




Plants 2023, 12(15), 2807; https://doi.org/10.3390/plants12152807 - 28 Jul 2023
Viewed by 698
Abstract
►
Show Figures
Common vetch (Vicia sativa L.) is one of the most cultivated feed crops with extensive agricultural diversity and numerous cultivars. This study concerns the first-time investigation of the dry plant biomass and grains of six vetch cultivars to define the detailed fingerprint
[...] Read more.
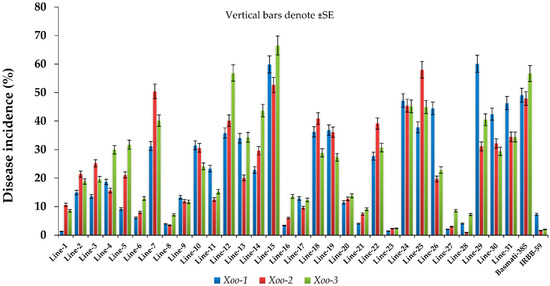
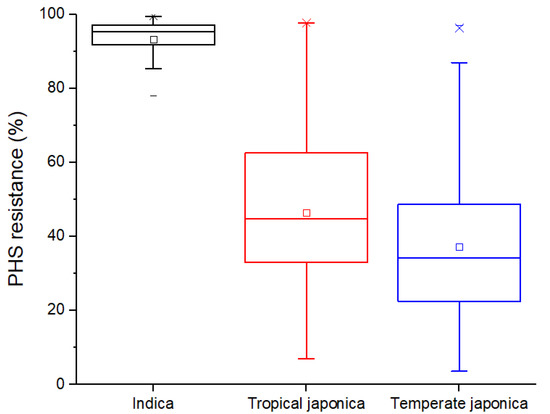
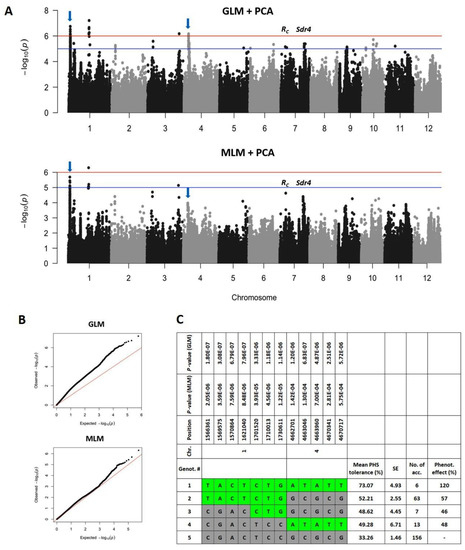
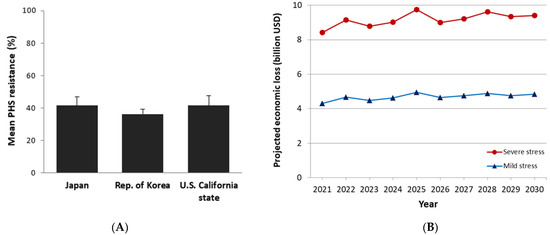
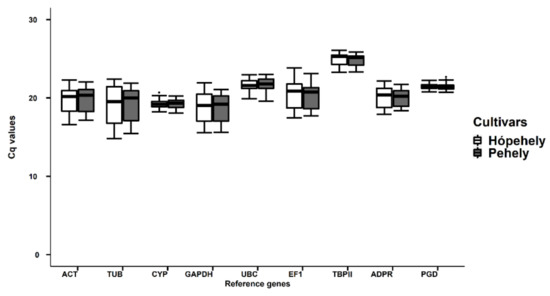
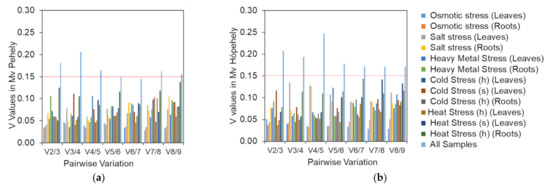
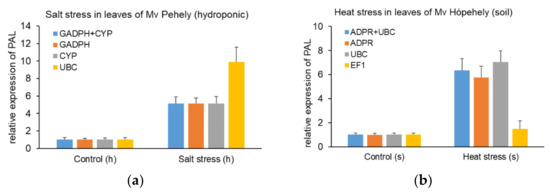
Common vetch (Vicia sativa L.) is one of the most cultivated feed crops with extensive agricultural diversity and numerous cultivars. This study concerns the first-time investigation of the dry plant biomass and grains of six vetch cultivars to define the detailed fingerprint of their phenolic and fatty acid content, along with their respective antioxidant potencies. The results revealed a substantial variation in the feed quality traits among the tested Vicia sativa varieties, highlighting the crucial role and influence the genotype plays in the achievement of high-quality livestock nutrition. Among the six varieties tested, Istros and M-6900 displayed a particularly intriguing phytochemical profile characterized by elevated phenolic content, significant antioxidant potency and remarkably high fatty acid indices. These findings are indicative of the great potential of these varieties to function as suitable candidates for incorporation into farm animal diets either in the form of dry biomass (hay) or as a grain feed additive.
Full article
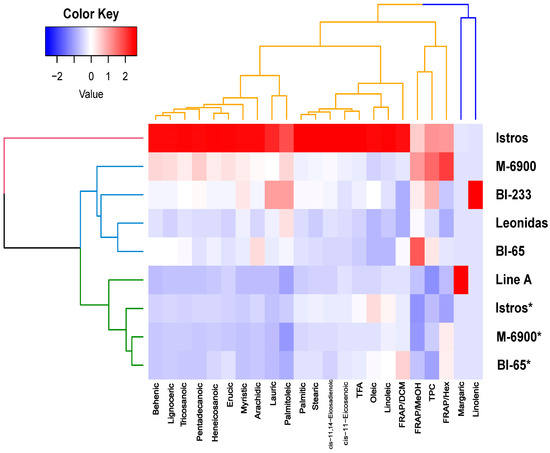
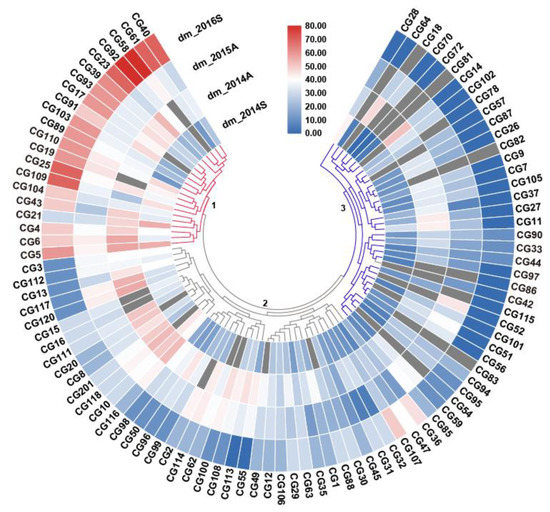
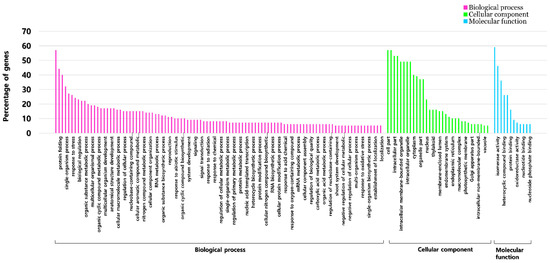
Figure 1


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}