The Preparation of New Phosphorus-Centered Functional Groups for Modified Oligonucleotides and Other Natural Phosphates

Abstract
:Introduction


| Entry | Compound | X | α (°) | a | b | a+b | pKa2 |
|---|---|---|---|---|---|---|---|
| 1 | 6a | O | 118.7 | 1.43 | 1.59 | 3.02 | 6.4 |
| 2 | 6b | CH2 | 112.1 | 1.807 | 1.51 | 3.32 | 7.6 |
| 3 | 6c | CHF | 113.3 | 1.82 | 1.50 | 3.32 | 6.5 |
| 4 | 6d | CF2 | 116.5 | 1.85 | 1.496 | 3.35 | 5.4 |




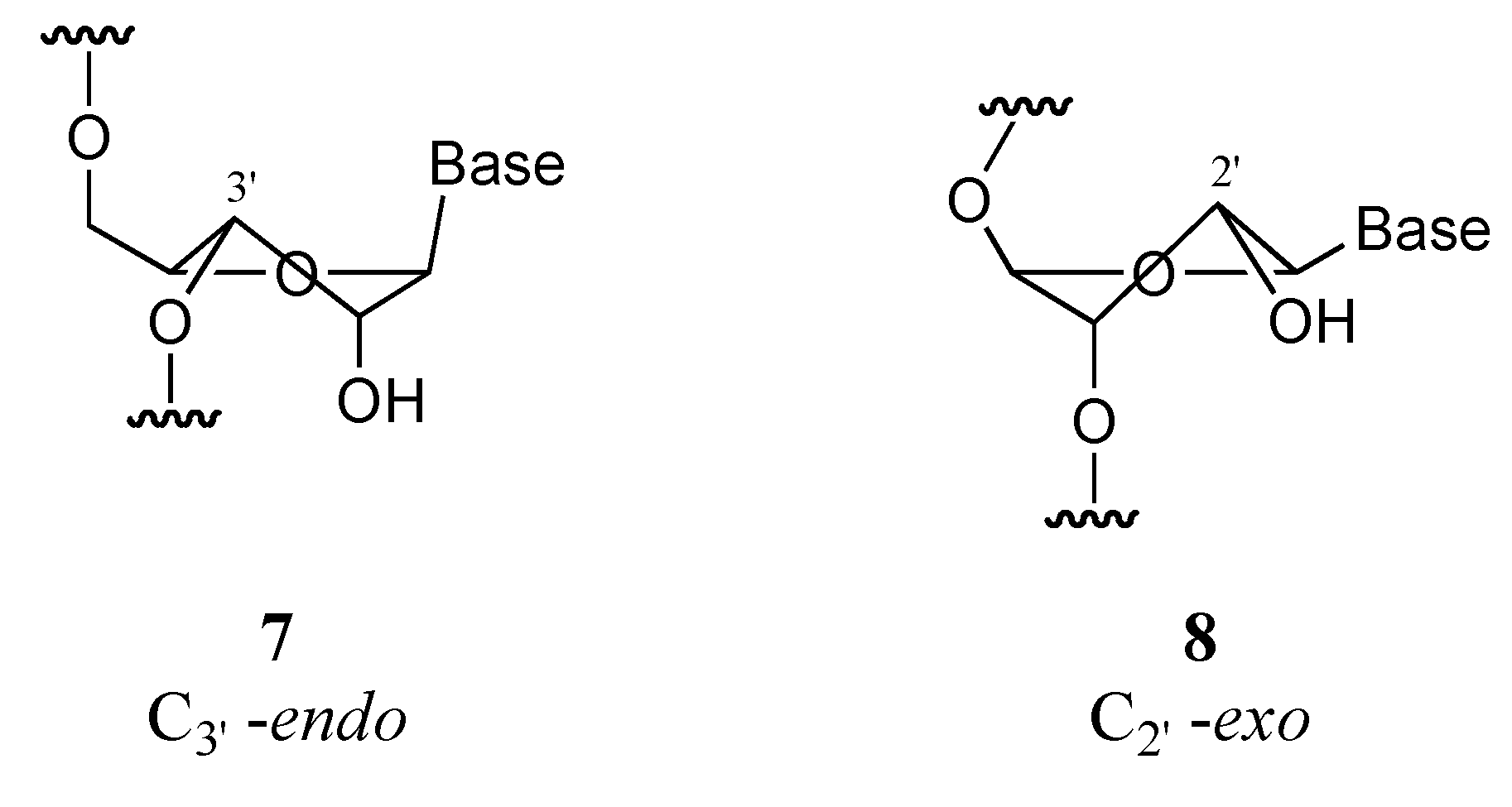
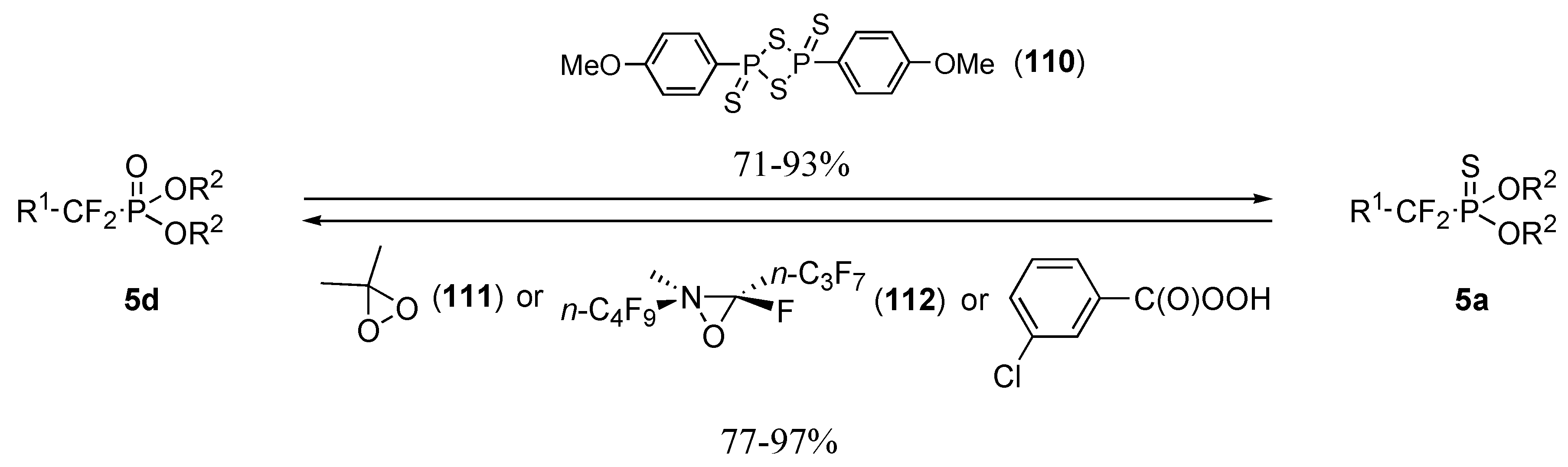
Results and Discussion


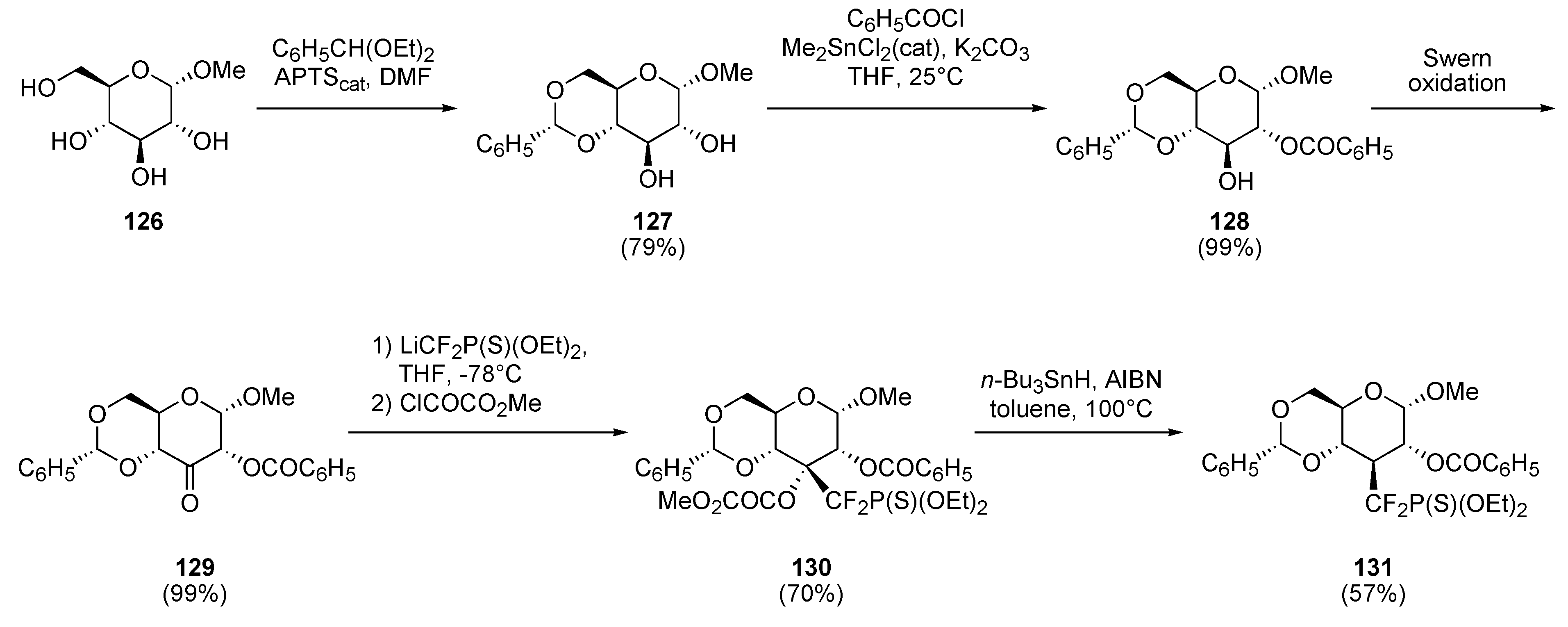
A) MONs
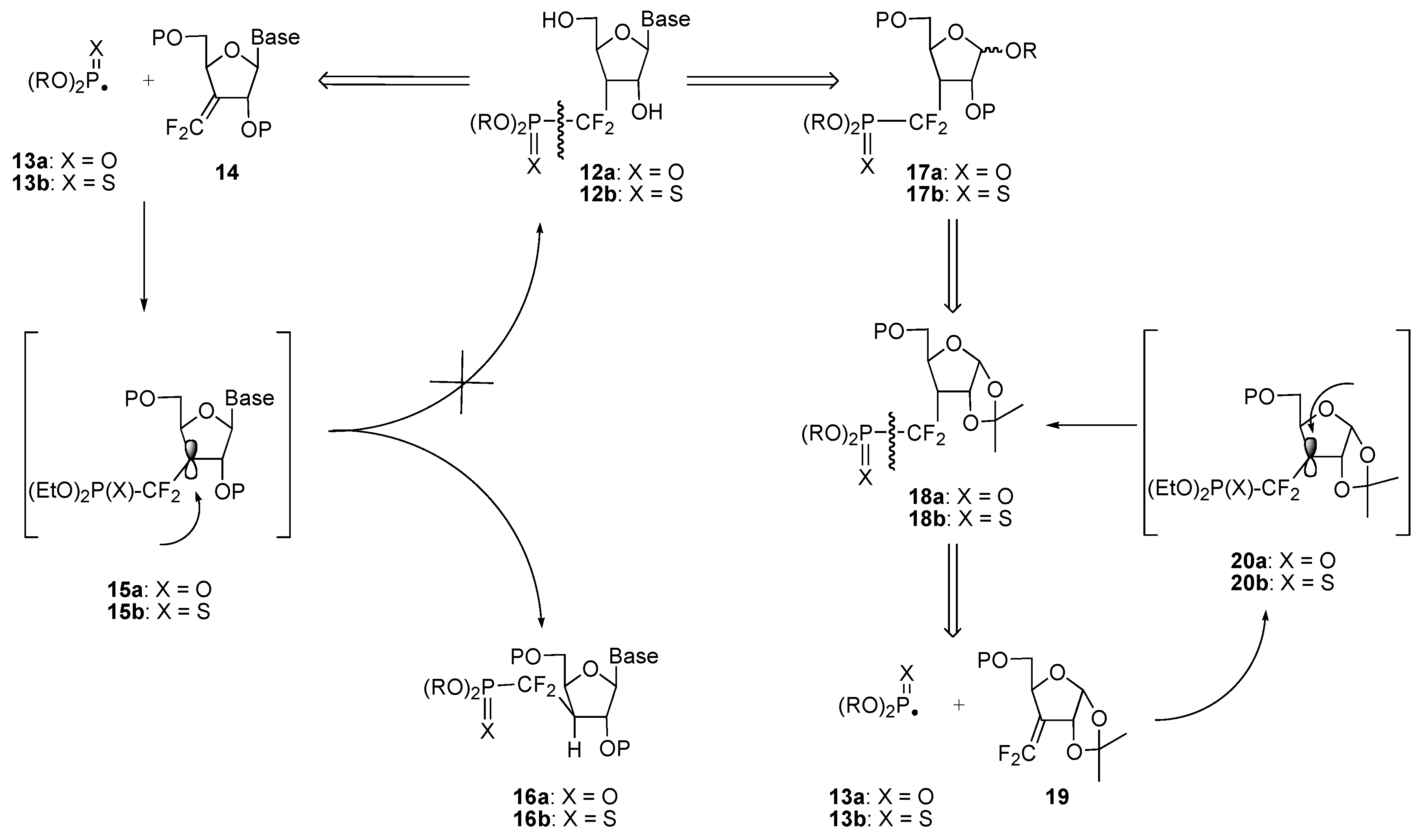
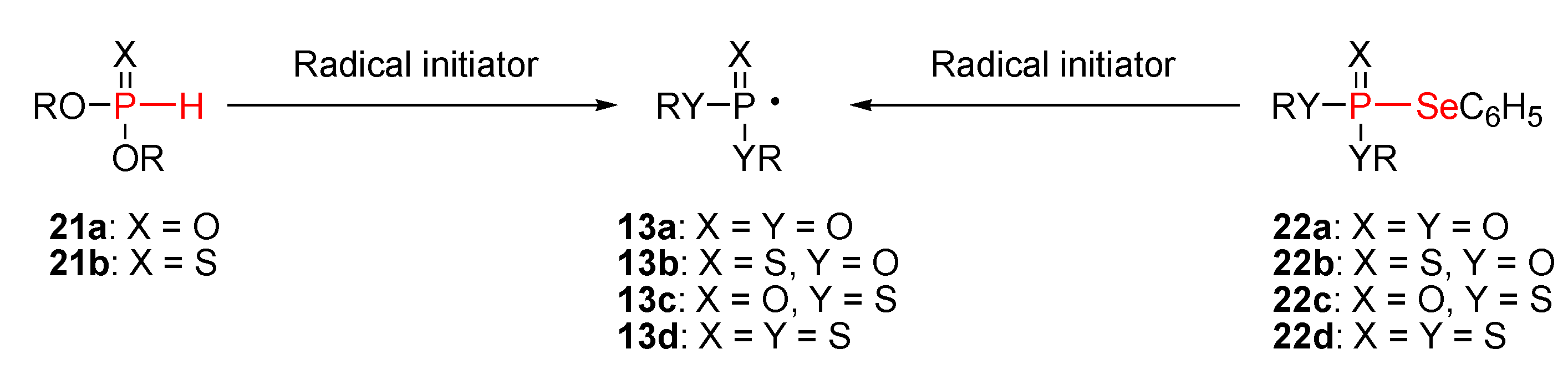
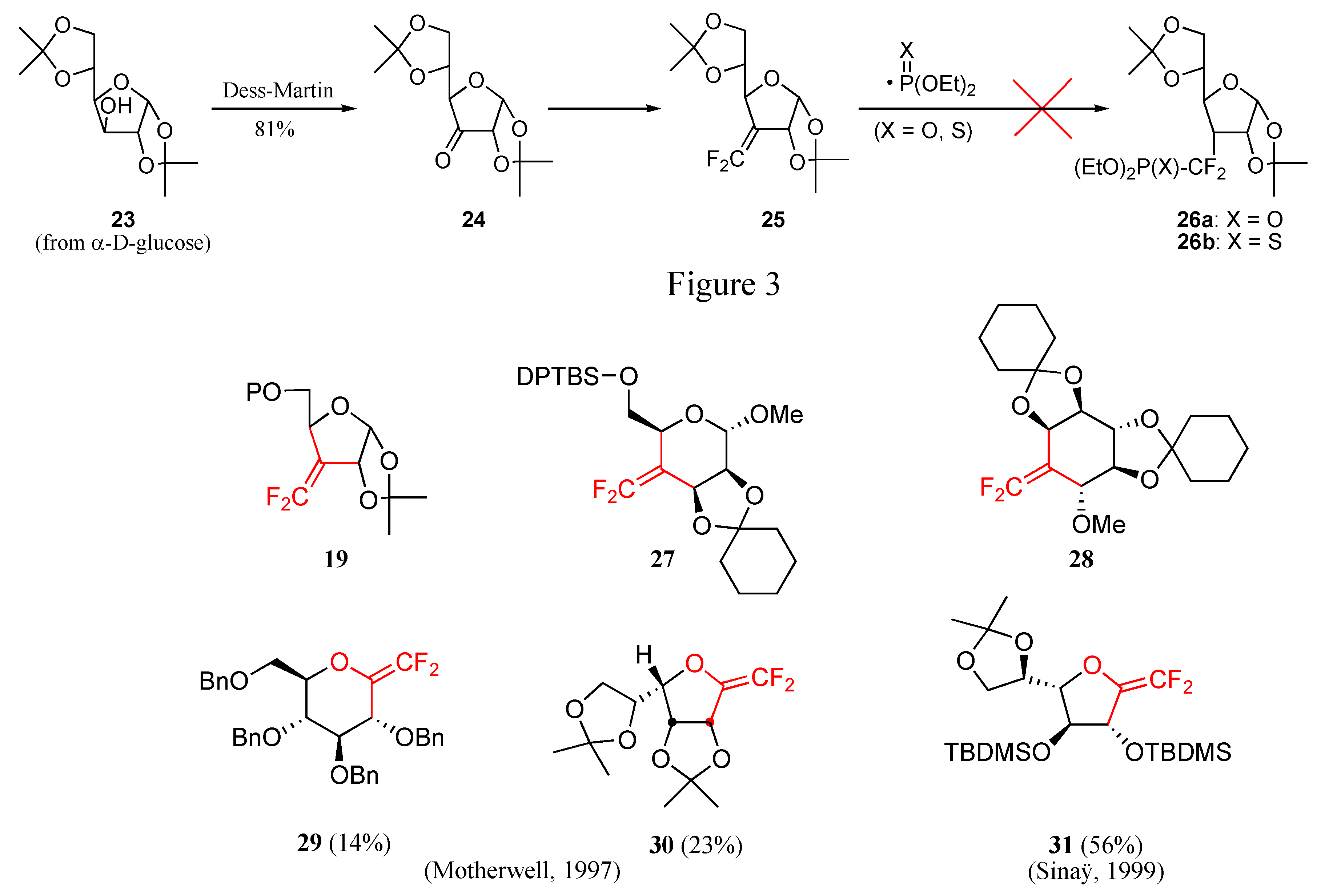
a) Radical approach













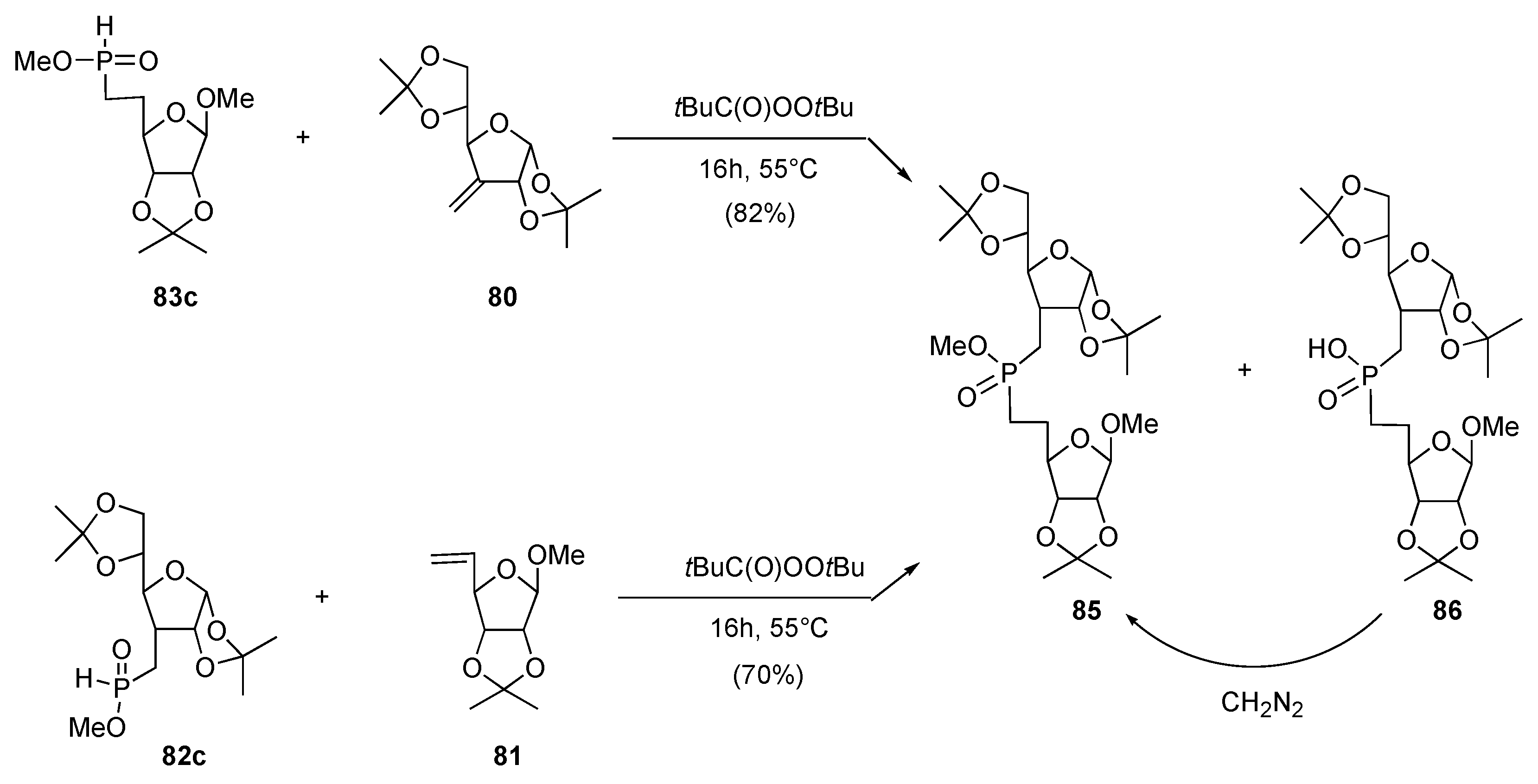
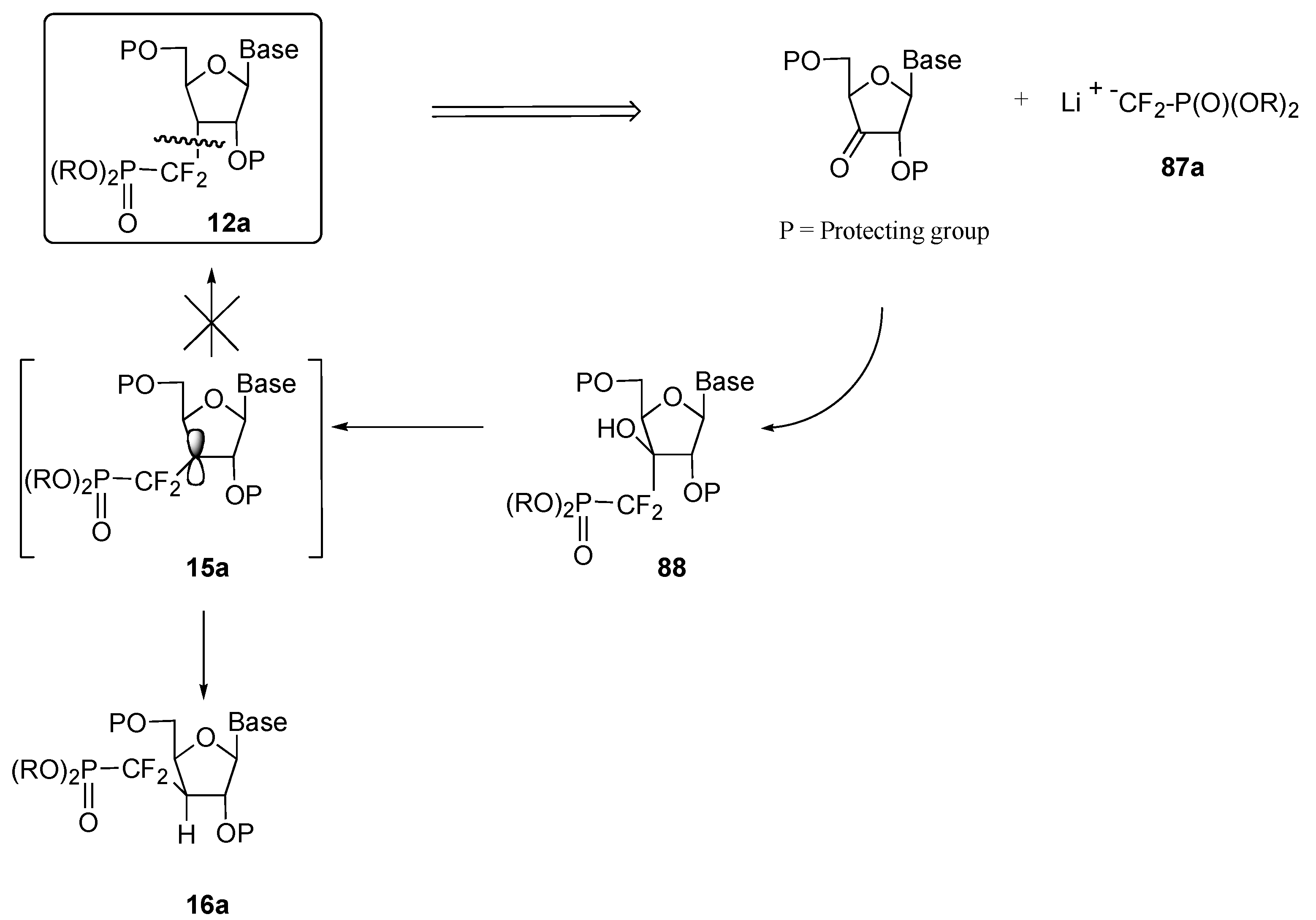
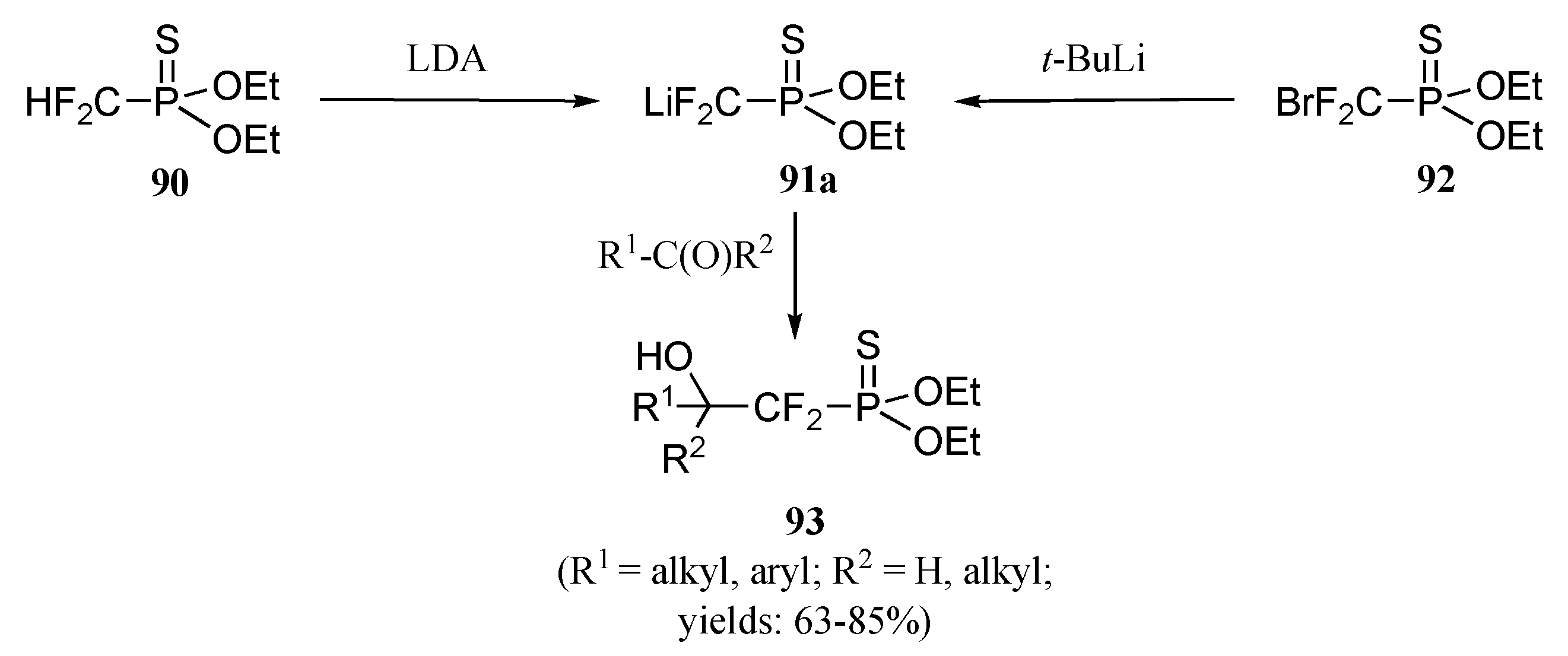
b) Ionic approach






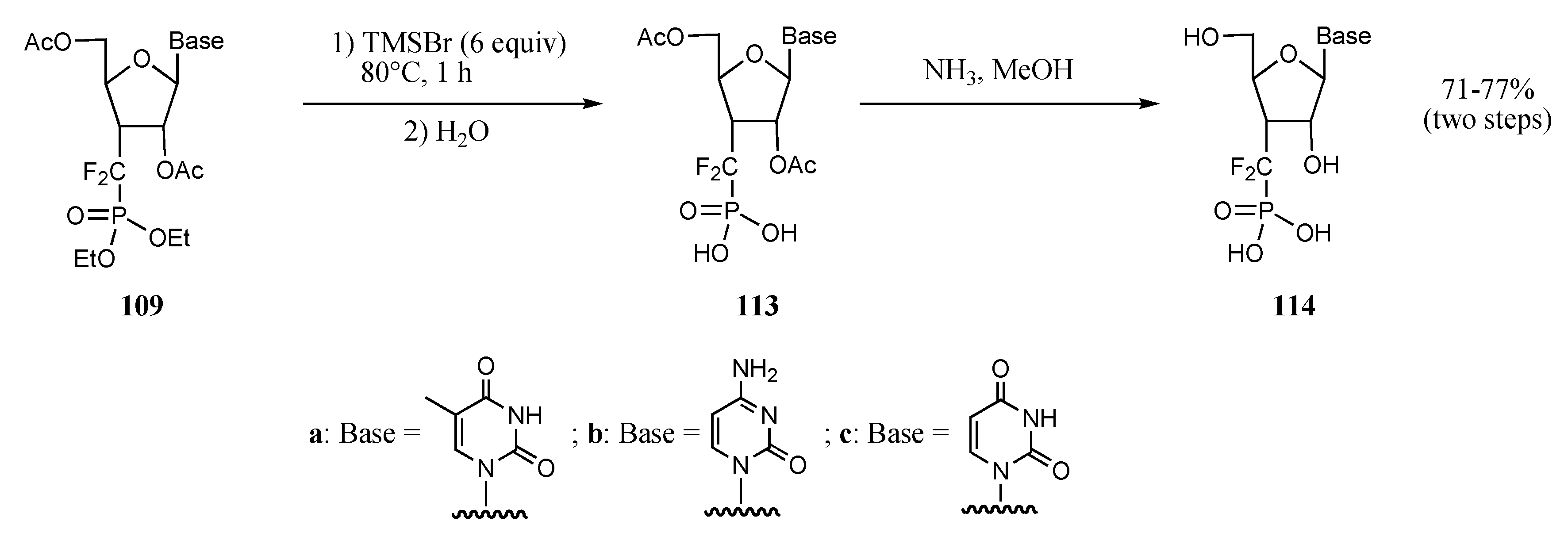
| Entry | Base | Reagent | Conditions | Products | Yields (%) | Products | Yields (%) |
|---|---|---|---|---|---|---|---|
| 1 |  |  | TMSOTf, ClCH2CH2C l, 25°C | 108a | 92 | 109a | 77 |
| 2 |  |  | TMSOTf, ClCH2CH2C l, 25°C | 108b | 81 | 109b | 54 |
| 3 |  |  | TMSOTf, ClCH2CH2C l, 80°C | 108c | 95 | 109c | 91 |
| 4 |  |  | SnCl4, CH3CN, 25°C | 108d | 87 | 109d | 73 |
| 5 |  |  | TMSOTf, ClCH2CH2C l, 80°C | 108e | 66 | 109e | 66 |




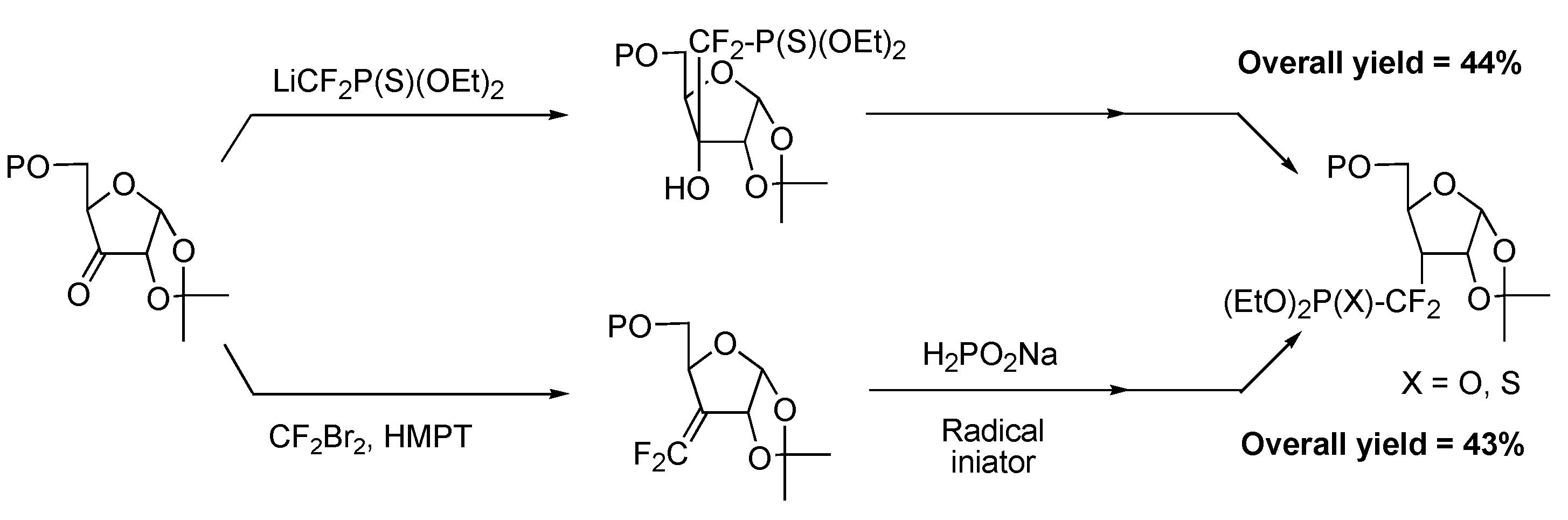
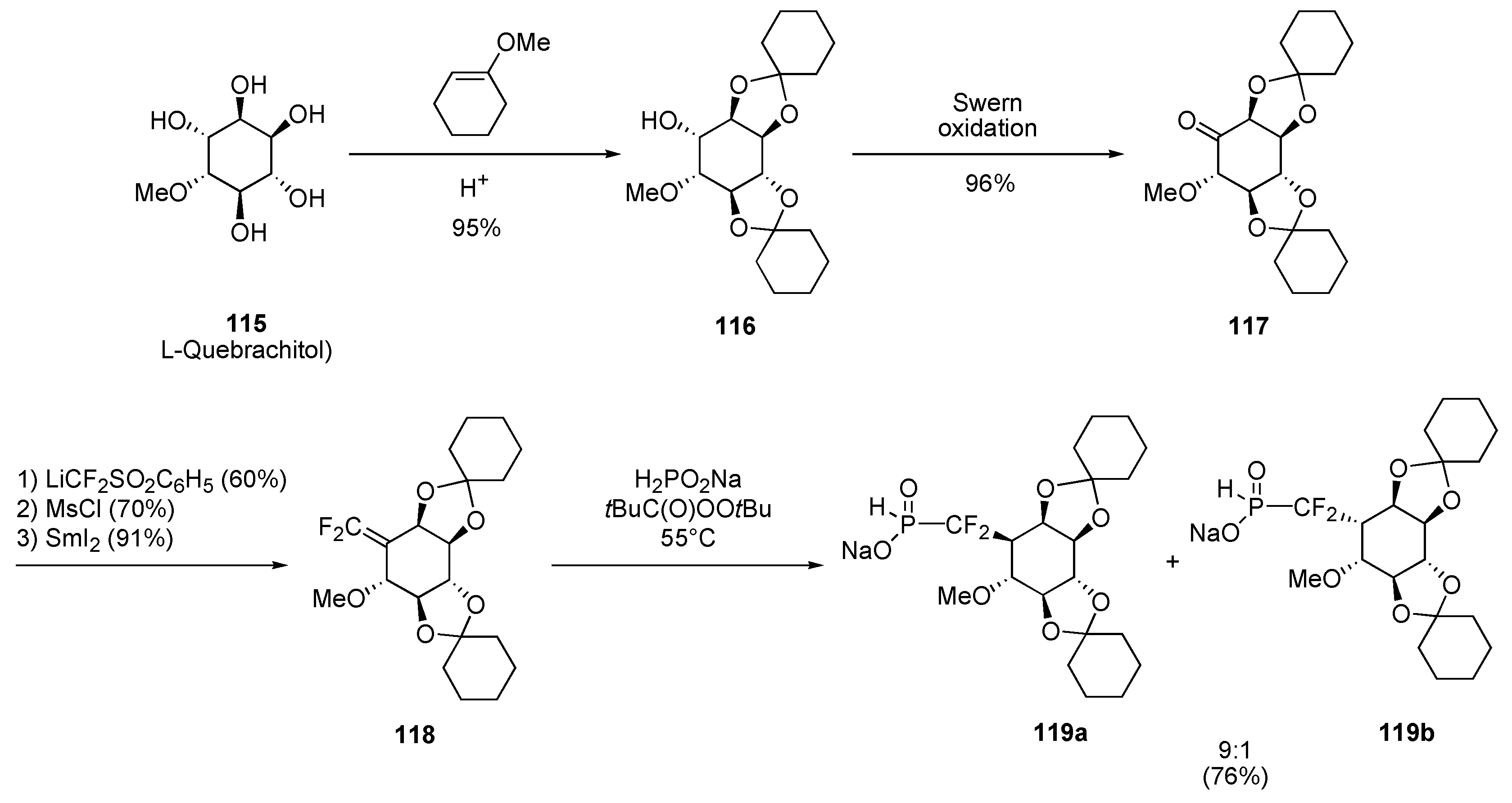
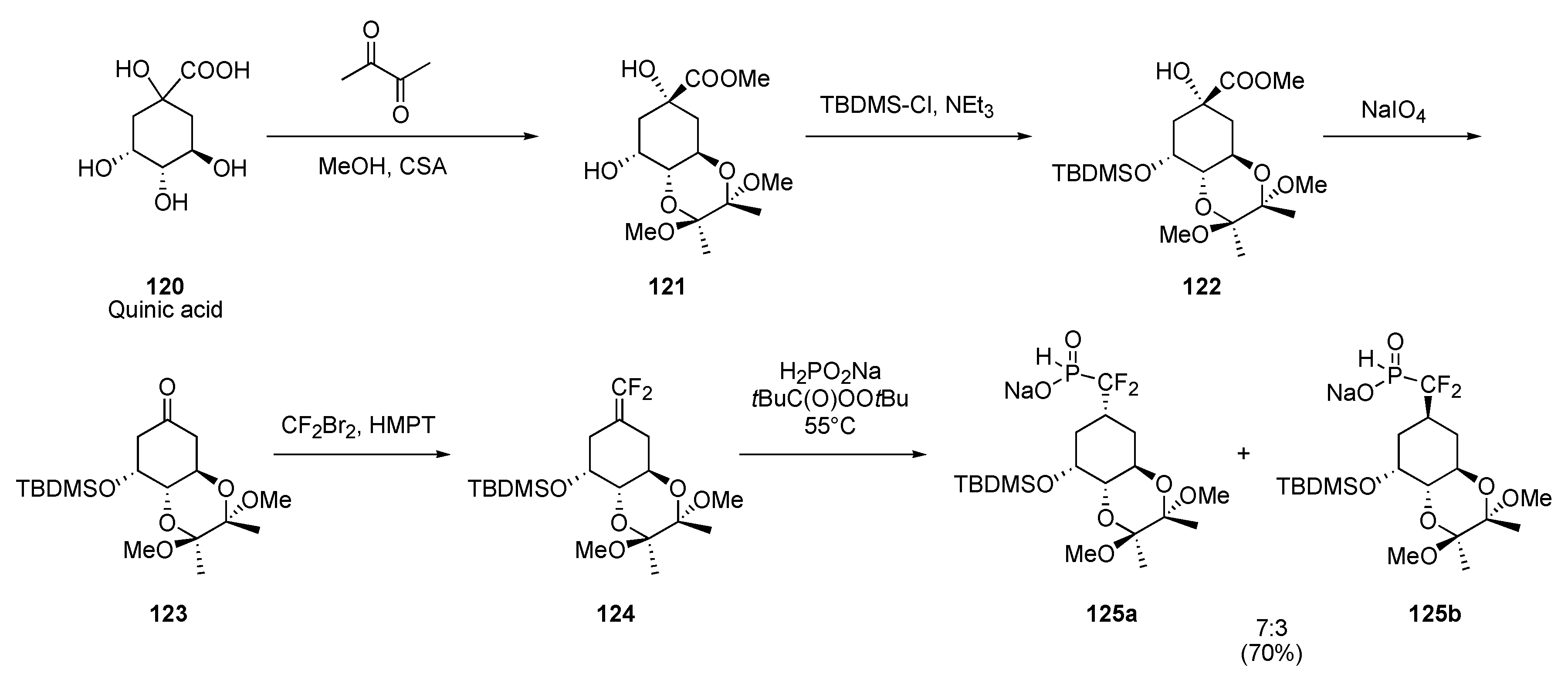
Cyclitols
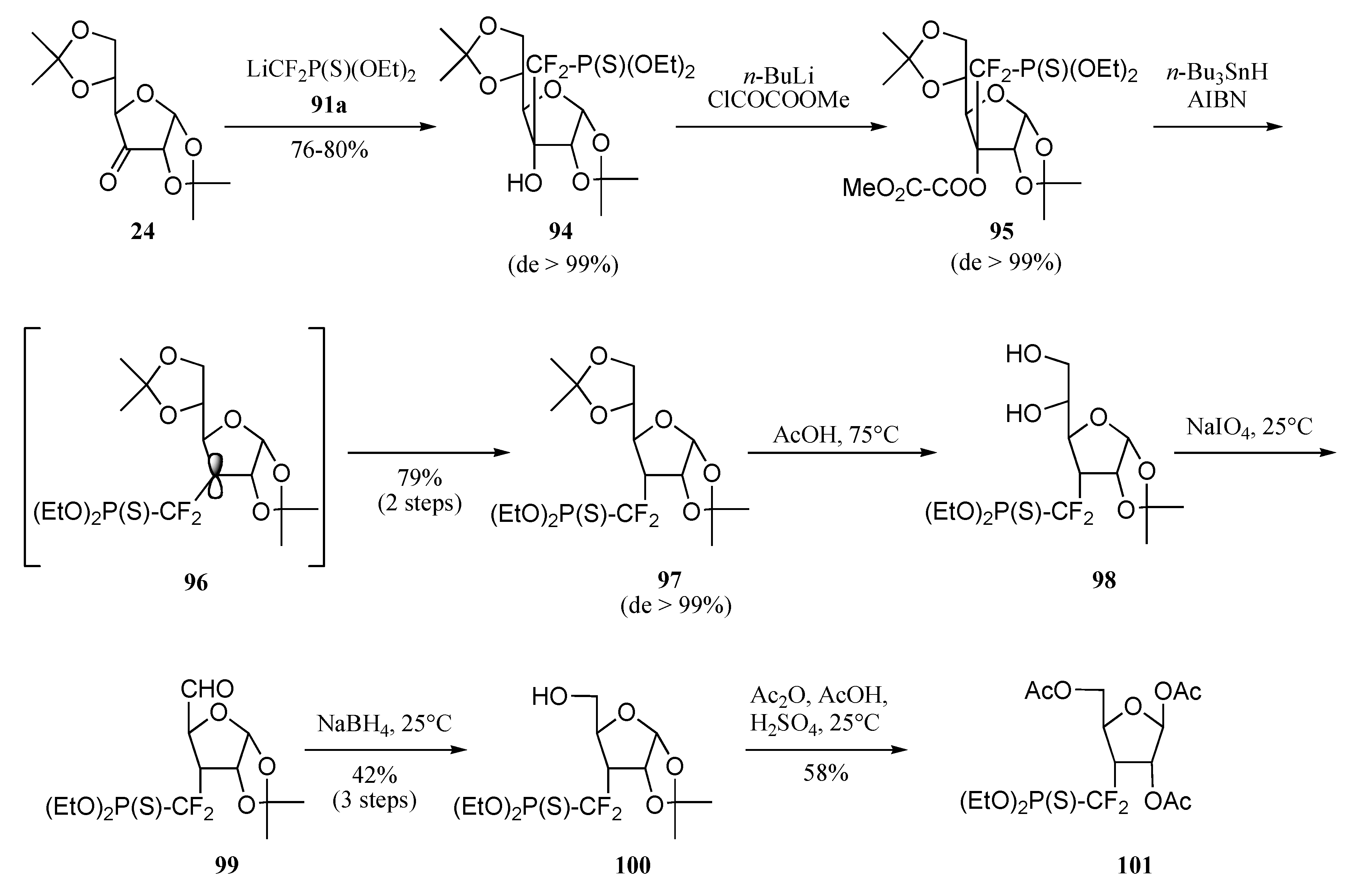
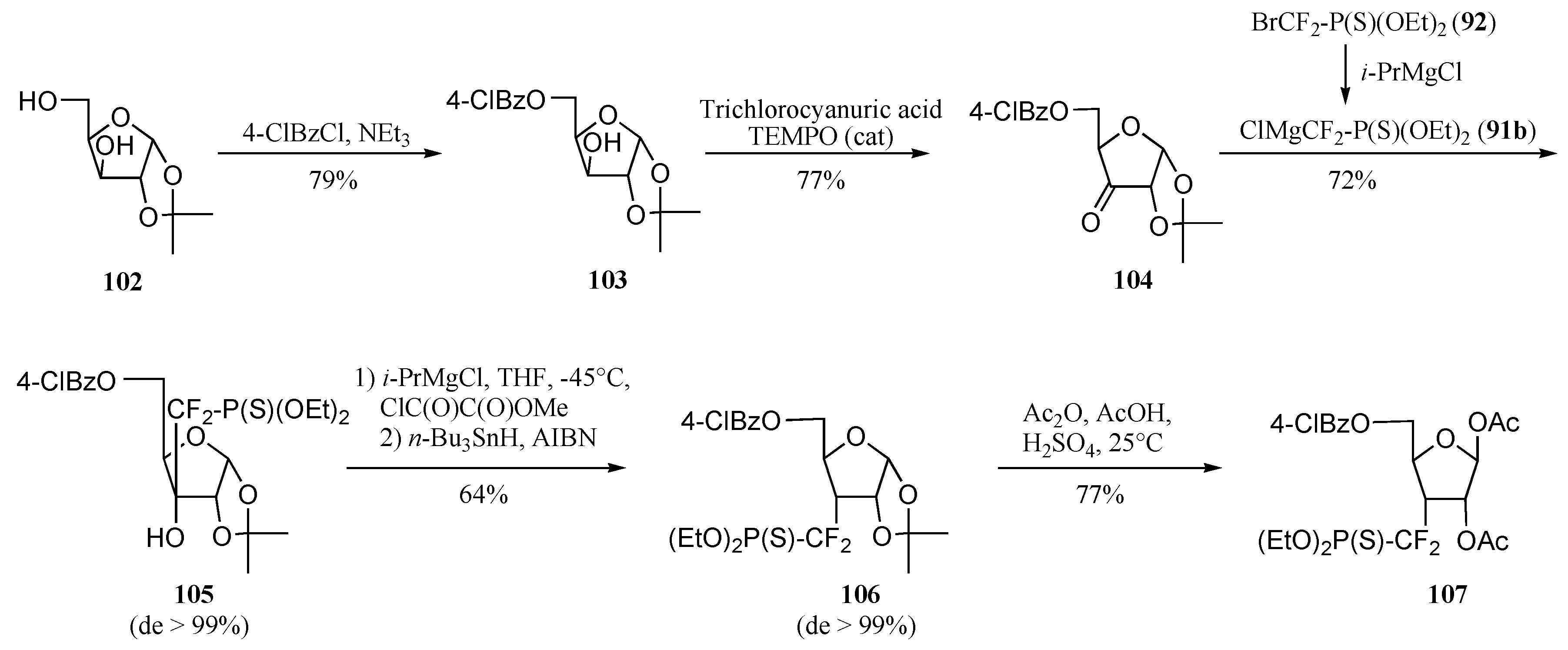
Radical approach


Ionic approach


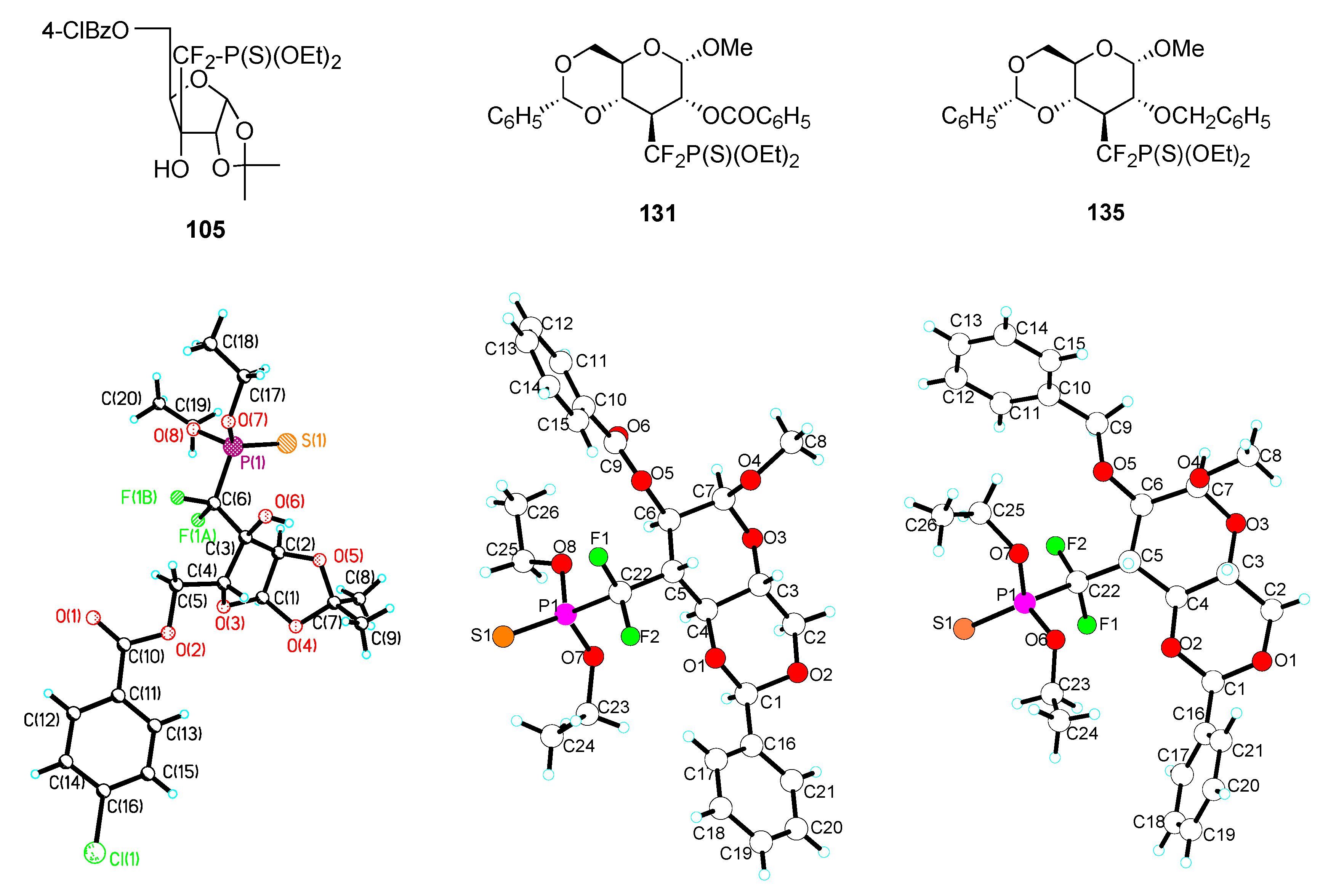
X–ray analyses

| Compound | X | Y | a (Å) | b (Å) | c (Å) | α (°) |
|---|---|---|---|---|---|---|
| 105 | CF2 | S | 1.88 | 1.51 | 1.90 | 120.3 |
| 131 | CF2 | S | 1.86 | 1.52 | 1.91 | 122.5 |
| 135 | CF2 | S | 1.86 | 1.55 | 1.91 | 117.5 |
| 6a | O | O | 1.59 | 1.43 | 1.52 | 118.5 |
| 6d | CF2 | O | 1.85 | 1.50 | 1.53 | 116.5 |

{kind=link}
{kind=link}
{kind=link}
{kind=link}
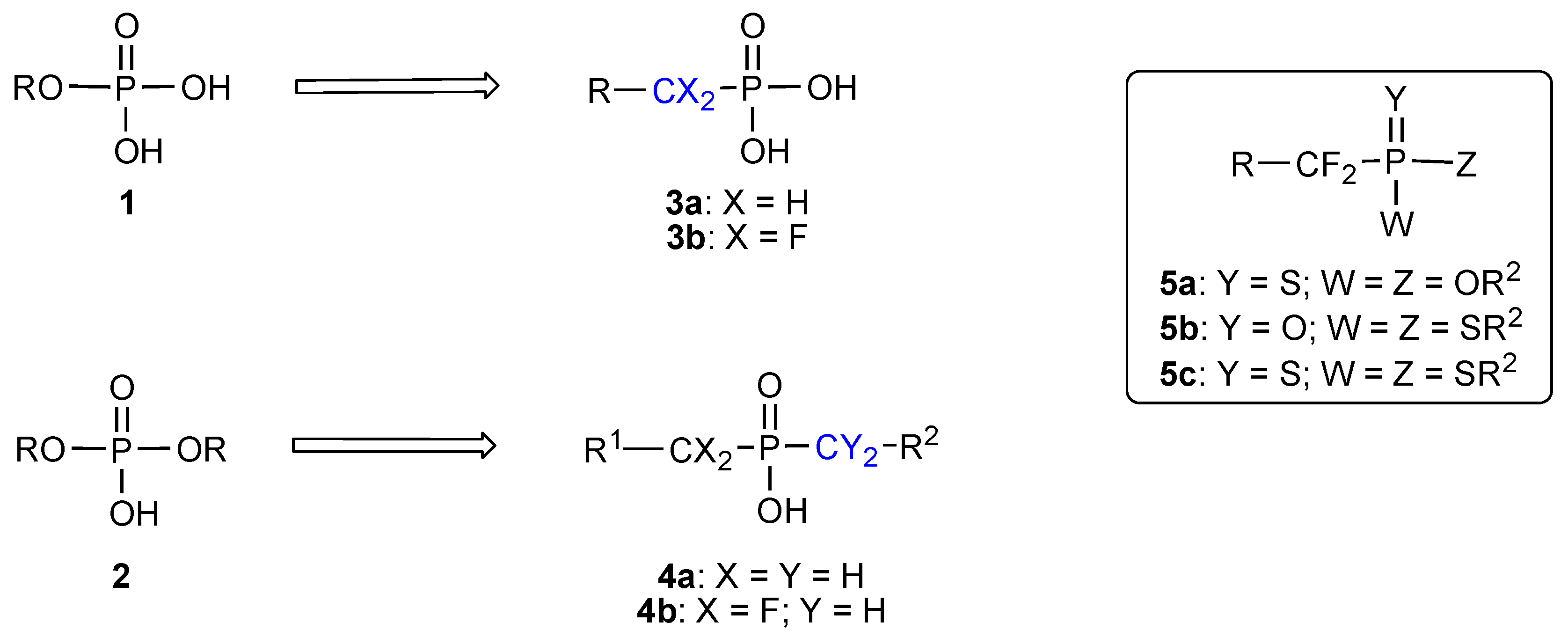{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
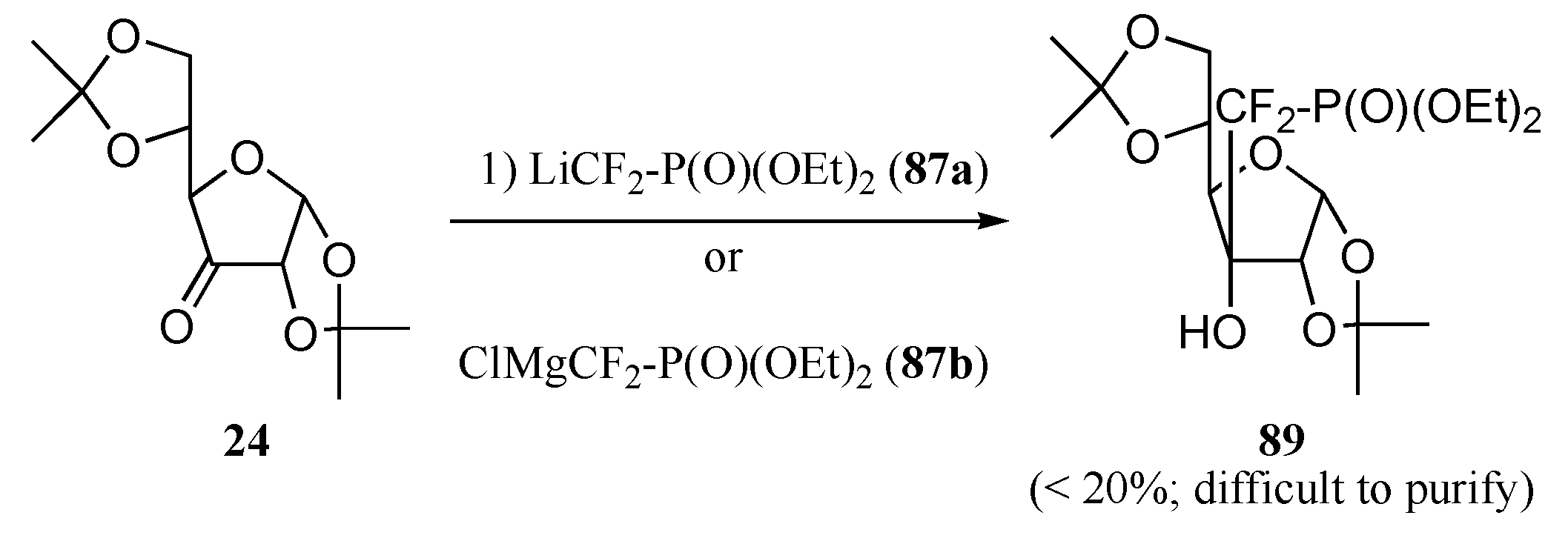{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
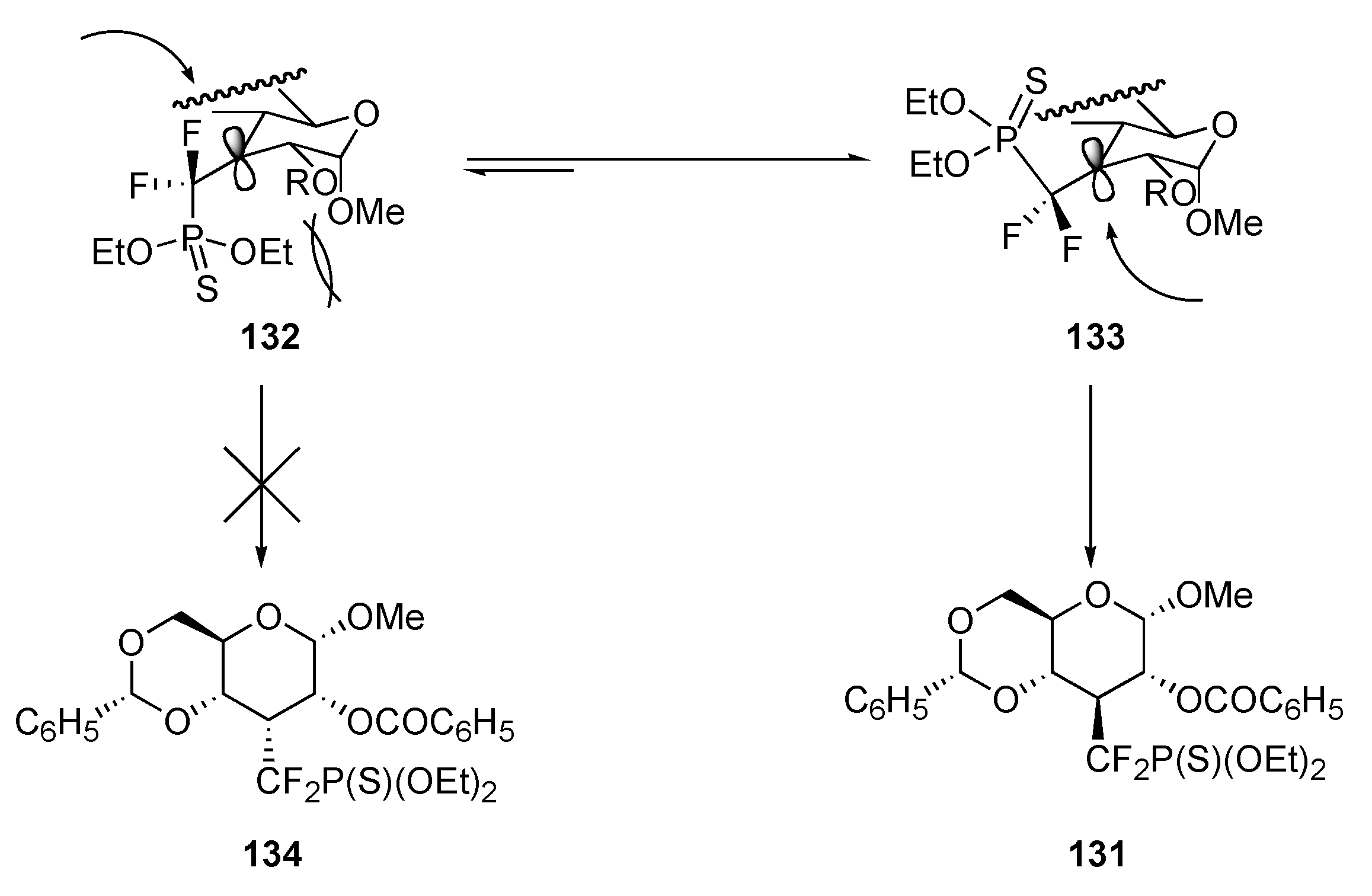{kind=link}
Conclusions
Acknowledgements
References and Notes
- Hartley, F. R. (Ed.) The Chemistry of Organophosphorus Compounds, Vol 4, Ter-and Quinque-valent Phosphorus Acids and Their Derivatives; John Wiley & Sons: Chichester, 1996.
- Blackburn, G. M.; England, D. A.; Kolmann, F. J. Chem. Soc., Chem. Commun. 1981, 930–932.Blackburn, G. M.; Kent, D. E.; Kolmann, F. J. Chem. Soc., Chem. Commun. 1981, 1188–1190.McKenna, C. E.; Shen, P.-D. J. Org. Chem. 1981, 46, 4573–4576.Blackburn, G. M.; Kent, D. E. J. Chem. Soc. Perkin Trans 1 1986, 913–917.Phillion, D. P.; Cleary, D. G. J. Org. Chem. 1992, 57, 2763–2764.Burke, T. R., Jr; Kole, H. K.; Roller, P. P. Biochem. Biophys. Res. Commun. 1994, 204, 129–134.Halazy, S.; Ehrhard, A.; Eggenspiller, A.; Berges-Gross, V.; Danzin, C. Tetrahedron 1996, 52, 177–184.Higashimoto, Y.; Saito, S.; Tong, X.-H.; Hong, A.; Sakaguchi, K.; Appella, E.; Anderson, C. W. J. Biol. Chem. 2000, 275, 23199–23203.
- Chambers, R. D.; O’Hagan, D.; Lamont, R. B.; Jain, S. C. Chem. Commun. 1990, 1053–1054.Thatcher, G. R. J.; Campbell, A. S. J. Org. Chem. 1993, 58, 2272–2281.Nieschalk, J. N.; Batsanov, A. S.; O’Hagan, D.; Chambers, R. D. Tetrahedron 1996, 52, 165–176.
- Burke, T. R., Jr.; Smyth, M. S.; Nomizu, M.; Otaka, A.; Roller, P. P. J. Org. Chem. 1993, 58, 1336–1340.Martin, S. F.; Wong, Y.-L.; Wagman, A. S. J. Org. Chem. 1994, 59, 4821–4831.Berkowitz, D. B.; Shen, Q.; Maeng, J.-H. Tetrahedron Lett. 1994, 35, 6445–6448.Burke, T. R., Jr.; Ye, B.; Akamatsu, M.; Ford, H., Jr.; Yan, X.; Kole, H. K.; Wolf, G.; Shoelson, S. E.; Roller, P. P. J. Med. Chem. 1996, 39, 1021–1027.Berkowitz, D. B.; Eggen, M.; Shen, Q.; Shoemaker, R. K. J. Org. Chem. 1996, 61, 4666–4675.Ye, B.; Burke, T. R., Jr. Tetrahedron 1996, 52, 9963–9970.Qabar, M. N.; Urban, J.; Khan, M. Tetrahedron 1997, 53, 11171–11178.Yokomatsu, T.; Abe, H.; Yamagishi, T.; Suemune, K.; Shibuya, S. J. Org. Chem. 1999, 64, 8413–8418.Burke, T. R., Jr.; Lee, K. Acc. Chem. Res.. 2003, 36, 426–433.
- Blackburn, G. M.; Kent, D. E. J. Chem. Soc., Chem. Commun. 1981, 511–513. Blackburn, G. M.; Kent, D. E.; Kolkmann, F. J. Chem. Soc., Perkin Trans. 1 1984, 1119–1125. Blackburn, G. M.; Guo, M.-J.; Langston, S. P.; Taylor, G. E. Tetrahedron Lett. 1990, 31, 5637–5640. Blackburn, G. M.; S. P. Langston, S. P. Tetrahedron Lett. 1991, 32, 6425–6428. Matulic-Adamic, J.; Usman, N. Tetrahedron Lett. 1994, 35, 3227–3230. Matulic-Adamic, J.; Haeberli, P.; Usman, N. J. Org. Chem. 1995, 60, 2563–2569. [CrossRef] Levvy, S. G.; Wasson, B.; Carson, D. A.; Cottam, H. B. Synthesis 1996, 843–846. Hamilton, C. J.; Roberts, S. M.; Shipitsin, A. J. Chem. Soc., Chem. Comm. 1998, 1087–1088. Hamilton, C. J.; Roberts, S. M. J. Chem. Soc., Perkin Trans. 1 1999, 1051–1056.
- Collingwood, S. P.; Baxter, A. D. Synlett 1995, 703.Regan, A. C.; Sciammetta, N.; Tattersall, P. I. Tetrahedron Lett. 2000, 41, 8211–8215. Centrone, C. A.; Lowary, T. L. J. Org. Chem. 2003, 68, 8115–8119. [PubMed]
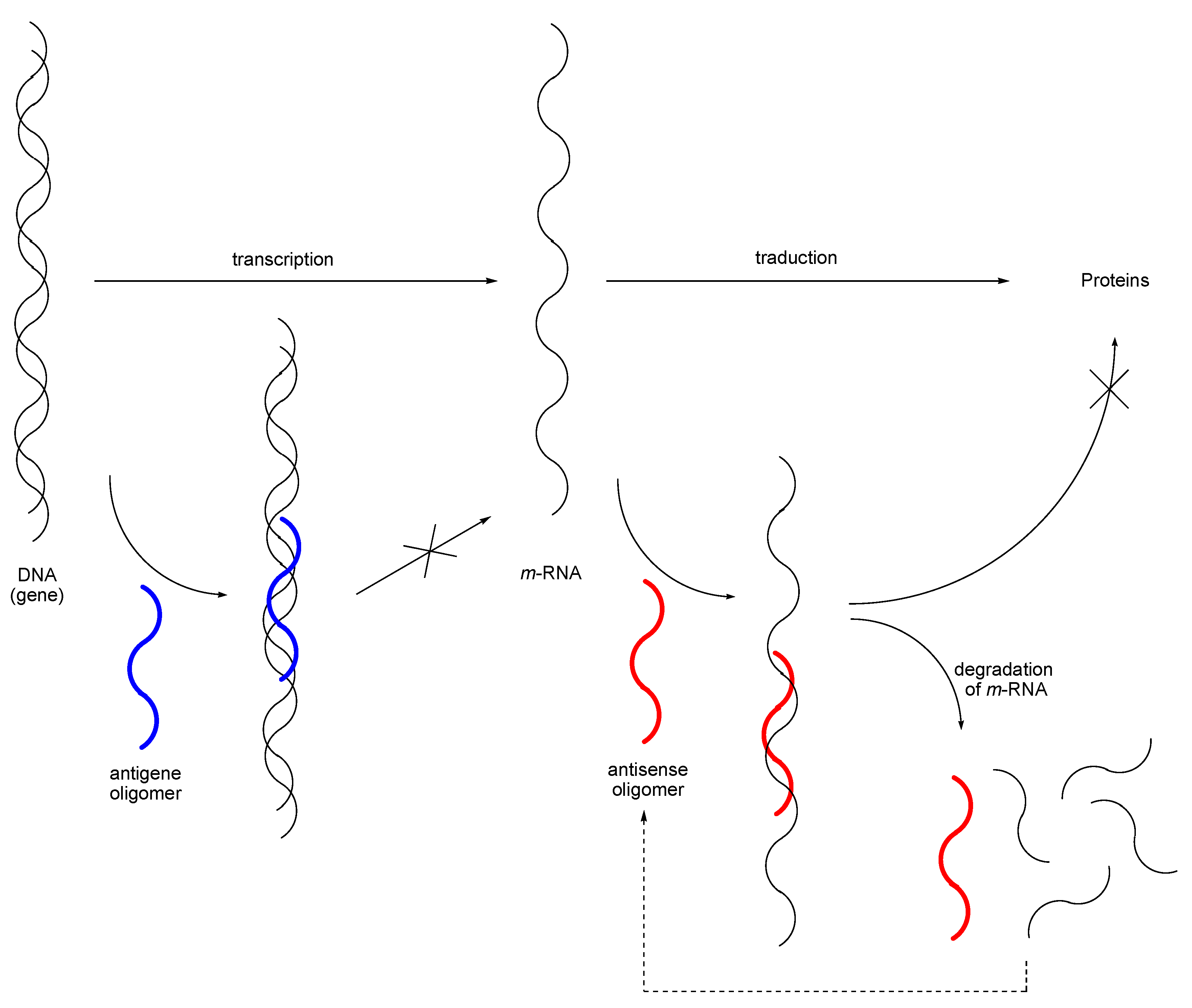
- For leading references on antisense and antigene strategies, see: De Mesmaeker, A.; Häner, R.; Martin, P.; Moser, H. E. Acc. Chem. Res. 1995, 28, 366–374. Roush, W. Science 1997, 276, 1192–1193. [PubMed] Gee, J. E.; Robbins, I.; van der Laan, A. C.; van Boom, J. H.; Colombier, C.; Leng, M.; Paible, A. M.; Nelson, J. S.; Lebleu, B. Antisense Nucleic Acid Drug Dev. 1998, 8, 103–111. [PubMed] Crooke, S. T. Biochim. Biophys. Acta 1999, 1489, 31–44. [PubMed] Marcusson, E. G.; Yacyshyn, B. R.; Shanahan, W. R., Jr.; Dean, N. M. Mol Biotechnol. 1999, 12, 1–11. [PubMed] Taylor, J. K.; Dean, N. M. Curr. Opin. Drug Dev. 1999, 2, 147–151. Holmlund, J. T.; Monia, B. P.; Kwoh, T. J.; Dorr, F. A. Curr. Opin. Mol. Ther. 1999, 1, 372–385. [PubMed] Dorr, F. A. Antisense Nucleic Acid Drug Dev. 1999, 9, 391–396. Lebedeva, I. V.; Stein, C. A. BioDrugs 2000, 13, 195–216. [PubMed]
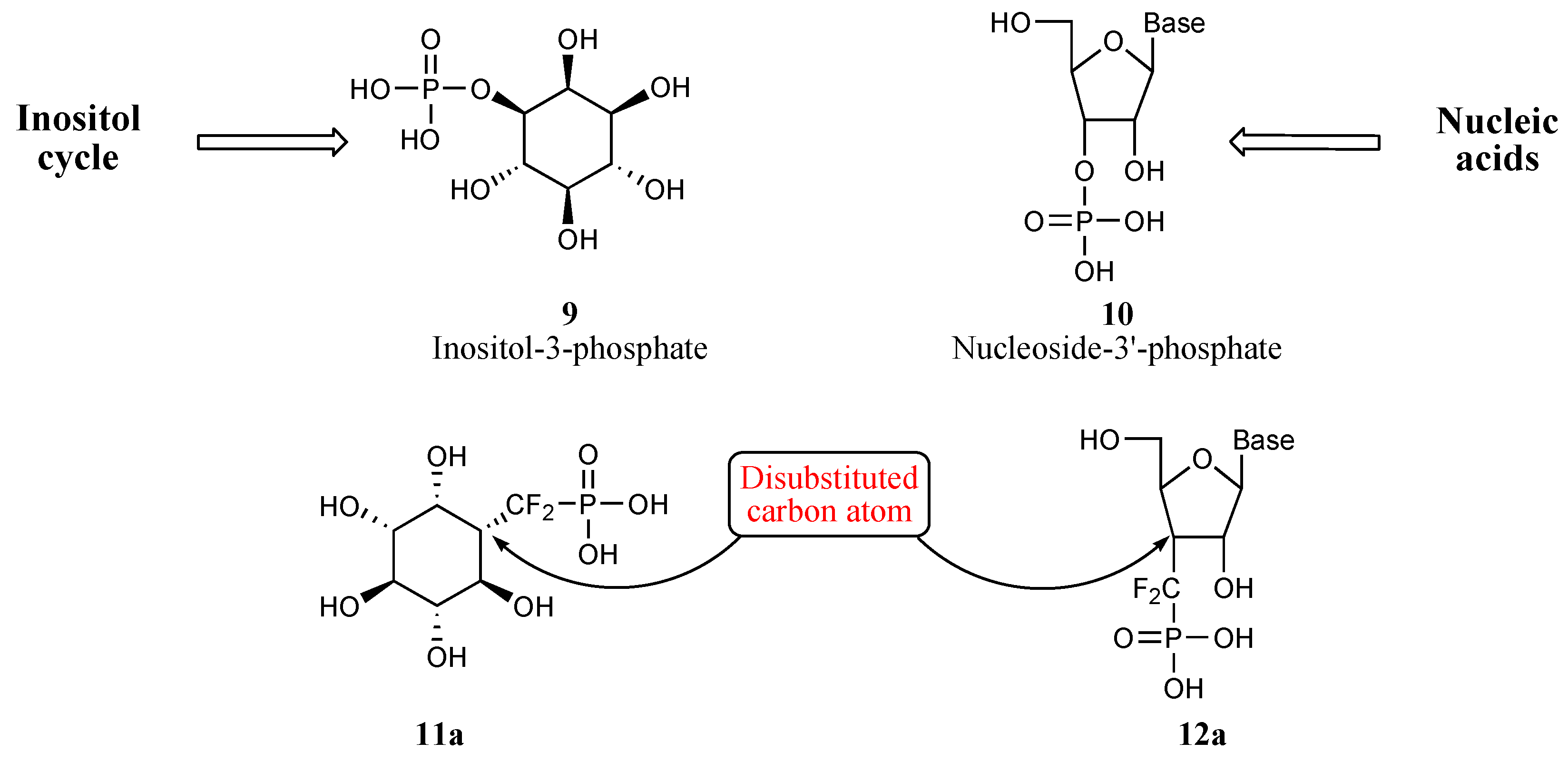
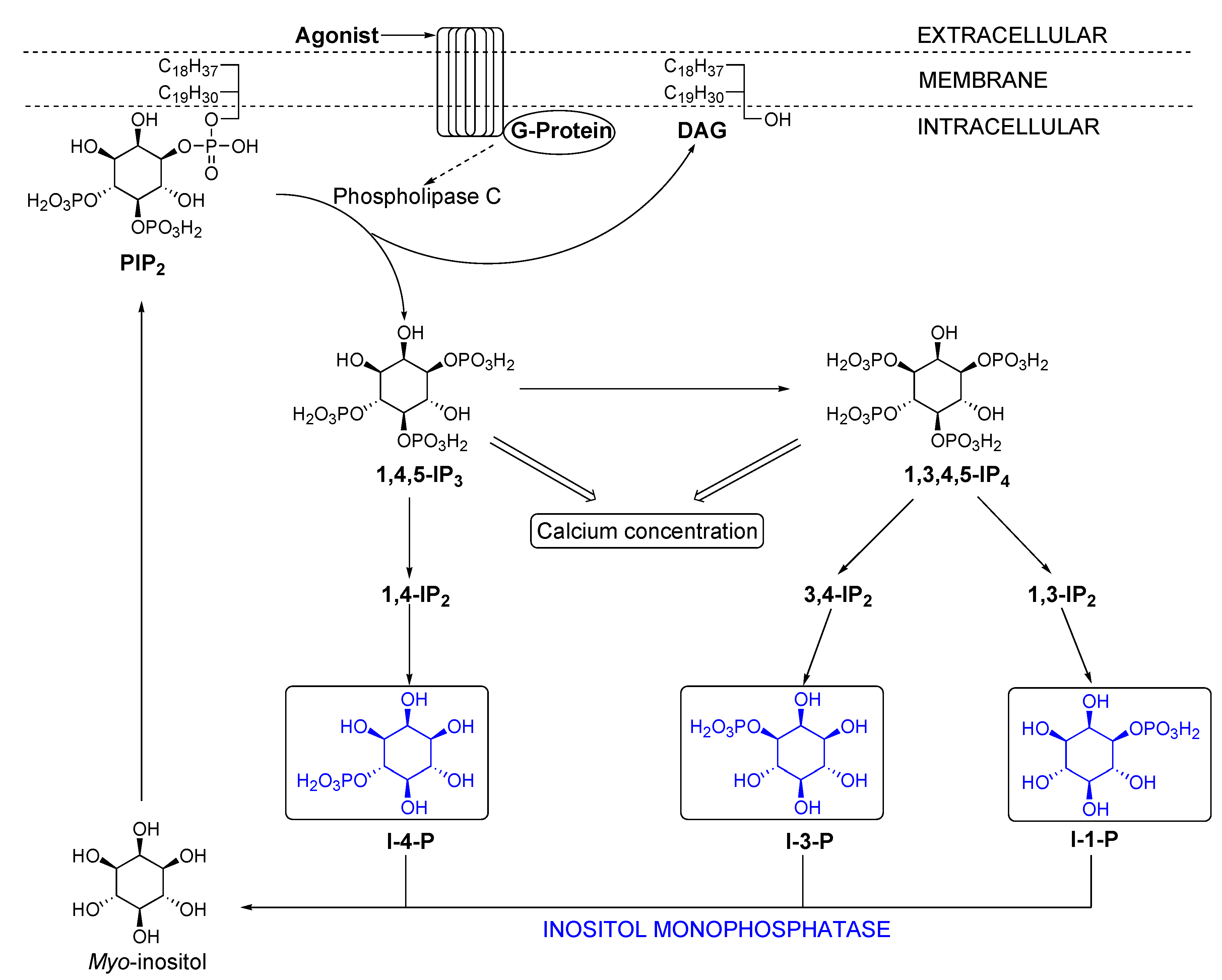
- For a leading reference on the inositol cycle, see: Billington, D. C. “The Inositol Phosphates. Chemical Synthesis and Biological Significance.”. VCH Publishers: Weinheim, 1991; pp. 9–21. [Google Scholar]
- McAllister, G.; Whitting, P.; Hammond, E. A.; Knowles, M. R.; Atack, J. R.; Bailey, F. J.; Maigetter, R.; Ragan, C. I. Biochem. J. 1992, 284, 749–754. [PubMed] Bone, R.; Springer, J. P.; Atack, J. R. Proc. Natl. Acad. Sci. USA 1992, 89, 1031–1035. Bone, R.; Frank, L.; Springer, J. P.; Pollack, S. J.; Osborn, S.; Atack, J. R.; Knowles, M. R.; McAllister, G.; Ragan, C. I.; Broughton, H. B.; Baker, R.; Fletcher, S. Biochemistry 1994, 33, 9460–9467. [PubMed] Bone, R.; Frank, L.; Springer, J. P.; Atack, J. R. Biochemistry 1994, 33, 9468–9476. [PubMed]
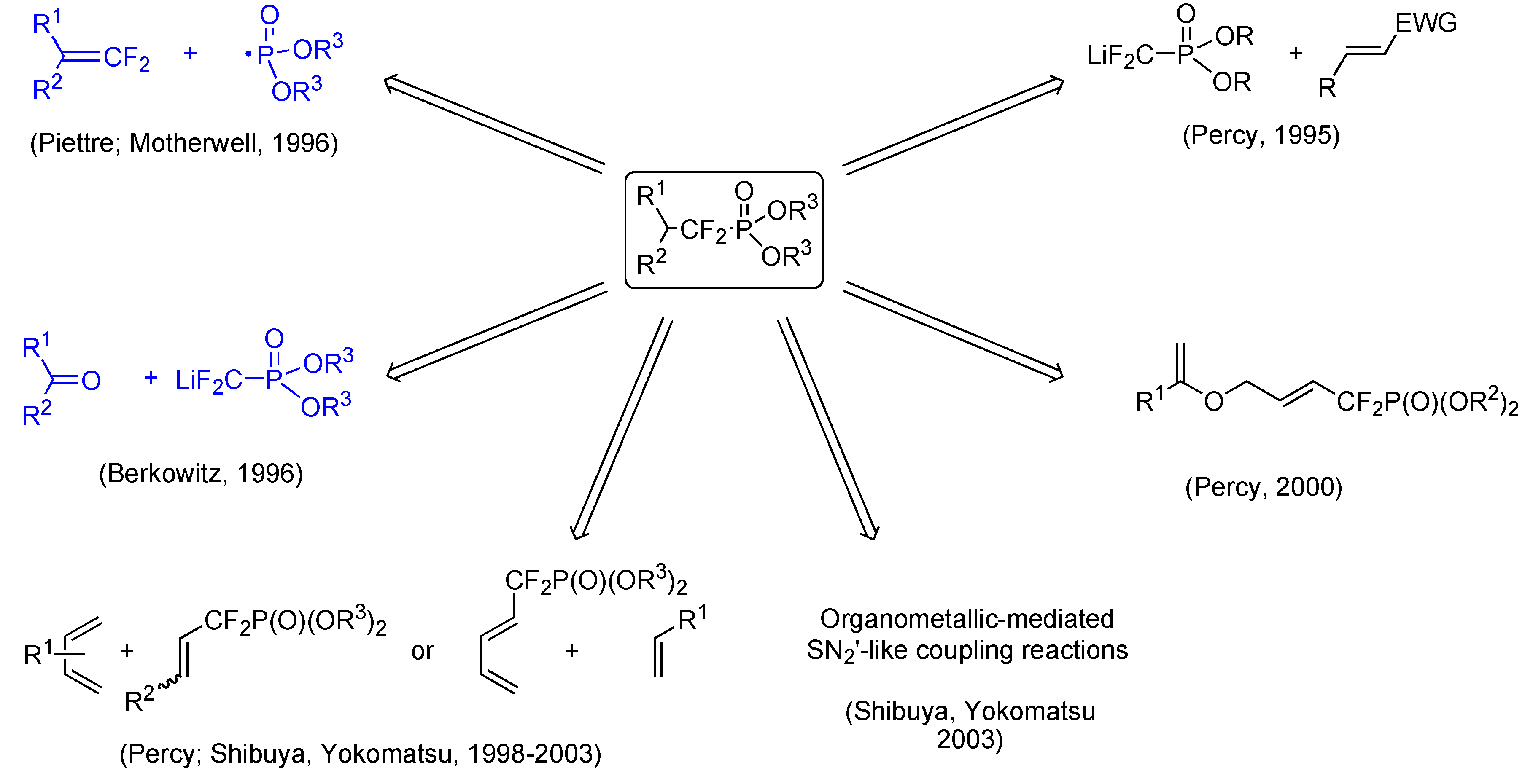
- Lequeux, T. P.; Percy, J. M. Synlett 1995, 361–362. Piettre, S. R. Tetrahedron Lett. 1996, 37, 2233–2236. Herpin, T. F.; Houlton, J. S.; Motherwell, W. B.; Roberts, B. P.; Weibel, J.-M. J. Chem. Soc., Chem. Comm. 1996, 613–614. Berkowitz, D. B.; Eggen, M.; Shen, Q.; Shoemaker, R. K. J. Org. Chem. 1996, 61, 4666–4675. [PubMed] Blades, K.; Lequeux, T. P.; Percy, J. M. Chem. Commun. 1996, 1457–1458. Blades, K.; Cockerill, G. S.; Easterfield, H. J.; Lequeux, T. P.; Percy, J. M. Chem. Commun. 1996, 1615–1616. Blades, K.; Lapôtre, D.; Percy, J. M. Tetrahedron Lett. 1997, 38, 5895–5898. Herpin, T. F.; Motherwell, W. B.; Roberts, B. P.; Roland, S.; Weibel, J.-M. Tetrahedron 1997, 53, 15085–15100. Blades, K.; Percy, J. M. Tetrahedron Lett. 1998, 39, 9085–9088. Kovensky, J.; McNeil, M.; Sinaÿ, P. J. Org. Chem. 1999, 64, 6202–6205. Butt, A. H.; Percy, J. M.; Spencer, N. S. Chem. Commun. 2000, 1691–1692. Yokomatsu, T.; Katayama, S.; Shibuya, S. Chem. Commun. 2001, 1878–1879. Yokomatsu, T.; Suemune, K.; Murano, T.; Shibuya, S. Heterocycles 2002, 56, 273–282. Butt, A. H.; Kariuki, B. M; Percy, J. M.; Spencer, N. S. Chem. Commun. 2002, 682–683. Murano, T.; Muroyama, S.; Yokomatsu, T.; Shibuya, S. Synlett 2002, 1657–1660. Yokomatsu, T.; Kato, J.; Sakuma, C.; Shibuya, S. Synlett 2003, 1407–1410. Murano, T.; Yuasa, Y.; Muroyama, S.; Yokomatsu, T.; Shibuya, S. Tetrahedron 2003, 59, 9059–9073. Murano, T.; Yuasa, Y.; Kobayakawa, H.; Yokomatsu, T.; Shibuya, S. Tetrahedron 2003, 59, 10223–10230.
- Rees, R. D.; James, K.; Tatchell, A. R.; Williams, R. H. J. Chem. Soc. (C) 1968, 276–2721. Sowa, W. Can. J. Chem. 1968, 46, 1586–1589. Bourgeois, J. M. Helv. Chim. Acta 1975, 53, 363–372. Yoshimura, J. Adv. Carbohydr. Chem. Biochem. 1984, 42, 69–134. Giese, B.; Gonzàlez-Gomez, J. A.; Witzel, T. Angew. Chem. Chem. Int. Ed. 1984, 23, 69–70. Giese, B. Angew. Chem. Chem. Int. Ed. 1989, 28, 969–980. Schmit, C. Synlett 1994, 241–242. Johnson, C. R.; Bhumralkar, D. R.; De Clercq, E. Nucleosides & Nucleotides 1995, 14, 185–194. Lavaire, S.; Plantier-Royon, R.; Portella, C. J. Carbohydr. Chem. 1996, 15, 361–370. Lavaire, S.; Plantier-Royon, R.; Portella, C. Tetrahedron: Asymmetry 1998, 9, 213–226. Lopin, C.; Gautier, A.; Gouhier, G.; Piettre, S. R. Tetrahedron Lett. 2000, 41, 10195–10200. Gautier, A.; Garipova, G.; Dubert, O.; Oulyadi, H.; Piettre, S. R. Tetrahedron Lett. 2001, 42, 5673–5676.
- Lopin, C.; Gouhier, G.; Gautier, A.; Piettre, S. R. J. Org. Chem. 2003, 68, 9916–9923. [PubMed] and references cited therein.
- Dess, D. B.; Martin, J. C. J. Am. Chem. Soc. 1991, 113, 7277–7274. Boeckman, R. K.; Shao, P.; Mullins, J. J. Org. Synth 2000, 77, 141–152.
- Serafinowski, P. J.; Barnes, C. L. Synthesis 1997, 225–228.
- Lopin, C.; Gautier, A.; Gouhier, G.; Piettre, S. R. J. Am. Chem. Soc. 2002, 124, 14668–14675. [PubMed]
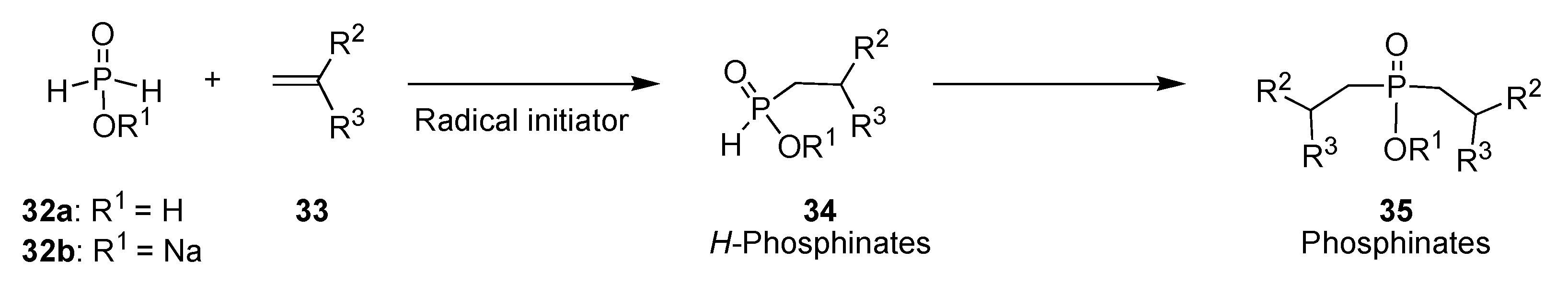
- Nifant’ev, E. E.; Koroteev, M.P. Zh. Obshch. Khim 1967, 37, 1366–1371. Nifant’ev, E. E.; Magdeeva, R. K.; Shchepet’eva, N. P. Zh. Obshch. Khim 1980, 50, 1416–1422. Nifant’ev, E. E.; Solovetskaya, L. A.; Maslennikova, V. I.; Magdeeva, R. K.; Sergeev, N. M. Zh. Obshch. Khim 1986, 56, 680–688. Deprele, S.; Montchamps, J.-L. J. Org. Chem. 2001, 66, 6745–6755. [PubMed]
- Gautier, A.; Garipova, G.; Salcedo, C.; Balieu, S.; Piettre, S. R. Angew. Chem. Int. Ed. 2004, 43. In Press.
- Stawinski, J.; Kraszewski, A. Acc. Chem. Res. 2002, 35, 952–960. [PubMed] Carruthers, M. H. Acc. Chem. Res. 1991, 24, 278–284. (c)Stawinski, J. “Some Aspects of H-Phosphonate Chemistry”. In Handbook of Organophosphorus Chemistry; Engel, R., Ed.; Marcel Dekker: New York, 1992; pp. 377–434. [Google Scholar]
- For examples of the Pudovik reaction, see: Pudovik, A. N.; Konovalova, I. V. Zh. Obshch. Khim. 1959, 29, 3342–3346. Pudovik, A. N.; Konovalova, I. V. Zh. Obshch. Khim. 1960, 30, 2348–2352. Pudovik, A. N.; Kusovleva, R. G. Zh. Obshch. Khim. 1965, 35, 354–358. Pudovik, A. N.; Batyeva, E. S. Zh. Obshch. Khim. 1969, 334–337. Semenzin, D.; Etemad-Moghadam, G.; Albouy, D.; Diallo, O.; Koenig, M. J. Org. Chem. 1997, 62, 2414–2422. [PubMed]
- For examples of the Abramov reaction, see: Abramov, V. S. Dokl. Akad. Nauk S.S.S.R 1950, 73, 487–489. Abramov, V. S.; Semenova, L. P.; Semenova, L. G. Dokl. Akad. Nauk S.S.S.R. 1952, 84, 281–284. Abramov, V. S.; Nazmutdinova, A. S. Zh. Obshch. Khim. 1955, 25, 1141–1146. Thiem, J.; Gunther, M.; Paulsen, H.; Kopf, J. Chem. Ber. 1977, 110, 3190–3200. Wroblewski, A. E. Tetrahedron 1986, 42, 3595–3606. Molin, H.; Noren, J. O.; Claesson, A. Carbohydr. Res 1989, 194, 209–221. [PubMed] (g)Engel, R. Handbook of Organophosphorus Chemistry; Marcel Dekker: New York, 1992. [Google Scholar] Darrow, J. W.; Drueckhammer, D. G. J. Org. Chem. 1994, 59, 2976–2985. Hanessian, S.; Galeotti, N.; Rosen, P.; Oliva, G.; Babu, S. Bioorg. Med.Chem. Lett. 1994, 4, 2763–2768. Darrow, J. W.; Drueckhammer, D. G. Bioorg. Med. Chem. 1996, 4, 1341–1348. [PubMed] Harvey, T. C.; Simiand, C.; Weiler, L.; Withers, S. G. J. Org. Chem. 1997, 62, 6722–6725. Hanessian, S.; Rogel, O. Bioorg. Med. Chem. Lett. 1999, 9, 2441–2446. Hanessian, S.; Rogel, O. J. Org. Chem. 2000, 65, 2667–2674. [PubMed]
- Dubert, O.; Gautier, A.; Condamine, E.; Piettre, S. R. Org. Lett. 2002, 4, 359–362. [PubMed]
- Waschbüsch, R.; Samadi, M.; Savignac, P. J. Organomet. Chem. 1997, 529, 267–278.
- Piettre, S. R.; Raboisson, P. Tetrahedron Lett. 1996, 37, 2229–2232.
- Obayashi, M.; Ito, E.; Kondo, K. Tetrahedron Lett. 1982, 23, 2323–2326.
- Lopin, C.; Gouhier, G.; Piettre, S. Tetrahedron Lett. 2003, 44, 8837–8840.
- Nomura, M.; Sato, T.; Washinosu, M.; Tanaka, M.; Asao, T.; Shuto, S.; Matsuda, A. Tetrahedron 2002, 58, 1279–1288.
- DeLucca, L.; Giacomelli, G.; Porcheddu, A. Org. Lett. 2001, 3, 3041–3043.
- Kalinina, I.; Gautier, A.; Salcedo, C.; Valnot, J.-Y.; Piettre, S. R. Tetrahedron 2004, 60, 4895–4900.
- Niedballa, U.; Vorbrüggen, H. Angew. Chem. Int. Ed. 1970, 9, 461–462. Niedballa, U.; Vorbrüggen, H. J. Org. Chem. 1974, 39, 3654–3660. [PubMed] ibid 3660–3663. ibid 3664–3667. ibid 3668–3671. ibid 3672–3674. Niedballa, U.; Vorbrüggen, H. J. Org. Chem. 1976, 41, 2084–2086. [PubMed] Vorbrüggen, H.; Bennua, B. Tetrahedron Lett. 1978, 39, 1339–1342. Vorbrüggen, H.; Krolikiewicz, K.; Bennua, B. Chem. Ber. 1981, 114, 1234–1255. Vorbrüggen, H.; Hölfe, G. Chem. Ber. 1981, 114, 1256–1268.
- Saneyoshi, M.; Satoh, E. Chem. Pharm. Bull. 1978, 27, 2518. Zou, R.; Robins, M. J. Can.J. Chem. 1987, 65, 1436–1437. Robins, M. J.; Zou, R.; Guo, Z.; Wnuk, S. F. J. Org. Chem. 1996, 61, 9207–9212.
- Yokomatsu, T.; Sada, T.; Shimizu, T.; Shibuya, S. Tetrahedron Lett. 1998, 39, 6299–6302.
- Piettre, S. R. Tetrahedron Lett. 1996, 37, 4707–4710.
- Kiddle, J. J. Chem. Rev. 1995, 95, 2189–2202.
- Sabol, J. S.; McCarthy, J. R. Tetrahedron Lett. 1992, 33, 3101–3104.
- Marco-Contelles, J.; Molina, M. T.; Anjum, S. Chem. Rev. 2004, 104, 2857–2899. [PubMed]
- Ferrier, R. J. Chem. Soc., Perkin Trans. 1 1979, 1455–1458. Ferrier, R.; Middleton, S. Chem. Rev. 1993, 93, 2779–2831. Dalko, P.; Sinaÿ, P. Angew. Chem., Int. Ed. 1999, 38, 773–777. Sollogoub, M.; Mallet, J.-M.; Sinaÿ, P. Angew. Chem., Int. Ed. 2000, 39, 362–364.
© 2005 by MDPI (http://www.mdpi.org). Reproduction is permitted for noncommercial purposes.
Share and Cite
Gautier, A.; Lopin, C.; Garipova, G.; Dubert, O.; Kalinina, I.; Salcedo, C.; Balieu, S.; Glatigny, S.; Valnot, J.; Gouhier, G.; et al. The Preparation of New Phosphorus-Centered Functional Groups for Modified Oligonucleotides and Other Natural Phosphates. Molecules 2005, 10, 1048-1073. https://doi.org/10.3390/10091048
Gautier A, Lopin C, Garipova G, Dubert O, Kalinina I, Salcedo C, Balieu S, Glatigny S, Valnot J, Gouhier G, et al. The Preparation of New Phosphorus-Centered Functional Groups for Modified Oligonucleotides and Other Natural Phosphates. Molecules. 2005; 10(9):1048-1073. https://doi.org/10.3390/10091048
Chicago/Turabian StyleGautier, A., C. Lopin, G. Garipova, O. Dubert, I. Kalinina, C. Salcedo, S. Balieu, S. Glatigny, J. Valnot, G. Gouhier, and et al. 2005. "The Preparation of New Phosphorus-Centered Functional Groups for Modified Oligonucleotides and Other Natural Phosphates" Molecules 10, no. 9: 1048-1073. https://doi.org/10.3390/10091048