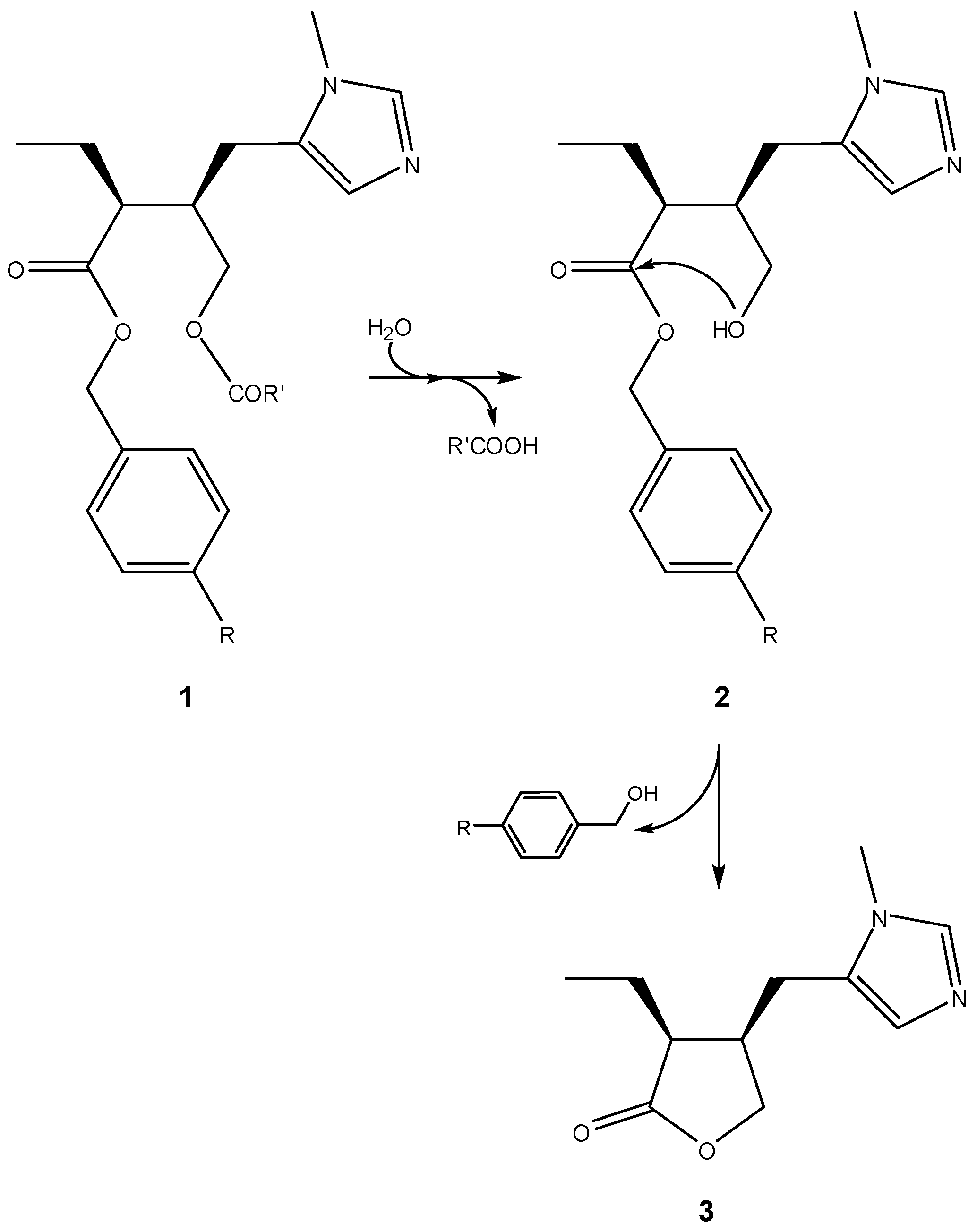
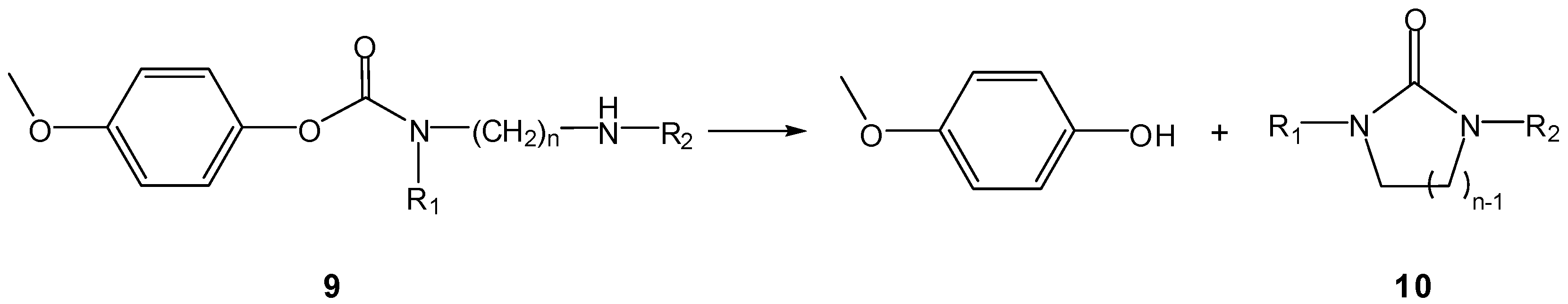
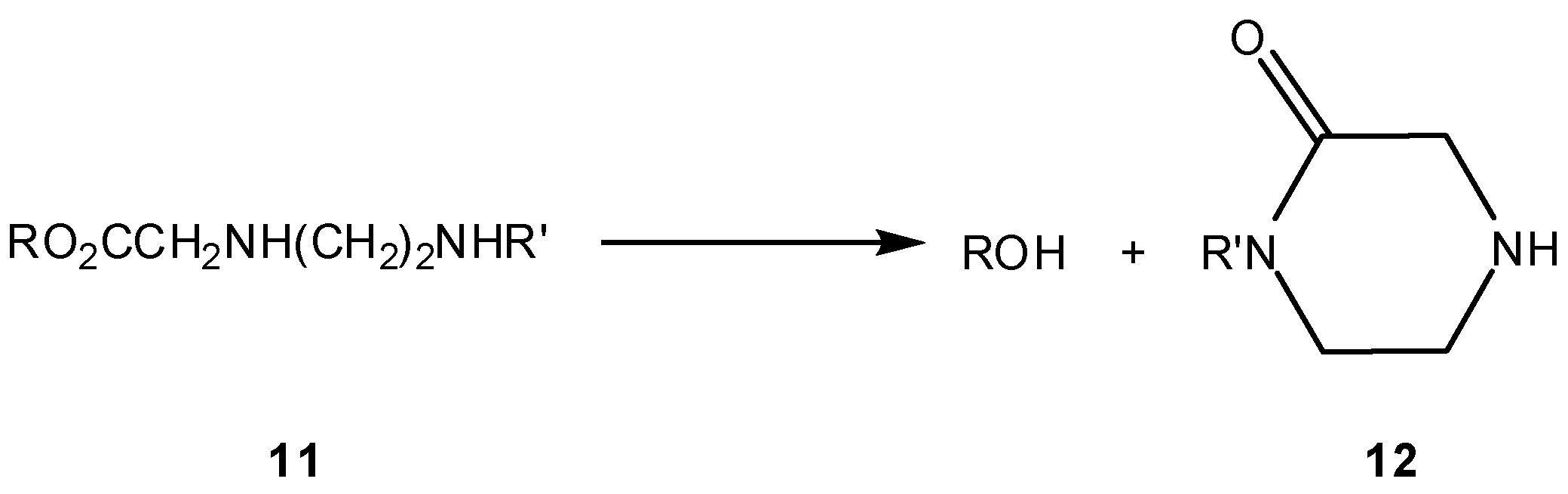
3.2.1. General basic amine carriers



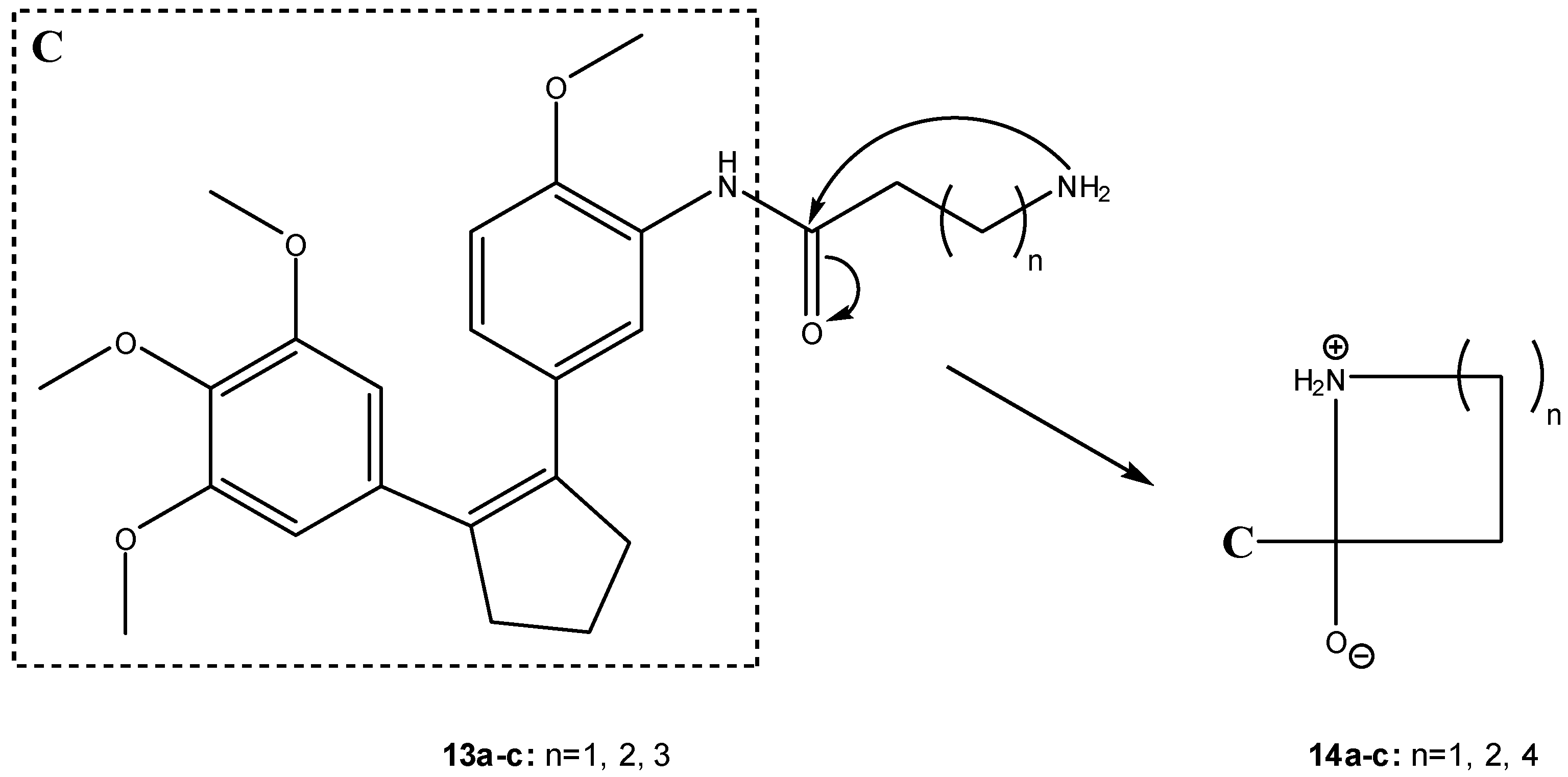
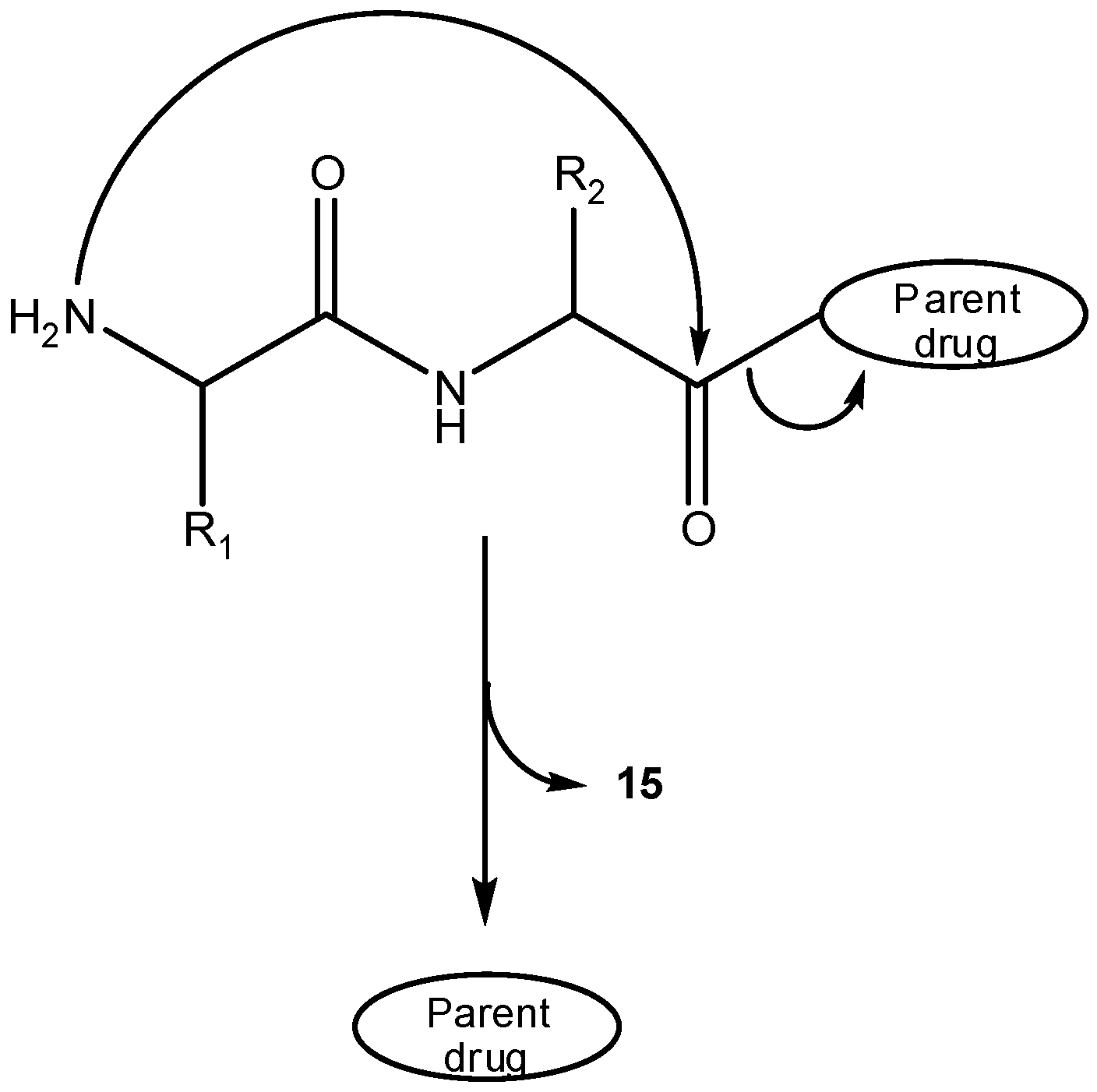
3.2.2. Peptide carriers
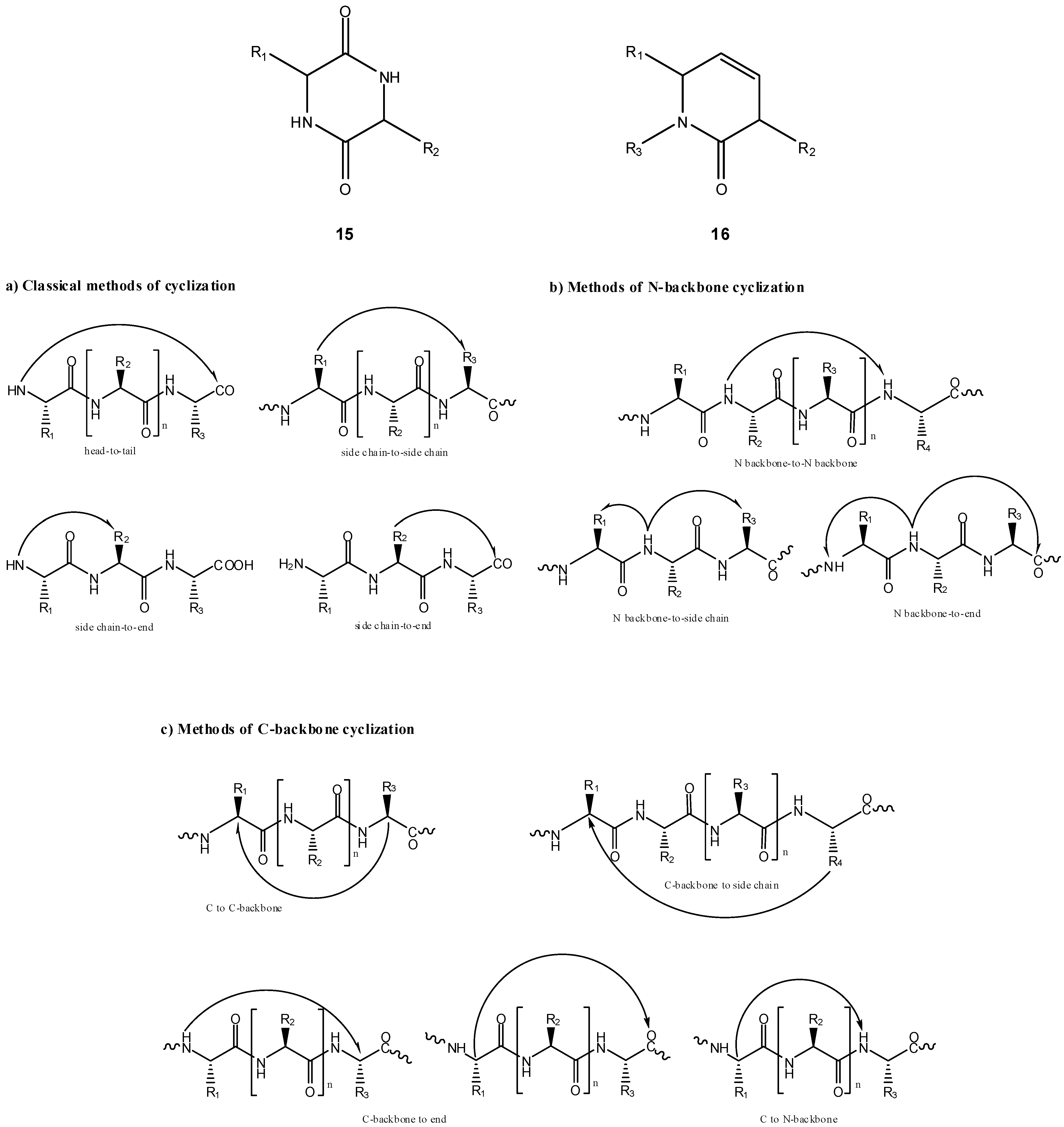
a) Peptide cyclization and prodrug design

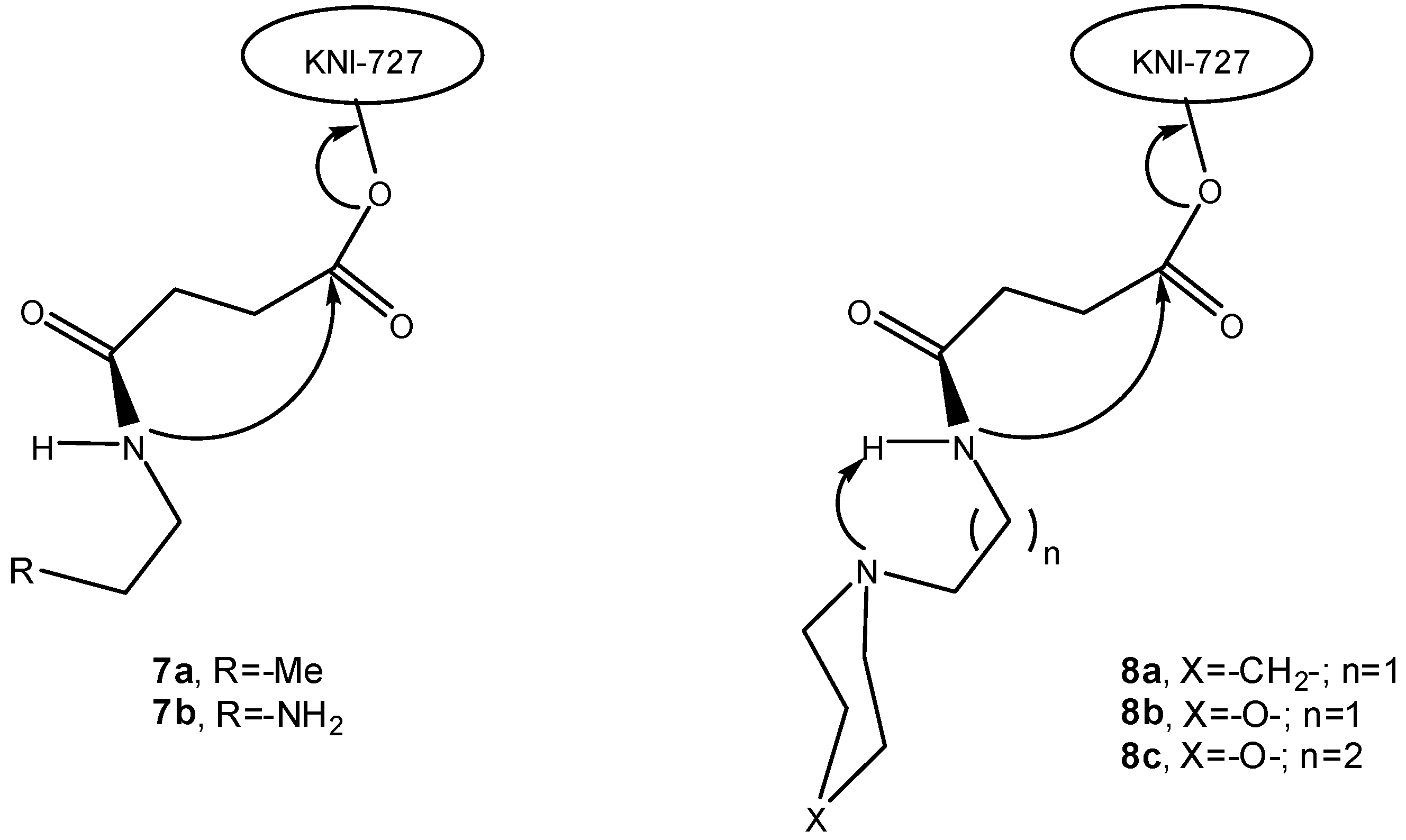{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}

b) Prodrug activation via DKP formation

4. Two-step Activation





5. Concluding Remarks
Acknowledgments
References and Notes
- Salama, N.N.; Fasano, A.; Thakar, M.; Eddington, N.D. The impact of ΔG on the oral bioavailability of low bioavailable therapeutic agents. J. Pharmacol. Exp. Ther. 2005, 312, 199–205. [Google Scholar]
- Lin, J.H.; Lu, A.Y.H. Role of pharmacokinetics and metabolism in drug discovery and development. Pharmacol. Rev. 1997, 49, 403–449. [Google Scholar]
- Han, H.K.; Amidon, G.L. Targeted prodrug design to optimize drug delivery. AAPS PharmSci. 2000, 2, E6. [Google Scholar]
- Sinkula, A.A.; Yalkowsky, S.H. Rationale for design of biologically reversible drug derivatives: prodrugs. J. Pharm. Sci. 1975, 64, 181–210. [Google Scholar]
- Yu, L.X.; Straughn, A.B.; Faustion, P.J.; Yang, Y.; Parekh, A.; Ciavarella, A.B.; Asafu-Adjaye, E.; Mehta, M.U.; Conner, D.P.; Lesko, L.J.; Hussain, A.S. The effect of food on the relative bioavailability of rapidly dissolving immediate-release solid oral products containing highly soluble drugs. Mol. Pharm. 2004, 1, 357–362. [Google Scholar]
- Shan, D.; Nicolaou, M.G.; Borchardt, R.T.; Wang, B. Prodrug strategies based on intramolecular cyclization reactions. J. Pharm. Sci. 1997, 86, 765–767. [Google Scholar]
- Testa, B.; Mayer, J.M. Design of intramolecularly activated prodrugs. Drug Metab. Rev. 1998, 30, 787–807. [Google Scholar]
- Wang, W.; Jiang, J.; Ballard, C.E.; Wang, B. Prodrug approaches to the improved delivery of peptide drugs. Curr. Pharm. Des. 1999, 5, 265–287. [Google Scholar]
- Vinšová, J.; Imramovský, A. Intramolekulární cycklizace využívané k uvolňování účinných látek z proléčiv. Chem. Listy 2005, 99, 21–29. [Google Scholar]
- Papot, S.; Tranoy, I.; Tillequin, F.; Florent, J.-C-; Gesson, J.-P. Design of selectively activated anticancer prodrugs: elimination and cyclization strategies. Curr. Med. Chem. – Anti-Cancer Agents 2002, 2, 155–185. [Google Scholar]
- Testa, B. Prodrug research: futile or fertile? Biochem. Pharmacol. 2004, 68, 2097–2106. [Google Scholar]
- Stella; Borchardt; Hageman; Oliyai; Maag; Tilley (Eds.) Prodrugs: challenges and rewards. Part 1 and 2; Springer-AAPS Press: New York, 2007; (the initial sections of Part 2 are particularly relevant in the present context).
- Bundgaard, H.; Falch, E.; Larsen, C.; Mesher, G.L.; Mikkelson, T. Pilocarpic acid esters as novel sequentially labile pilocarpine prodrugs for improved ocular delivery. J. Med. Chem. 1985, 28, 979–981. [Google Scholar]
- Song, L.; Bevins, R.; Anderson, B.D. Kinetics and mechanisms of activation of α-amino acid ester prodrugs of camptothecins. J. Med. Chem. 2006, 49, 4344–4355. [Google Scholar]
- Bom, D.; Curran, D.P.; Kruszewski, S.; Zimmer, S.G.; Strode, J.T.; Kohlhagen, G.; Du, W.; Chavan, A.J.; Fradey, K.A.; Bingcang, A.L.; Latus, L.J.; Pommier, Y.; Burke, T.G. The novel silatecan 7-tert-butyldimethylsilyl-10-hydroxycamptothecin displays high lipophilicity, improved human blood stability, and potent anticancer activity. J. Med. Chem. 2000, 43, 3970–3980. [Google Scholar]
- Vigroux, A.; Bergon, M.; Zedde, C. Cyclization-activated prodrugs: N-(substituted 2 hydroxyphenyl and 2-hydroxypropyl)carbamates based on ring-opened derivatives of active benzoxazolones and oxazolidinones as mutual prodrugs of acetaminophen. J. Med. Chem. 1995, 38, 3983–3994. [Google Scholar]
- Fredholt, K.; Mork, N.; Begtrup, M. Hemiesters of aliphatic dicarboxylic acids as cyclization-activated prodrug forms for protecting phenols against first-pass metabolism. Int. J. Pharm. 1995, 123, 209–216. [Google Scholar]
- Bruice, T.C.; Pandit, U.K. The effect of germinal substitution, ring size and rotamer distribution on the intramolecular nucleophilic catalysis of the hydrolysis of monophenyl esters of dibasic acids and the solvolysis of the intermediate anhydrides. J. Am. Chem. Soc. 1960, 82, 5858–5865. [Google Scholar]
- Thomsen, K.F.; Bundgaard, H. Cyclization-activated phenyl carbamate prodrug forms for protecting phenols against first-pass metabolism. Int. J. Pharm. 1993, 91, 39–49. [Google Scholar]
- Matsumoto, H.; Sohma, Y.; Kimura, T.; Hayashi, Y.; Kiso, Y. Controlled drug release: new water-soluble prodrugs of an HIV protease inhibitor. Bioorg. Med. Chem. Lett. 2001, 11, 605–609. [Google Scholar]
- Sohma, Y.; Hayashi, Y.; Ito, T.; Matsumoto, H.; Kimura, T.; Kiso, Y. Development of water-soluble prodrugs of HIV-1 protease inhibitor KNI-727: importance of the conversion time for higher gastrointestinal absorption of prodrugs based on spontaneous chemical cleavage. J. Med. Chem. 2003, 46, 4124–4135. [Google Scholar]
- Matsumoto, H.; Kimura, T.; Hamawaki, T.; Kumagai, A.; Goto, T.; Sano, K.; Hayashi, Y.; Kiso, Y. Design, synthesis, and biological evaluation of anti-HIV double-drugs: conjugates of HIV protease inhibitors with a reverse transcriptase inhibitor through spontaneously cleavable linkers. Bioorg. Med. Chem. 2001, 9, 1589–1600. [Google Scholar]
- Santos, C.; Mateus, M.L.; Santos, A.P.; Moreira, R.; Oliveira, E.; Gomes, P. Cyclization-activated prodrugs. Synthesis, reactivity and toxicity of dipeptide esters of paracetamol. Bioorg. Med. Chem. Lett. 2005, 15, 1595–1598. [Google Scholar]
- Gomes, P.; Gomes, J.R.B.; Rodrigues, M.; Moreira, R. Amino acids as selective sulfonamide acylating agents. Tetrahedron 2003, 59, 7473–7480. [Google Scholar]
- Saari, W.S.; Schwering, J.E.; Lyle, P.A.; Smith, S.J.; Engelhardt, E.L. Cyclization-activated prodrugs. Basic carbamates of 4-hydroxyanisole. J. Med. Chem. 1990, 33, 97–101. [Google Scholar]
- Saari, W.S.; Schwering, J.E.; Lyle, P.A.; Smith, S.J.; Engelhardt, E.L. Cyclization-activated prodrugs. Basic esters of 5-bromo-2’-deoxyuridine. J. Med. Chem. 1990, 33, 2590–2595. [Google Scholar] [CrossRef]
- Nam, N.H.; Kim, Y.; You, Y.J.; Hong, D.H.; Kim, H.M.; Ahn, B.Z. Water soluble prodrugs of the antitumor agent 3-[(3-amino-4-methoxy)phenyl]-2-(3,4,5-trimethoxyphenyl)cyclopent-2-ene-1-one. Bioorg. Med. Chem. 2003, 11, 1021–1029. [Google Scholar]
- Besser, D.; Müller, B.; Agricola, I.; Reissmann, S. Synthesis of differentially protected N-acylated reduced pseudodipeptides as building units for backbone cyclic peptides. J. Peptide Sci. 2000, 6, 130–138. [Google Scholar]
- Besser, D.; Müller, B.; Kleinwächter, P.; Greiner, G.; Seyfarth, L.; Steinmetzer, T.; Arad, O.; Reissmann, S. Synthesis and characterization of octapeptide somatostatin analogues with backbone cyclization: comparison of different strategies, biological activities and enzymatic stabilities. J. Prakt. Chem. 2000, 342, 537–545. [Google Scholar]
- Kaul, R.; Surprenant, S.; Lubell, W.D. Systematic study of the synthesis of macrocyclic dipeptide β-turn mimics possessing 8-, 9-, and 10- membered rings by ring-closing metathesis. J. Org. Chem. 2005, 70, 3838–3844. [Google Scholar]
- Poteau, R.; Trinquier, G. All-cis cyclic peptides. J. Am. Chem. Soc. 2005, 127, 13875–13889. [Google Scholar]
- Nielsen, T.E.; Quement, S.L.; Meldal, M. Solid-phase synthesis of bicyclic dipeptide mimetics by intramolecular cyclization of alcohols, thiols, amines, and amides with N-acyliminium intermediates. Org. Lett. 2005, 7, 3601–3604. [Google Scholar]
- Liederer, B.M.; Fuchs, T.; Velde, D.V.; Siahaan, T.J.; Borchardt, R.T. Effects of amino acid chirality and the chemical linker on the cell permeation characteristics of cyclic prodrugs of opioid peptides. J. Med. Chem. 2006, 49, 1261–1270. [Google Scholar]
- Keramida, M.; Tselios, T.; Mantzourani, E.; Papazisis, K.; Mavromoustakos, T.; Klaussen, C.; Agelis, G.; Deraos, S.; Friligou, I.; Habibi, H.; Matsoukas, J. Design, synthesis, and molecular modeling of a novel amide-linked cyclic GnRH analogue cyclo(4-9)[Lys4,d-Trp6,Glu9]GnRH: stimulation of gonadotropin gene expression. J. Med. Chem. 2006, 49, 105–110. [Google Scholar]
- Flora, D.; Mo, H.; Mayer, J.P.; Khan, M.A.; Yan, L.Z. Detection and control of aspartimide formation in the synthesis of cyclic peptides. Bioorg. Med. Chem. Lett. 2005, 15, 1065–1068. [Google Scholar]
- Che, Y.; Marshall, G.R. Engineering cyclic tetrapeptides containing chimeric amino acids as preferred reverse-turn scaffolds. J. Med. Chem. 2006, 49, 111–124. [Google Scholar]
- Arnott, G.; Clayden, J.; Hamilton, S.D. Azabicyclic amino acids by stereoselective dearomatizing cyclization of the enolates of N-nicotinoyl glycine derivatives. Org. Lett. 2006, 8, 5325–5328. [Google Scholar]
- Norgren, A.S.; Büttner, F.; Prabpai, S.; Kongsaeree, P.; Arvidsson, P.I. β2-aminoacids in the design of conformationally homogeneous cyclo-peptide scaffolds. J. Org. Chem. 2006, 71, 6814–6821. [Google Scholar]
- Gilon, C.; Halle, D.; Chorev, M.; Selinger, Z.; Byk, G. Backbone cyclization: a new conformational constraint on peptides. Biopolymers 1991, 31, 745–750. [Google Scholar]
- Al-Obeidi, F.; Castrucci, A.M.L.; Hadley, M.E.; Hruby, V.J. Potent and prolonged acting cyclic lactam analogues off α-melanotropin: design based on molecular dynamics. J. Med. Chem. 1989, 32, 2555–2561. [Google Scholar]
- Charpentier, B.; Dor, A.; Roy, P.; England, P.; Pham, H.; Durieux, C.; Roques, B.P. Synthesis and binding affinities of cyclic and related linear analogues of CCK8 selective for central receptors. J. Med. Chem. 1989, 32, 1184–1190. [Google Scholar]
- Reissmann, S.; Imhof, D. Development of conformationally restricted analogues of bradykinin and somatostain using constrained amino acids and different types of cyclization. Curr. Med. Chem. 2004, 11, 2823–2844. [Google Scholar]
- Davies, J.S. The cyclization of peptides and depsipeptides. J. Peptide Sci. 2003, 9, 471–501. [Google Scholar]
- Bray, A.M.; Maeji, N.J.; Valerio, R.M.; Campbell, R.A.; Geysen, H.M. Direct cleavage of peptides from a solid support into aqueous buffer. Applications in simultaneous multiple peptide synthesis. J. Org. Chem. 1991, 56, 6659–6666. [Google Scholar]
- Capasso, S.; Vergara, A.; Mozzarella, l. Mechanism of 2,5-dioxopiperazine formation. J. Am. Chem. Soc. 1998, 120, 1990–1995. [Google Scholar]
- Besada, P.; Mamedova, L.; Thomas, C.J.; Costanzi, S.; Jacobson, K.A. Design and synthesis of new bicyclic diketopiperazines as scaffolds for receptor probes of structurally diverse functionality. Org. Biomol. Chem. 2005, 3, 2016–2025. [Google Scholar]
- Moyroud, J.; Gelin, J.; Chêne, A.; Mortier, J. Synthèse d’analogues structuraux de Thaxtomines, phytotoxines responsables de la gale de la pomme de terre. Tetrahedron 1996, 52, 8525–8534. [Google Scholar]
- Szardenings, A.K.; Burkoth, T.S. A simple procedure for the solid phase synthesis of diketopiperazine and diketomorpholine derivatives. Tetrahedron 1997, 53, 6573–6593. [Google Scholar]
- Sollis, S.L. Short and novel stereospecific synthesis of trisubstituted 2,5-diketopiperazines. J. Org. Chem. 2005, 70, 4735–4740. [Google Scholar]
- Niida, A.; Tanigaki, H.; Inokuchi, E.; Sasaki, Y.; Oishi, S.; Ohno, H.; Tamamura, H.; Wang, Z.; Peiper, S.C.; Kitaura, K.; Otaka, A.; Fujii, N. Stereoselective synthesis of 3,6-disubstituted-3,6-dihydropyridun-2-ones as potential diketopiperazine mimetics using organocopper-mediated anti-Sn2’ reactions and their use in the preparation of low-molecule CXCR4 antagonists. J. Org. Chem. 2006, 71, 3942–3951. [Google Scholar]
- Kanoh, K.; Kohno, S.; Katada, J.; Takahashi, J.; Uno, I.; Hayashi, Y. Synthesis and biological activies of phenylahistin derivatives. Bioorg. Med. Chem. 1999, 7, 1451–1457. [Google Scholar]
- Donkor, I.O.; Sanders, M.L. Synthesis of a reported calpain inhibitor isolated from Streptomyces griseus. Bioorg. Med. Chem. Lett. 2001, 11, 2647–2649. [Google Scholar]
- Nam, N.H.; Ye, G.; Sun, G.; Parang, K. Conformationally constrained peptide analogues of pTyr-Glu-Glu-Ile as inhibitors of the Src SH2 domain binding. J. Med. Chem. 2004, 47, 3131–3141. [Google Scholar]
- Borthwick, A.D.; Davies, D.E.; Exall, A.M.; Livermore, D.G.; Sollis, S.L.; Nerozzi, F.; Allen, M.J.; Perren, M.; Shabbir, S.S.; Woollard, P.M.; Wyatt, P.G. 2,5-Diketopiperazines as potent, selective, and orally bioavailable oxytocin antagonists. 2. Synthesis, chirality, and pharmacokinetics. J. Med. Chem. 2005, 48, 6956–6969. [Google Scholar] [CrossRef]
- Gomes, P.; Gomes, J.R.B.; Rodrigues, M.; Moreira, R. Amino acids as selective sulfonamide acylating agents. Tetrahedron 2003, 59, 7473–7480. [Google Scholar]
- Santos, C.; Moreira, R.; Gomes, P. Cyclization-activated prodrugs: dipeptide esters of paracetamol. In Peptides 2004; M. Flegel, M., Fridkin, C., Gilon e, J., Slaninova, Eds.; Kenes International: Tel A’viv, 2005. [Google Scholar]
- Brady, S.F.; Pawluczyk, J.M.; Lumma, P.K.; Feng, D.M.; Wai, J.M.; Jones, R.; DeFeo-Jones, D.; Wong, B.K.; Miller-Stein, C.; lin, J.H.; Oliff, A.; Freidinger, R.M.; Garsky, V.M. Design and synsthesis of a pro-drug of vinblastine targeted at treatment of prostate cancer with enhanced efficacy and reduced systemic toxicity. J. Med. Chem. 2002, 45, 4706–4715. [Google Scholar]
- Purdie, J. E.; Benoiton, N. L. Piperazinedione formation from esters of dipeptides containing glycine, alanine, and sarcosine: the kinetics in aqueous solution. J. Chem. Soc. Perkin II 1973, 13, 1845–1852. [Google Scholar]
- Meresaar, U.; Ågren, A. Hydrolysis of protolytic esters VI. Alkaline hydrolysis and ring closure of glycylglycine ethyl ester. Acta Pharm. Suec. 1968, 5, 85–94. [Google Scholar]
- Jensen, E.; Bundgaard, H. Peptide esters as water-soluble prodrugs for hydroxyl containing agents: chemical stability and enzymatic hydrolysis of benzyl esters of glycine, diglycine and triglycine. Int. J. Pharm. 1991, 71, 117–125. [Google Scholar]
- Larsen, S.W.; Ankersen, M.; Larsen, C. Kinetics of degradation and oil solubility of ester prodrug of a model dipeptide (Gly-Phe). Eur. J. Pharm. Sci. 2004, 22, 399–408. [Google Scholar]
- Goolcharran, C.; Borchardt, R. T. Kinetics of diketopiperazine formation using model peptides. J. Pharm. Sci. 1998, 87, 283–288. [Google Scholar]
- Shan, D.; Nicolaou, M.G.; Borchardt, R.T.; Wang, B. Prodrug strategies based on intramolecular cyclization reactions. J. Pharm. Sci. 1997, 86, 765. [Google Scholar]
- Bertoloni, A.; Ferrari, A.; Ottani, A.; Guerzoni, S.; Tacchi, R.; Leone, S. Paracetamol: new vistas of an old drug. CNS Drug Rev. 2006, 12, 250–275. [Google Scholar]
- Lloyd-Williams, P.; Albericio, F.; Giralt, E. Chemical Approaches to the Synthesis of Peptides and Proteins.; CRC Press: Boca Raton, 1997; p. 60. [Google Scholar]
- Wipf, P.; Li, W.L.; Adeyeye, C.M.; Rusnak, J.M.; Lazo, J.S. Synthesis of chemoreversible prodrugs of ara-C with variable time-release profiles. Biological evaluation of their apoptotic activity. Bioorg. Med. Chem. 1996, 4, 1585–1596. [Google Scholar]
- Hamel, A.R.; Hubler, F.; Carrupt, A.; Wenger, R.M.; Mutter, M. Cyclosporin A prodrugs: design, synthesis and biophysical properties. J. Peptide Res. 2004, 63, 147–154. [Google Scholar]
- Wei, Y.; Pei, D. Activation of antibacterial prodrugs by peptide deformylase. Bioorg, Med. Chem. Lett. 2000, 10, 1073–1076. [Google Scholar] [CrossRef]
- Jackson, D.Y.; Burnier, J.P.; Wells, J.A. Enzymatic cyclization of linear peptide esters using subtiligase. J. Am. Chem. Soc. 1995, 117, 819–820. [Google Scholar]
- Kohchi, Y.; Hattori, K.; Oikawa, N.; Mizuguchi, E.; Isshiki, Y.; Aso, K.; Yoshinari, K.; Shirai, H.; Miwa, M.; Inagaki, Y.; Ura, M.; Ogawa, K.; Okabe, H.; Ishitsuka, H.; Shimma, N. Design and synthesis of novel prodrugs of 2’-deoxy-2’-methylidenecytidine activated by membrane dipeptidase overexpressed in tumor tissues. Bioorg. Med. Chem. Lett. 2007, 17, 2241–2245. [Google Scholar]
- Atwell, G.; Sykes, B.M.; O’Connor, C.J.; Denny, W.A. Relationships between structure and kinetics of cyclization of 2-aminoaryl amides: potential prodrugs of cyclization-activated aromatic mustards. J. Med. Chem. 1994, 37, 371–380. [Google Scholar]
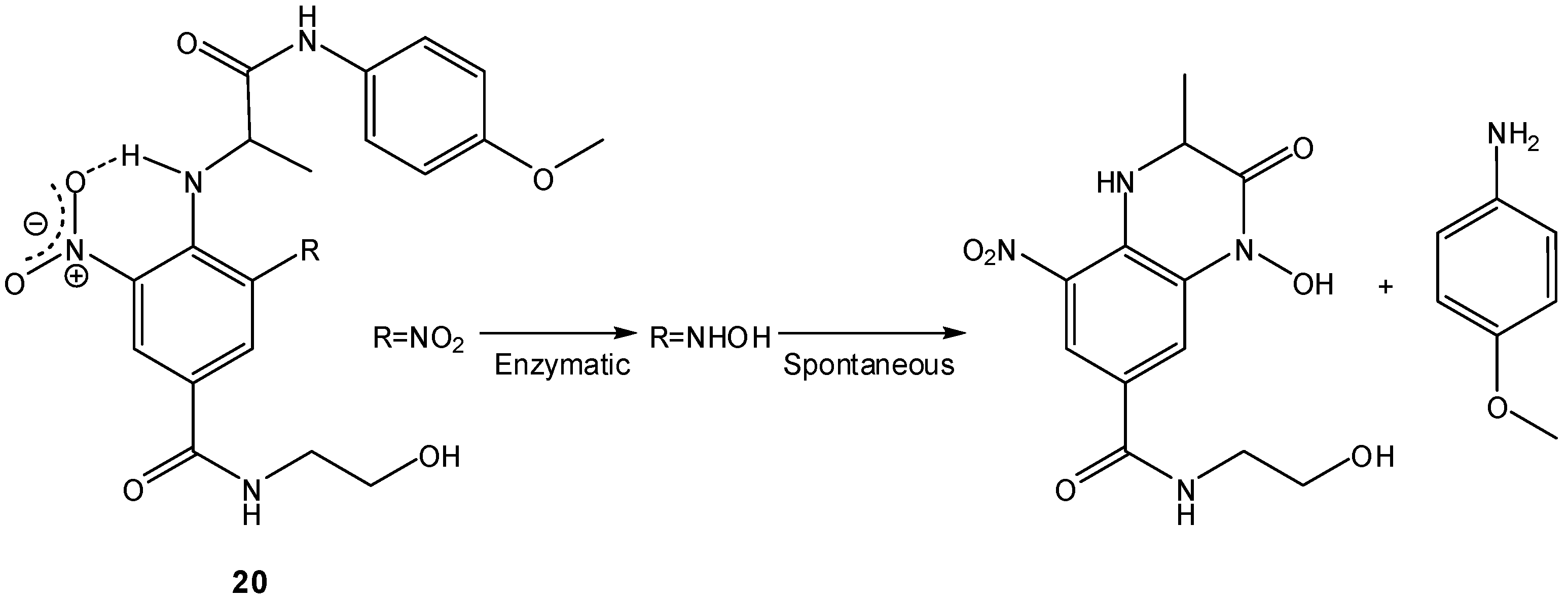
- Sykes, B.M.; Atwell, G.J.; Hogg, A.; Wilson, W.R.; O’Connor, C.J.; Denny, W.A. N-substituted 2-(2,6-dinitrophenylamino)propanamides: novel prodrugs that release a primary amine via nitroreduction and intramolecular cyclization. J. Med. Chem. 1999, 4, 346–355. [Google Scholar]
- Kirk, K.L.; Cohen, L.A. Intramolecular aminolysis of amides. Effects of electronic variation in the attacking and leaving groups. J. Am. Chem. Soc. 1972, 94, 8142–8147. [Google Scholar]
- Liu, B.; Hu, L. 5'-(2-nitrophenylalkanoyl)-2'-deoxy-5-fluorouridines as prodrugs of FUDR for reductive activation. Bioorg. Med. Chem. 2003, 11, 3889–3899. [Google Scholar]
- Milstien, S.; Cohen, L.A. Stereopopulation control. I. Rate enhancement in the lactonizations of o-hydroxyhydrocinnamic acids. J. Am. Chem. Soc. 1972, 94, 9158–9165. [Google Scholar] [CrossRef]
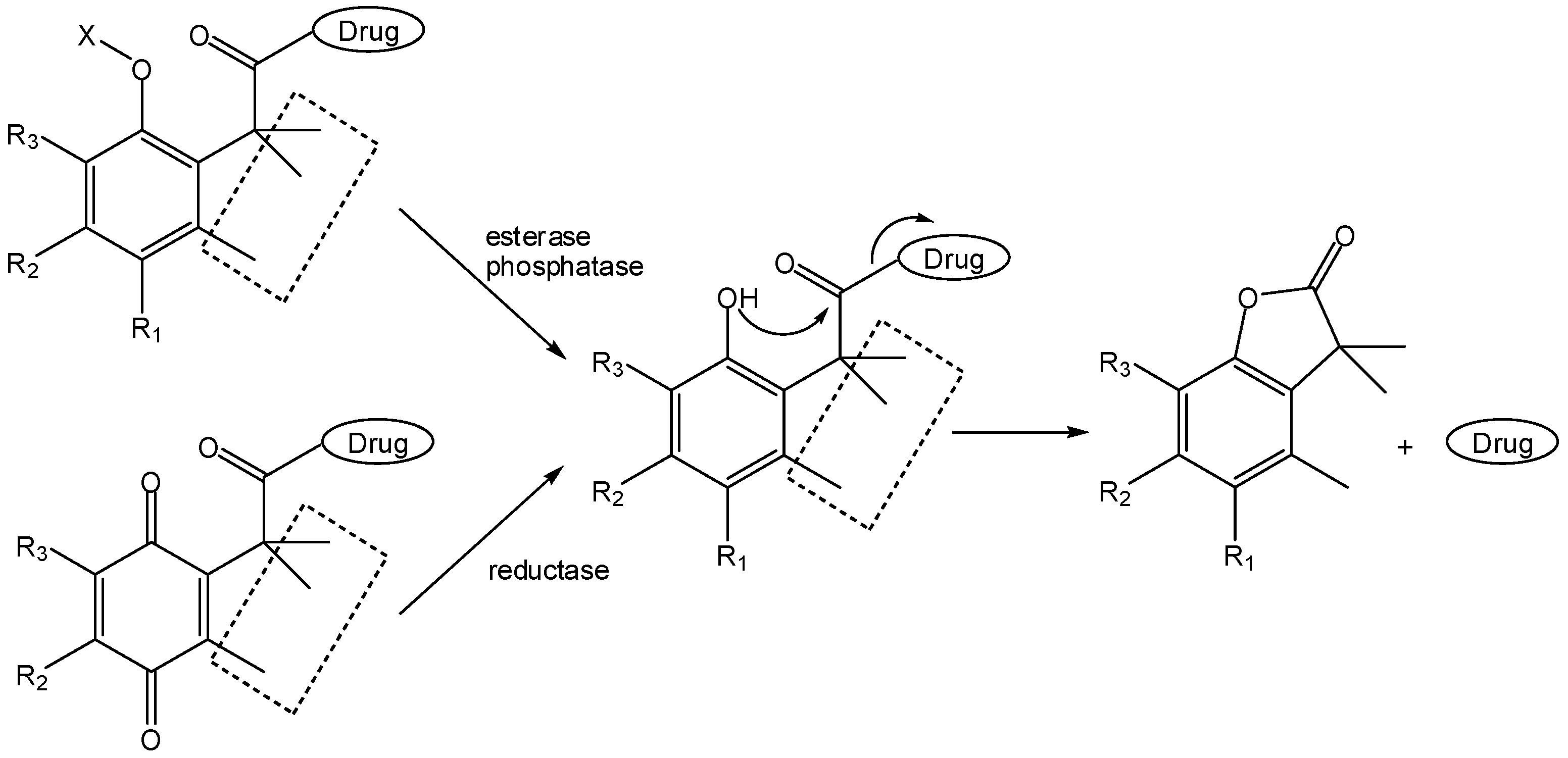
- Amsberry, K.L.; Borchardt, R.T. Amine prodrugs which utilize hydroxy amide lactonization. I. A potential redox-sensitive amide prodrug. Pharm. Res. 1991, 8, 323–330. [Google Scholar] [CrossRef]
- Wolfe, J.L.; Vander Velde, D.G.; Borchardt, R.T. Facilitated intramolecular conjugate addition of N-(p- methoxypheny1)-3-(3’,6’-dioxo-2’,4’-dimethylcyclohexa-1’,4’-dienyl)-3,3-dimethylpro-pionamide. 1. Product characterization. J. Org. Chem. 1992, 57, 6138–6142. [Google Scholar]
- Nicolaou, M.G.; Wolfe, J.L.; Schowen, R.L.; Borchardt, R.T. Facilitated intramolecular conjugate addition of amides of 3-(3’,6’-dioxo-2’,4’-dimethyl-1’,4’-cyclohexadienyl)-3,3-dimethylpropionic acid. 2. Kinetics of degradation. J. Org. Chem. 1996, 61, 6633–6638. [Google Scholar]
- Amsberry, K.L.; Gerstenberger, E.; Borchardt, R.T. Amine prodrugs which utilize hydroxy amide lactonization. II. A potential esterase-sensitive amide prodrug. Pharm. Res. 1991, 8, 455–461. [Google Scholar]
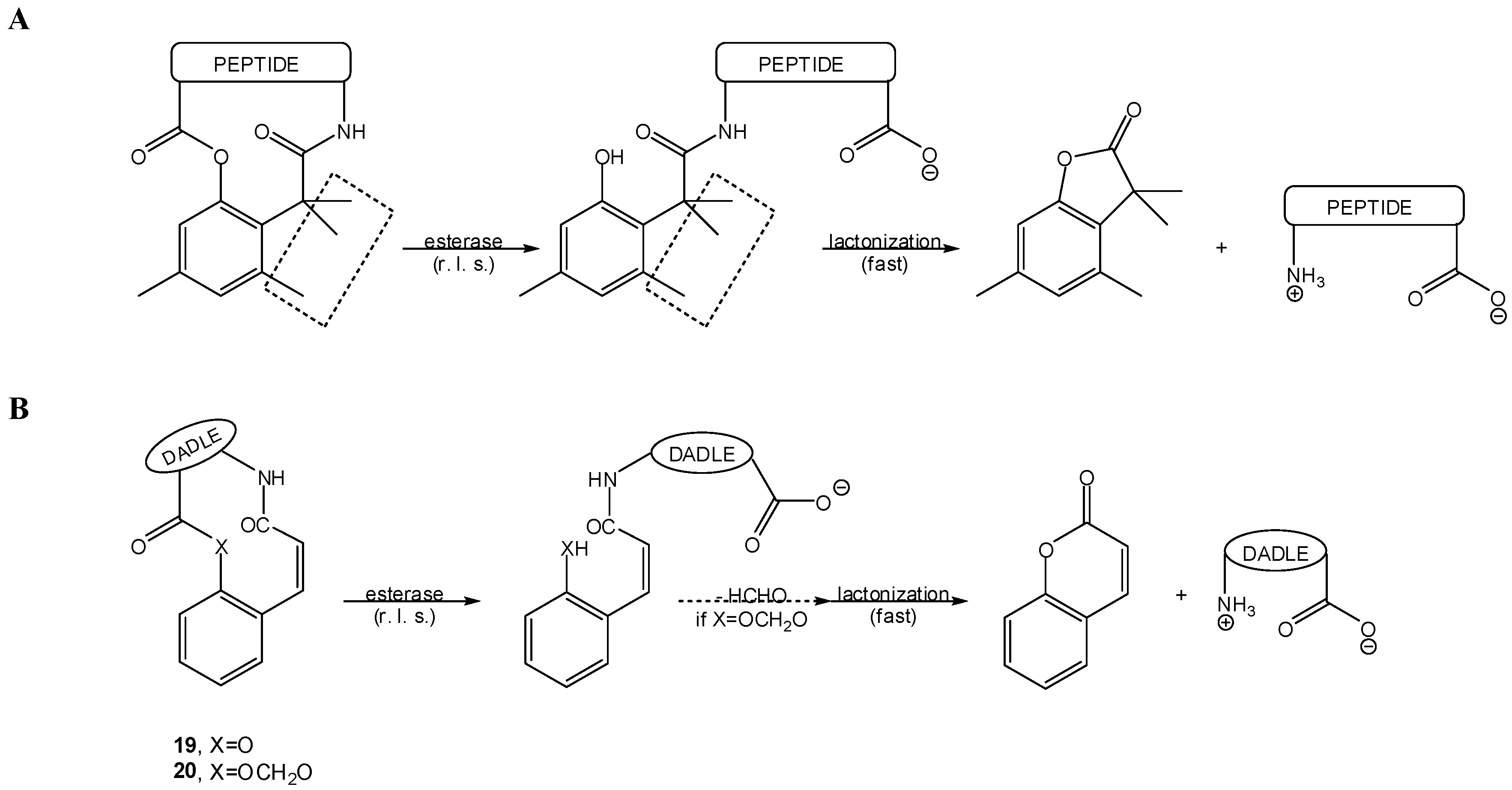
- Wang, B.; Gangwar, S.; Pauletti, G.M.; Siahaan, T.J.; Borchardt, R.T. Synthesis of a novel esterase-sensitive cyclic prodrug system for peptides that utilizes a “trimethyl lock”-facilitated lactonization reaction. J. Org. Chem. 1997, 62, 1363–1367. [Google Scholar]
- Pauletti, G.M.; Gangwar, S.; Wang, B.; Borchardt, R.T. Esterase-sensitive cyclic prodrugs of peptides: evaluation of a phenylpropionic acid promoiety in a model hexapeptide. Pharm. Res. 1997, 14, 11–17. [Google Scholar]
- Nicolaou, M.G.; Yuan, C.-S.; Borchardt, R.T. Phosphate prodrugs for amines utilizing a fast intramolecular hydroxy amide lactonization. J. Org. Chem. 1996, 61, 8636–8641. [Google Scholar]
- Hershfield, R.; Schmir, G.L. Lactonization of coumarinic acids. Kinetic evidence for three species of the tetrahedral intermediate. J. Am. Chem. Soc. 1973, 95, 8032–8040. [Google Scholar]
- Wang, B.; Zhang, H.; Wang, W. Chemical feasibility studies of a potential coumarin-based prodrug system. Bioorg. Med. Chem. Lett. 1996, 6, 945–950. [Google Scholar]
- Wang, B.; Wang, W.; Zhang, H.; Shan, D.; Smith, T.D. Coumarin-based prodrugs 2. Synthesis and bioreversibility studies of an esterase-sensitive cyclic prodrug of dadle, an opioid peptide. Bioorg. Med. Chem. Lett. 1996, 6, 2823–2826. [Google Scholar]
- Ouyang, H.; Borchardt, R.T.; Siahaan, T.J.; Vander Velde, D.G. Synthesis and conformational analysis of a coumarinic acid-based cyclic prodrug of an opioid peptide with modified sensitivity to esterase-catalyzed bioconversion. J. Peptide Res. 2002, 59, 183–195. [Google Scholar]
- Tang, F.; Borchardt, R.T. Characterization of the efflux transporter(s) responsible for restricting intestinal mucosa permeation of an acyloxyalkoxy-based cyclic prodrug of the opioid peptide DADLE. Pharm. Res. 2002, 19, 780–786. [Google Scholar]
- Tang, F.; Borchardt, R.T. Characterization of the efflux transporter(s) responsible for restricting intestinal mucosa permeation of a coumarinic acid-based cyclic prodrug of the opioid peptide DADLE. Pharm. Res. 2002, 19, 787–793. [Google Scholar]
- Yang, J.Z.; Chen, W.; Borchardt, R.T. In vitro stability and in vivo pharmacokinetic studies of a model opioid peptide, H-Tyr-D-Ala-Gly-Phe-D-Leu-OH (DADLE), and its cyclic prodrugs. J. Pharmacol. Exper. Ther. 2002, 303, 840–848. [Google Scholar]
- Chen, W.; Yang, J.Z.; Andersen, R.; Nielsen, L.H.; Borchardt, R.T. Evaluation of the permeation characteristics of a model opioid peptide, H-Tyr-D-Ala-Gly-Phe-D-Leu-OH (DADLE), and its cyclic prodrugs across the blood-brain barrier using an in situ perfused rat brain model. J. Pharmacol. Exper. Ther. 2002, 303, 849–857. [Google Scholar]
- Liederer, B.M.; Borchardt, R.T. Stability of oxymethyl-modified coumarinic acid cyclic prodrugs of diastereomeric opioid peptides in biological media from various animal species including human. J. Pharm. Sci. 2005, 94, 2198–2206. [Google Scholar]
- Camenish, G.P.; Wang, W.; Wang, B.; Borchardt, R.T. A comparison of the bioconverstion rates and the Caco-2 cell permeation characteristics of coumarin-based cyclic prodrugs and methylester-based linear prodrugs of RGD peptidomimetics. Pharm. Res. 1998, 15, 1174–1181. [Google Scholar]
- Dillon, M.P.; Cai, H.; Maag, H. Application of the "trimethyl lock” to ganciclovir, a pro-prodrug with increased oral bioavailability. Bioorg. Med. Chem. Lett. 1996, 6, 1653–1656. [Google Scholar]
- Greenwald, R.B.; Choe, Y.H.; Conover, C.D.; Shum, K.; Wu, D.; Royzen, M. Drug delivery systems based on trimethyl lock lactonization: poly(ethyleneglycol) prodrugs of amino-containing compounds. J. Med. Chem. 2000, 43, 475–487. [Google Scholar]
- Chandran, S.S.; Dickson, K.A.; Raines, R.T. Latent fluorophore based on the trimethyl lock. J. Am. Chem. Soc. 2005, 127, 1652–1653. [Google Scholar]
- Lavis, L.D.; Chao, T.-Y.; Raines, R.T. Latent blue and red fluorophores based on trimethyl lock. ChemBioChem. 2006, 7, 1151–1154. [Google Scholar]
- Weerapreeyakul, N.; Anorach, R.; Khuansawad, T.; Yenjai, C.; Isaka, M. Synthesis of bioreductive esters from fungal compounds. Chem. Pharm. Bull. 2007, 55, 930–935. [Google Scholar]
- Sample Availability: Not available.
© 2007 by MDPI (http://www.mdpi.org). Reproduction is permitted for noncommercial purposes.
Share and Cite
Gomes, P.; Vale, N.; Moreira, R. Cyclization-activated Prodrugs. Molecules 2007, 12, 2484-2506. https://doi.org/10.3390/12112484
Gomes P, Vale N, Moreira R. Cyclization-activated Prodrugs. Molecules. 2007; 12(11):2484-2506. https://doi.org/10.3390/12112484
Chicago/Turabian StyleGomes, Paula, Nuno Vale, and Rui Moreira. 2007. "Cyclization-activated Prodrugs" Molecules 12, no. 11: 2484-2506. https://doi.org/10.3390/12112484