




Molecules 2021, 26(3), 710; https://doi.org/10.3390/molecules26030710 - 29 Jan 2021
Cited by 2 | Viewed by 2258
Abstract
►
Show Figures
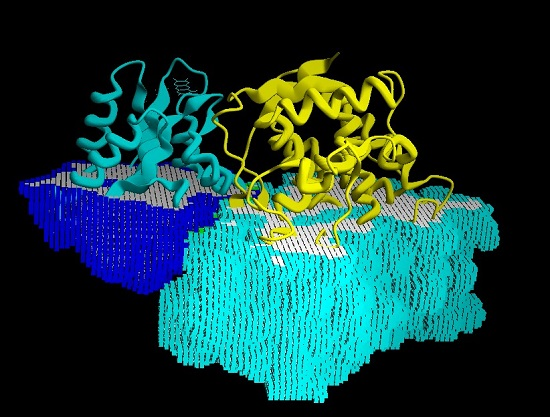
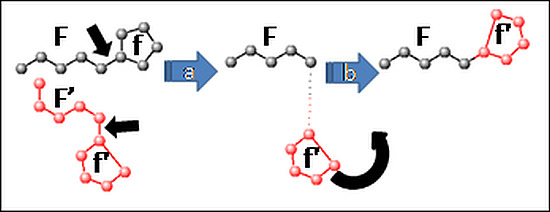
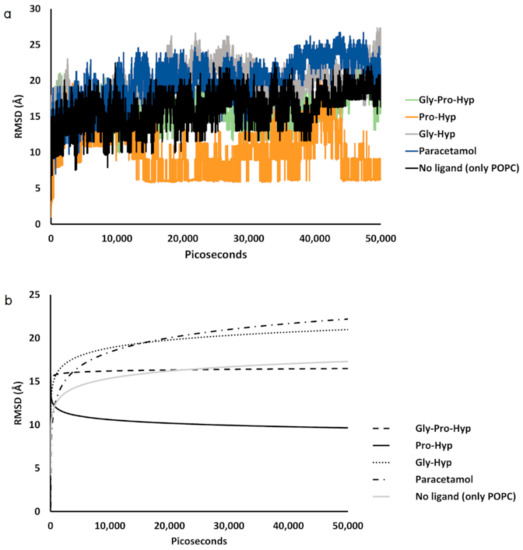
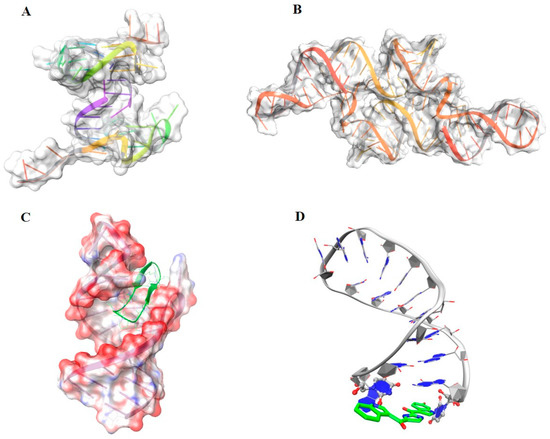
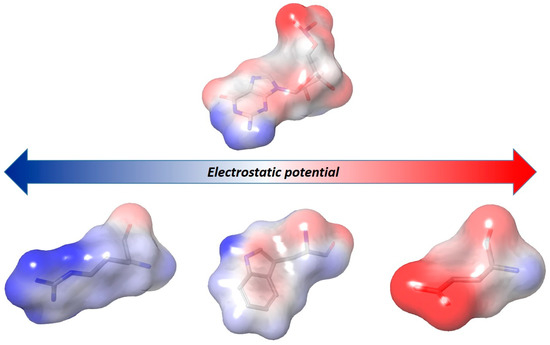
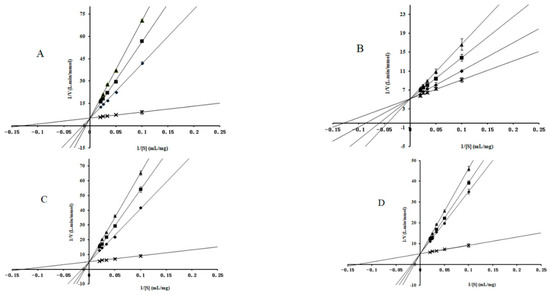
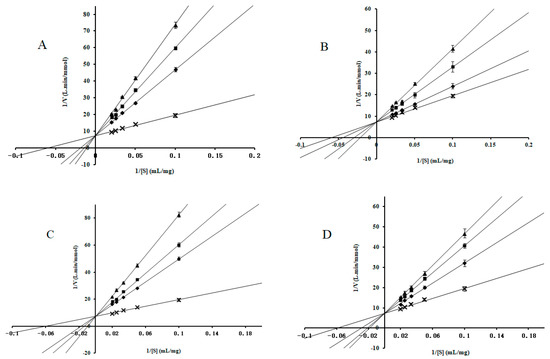
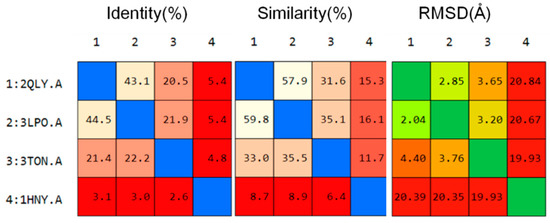
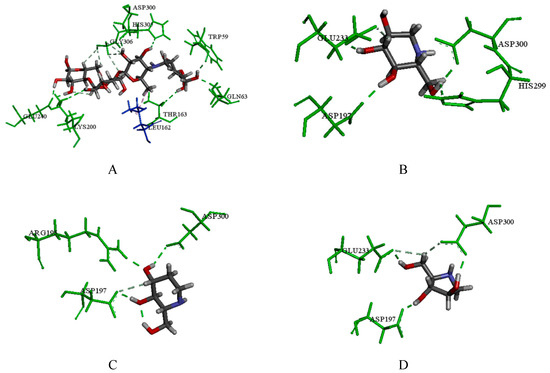
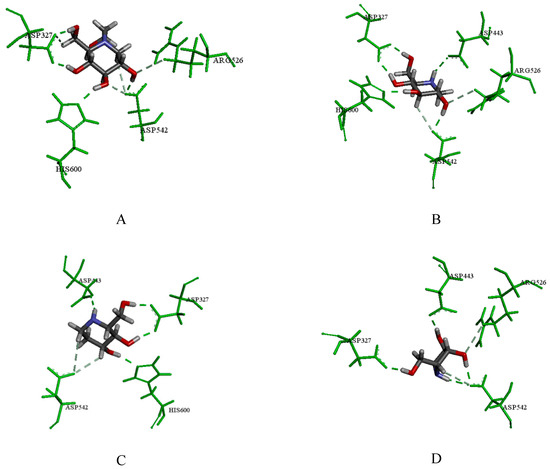
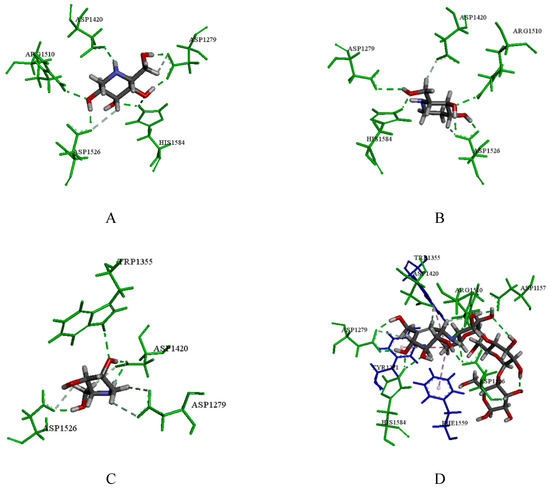
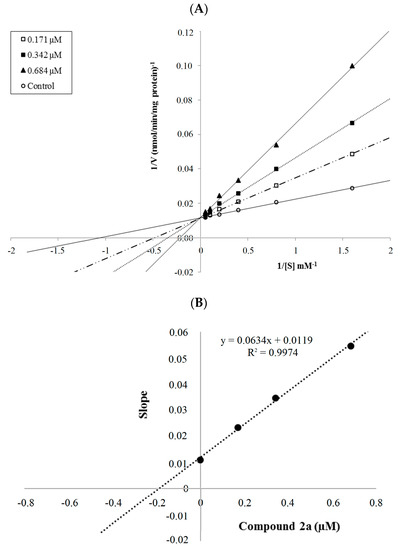
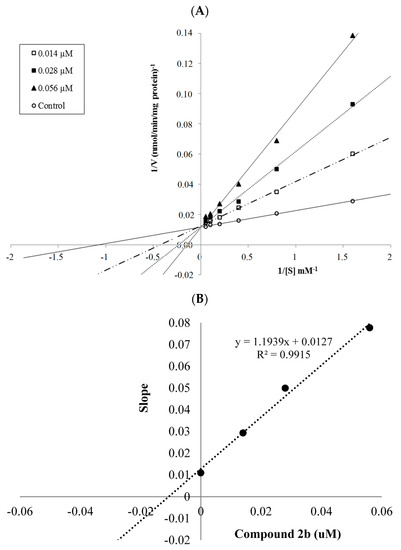
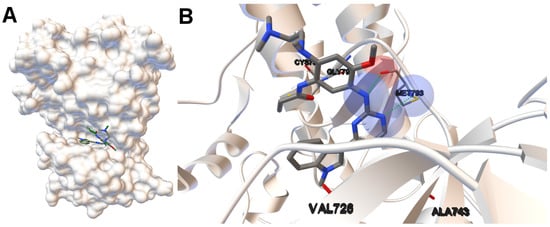
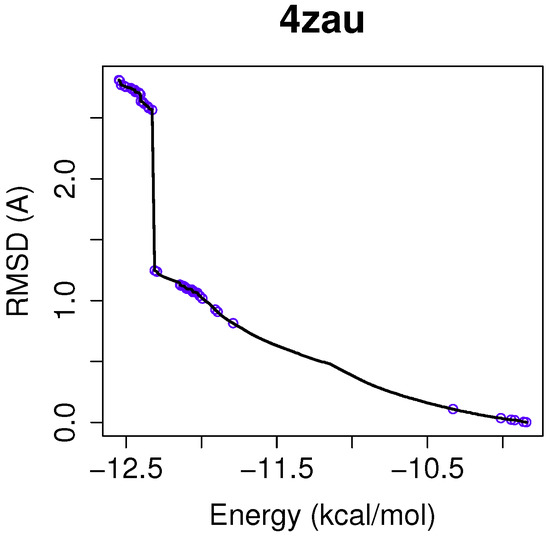
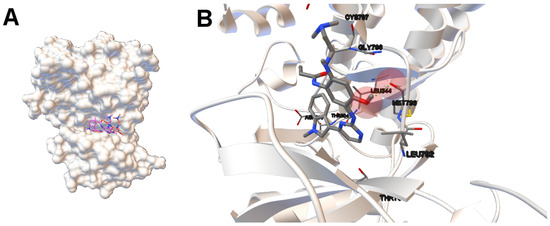
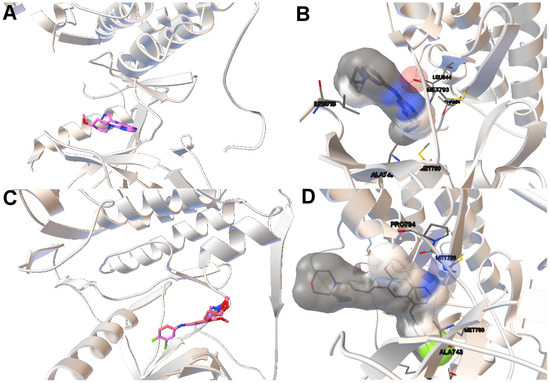
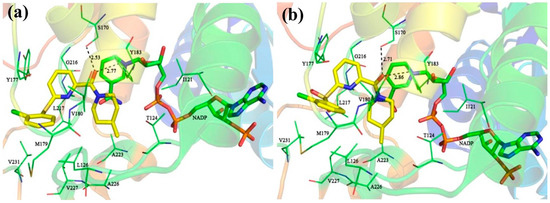
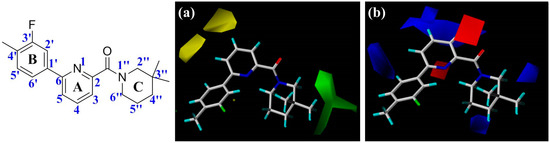
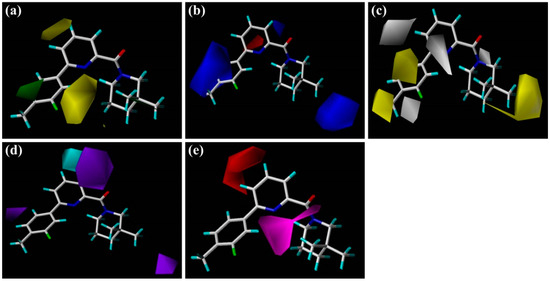
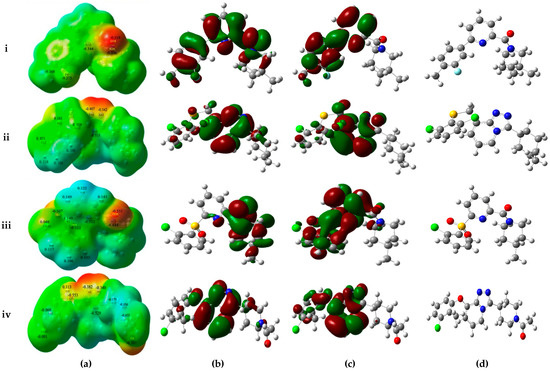
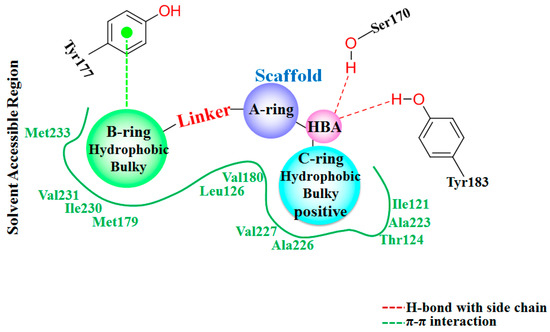
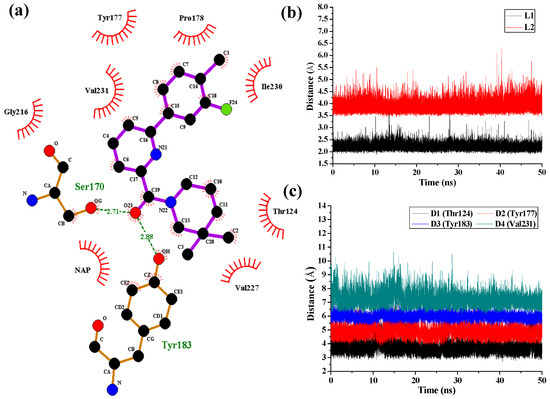
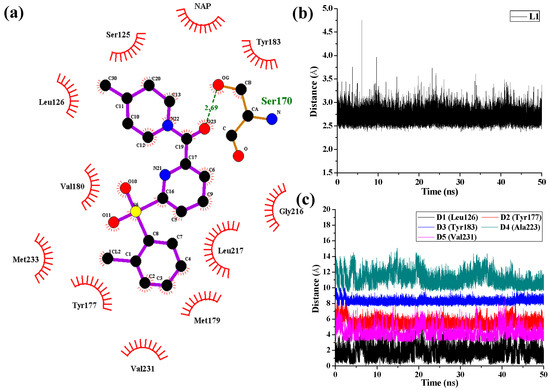
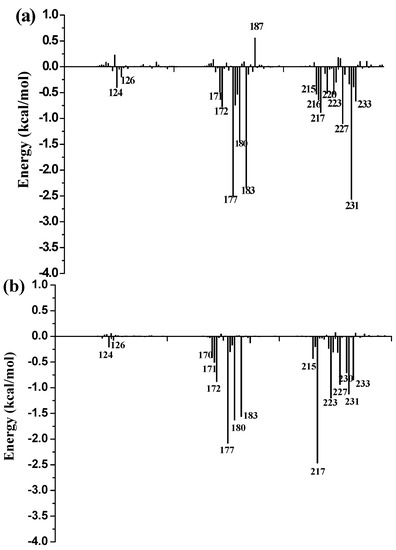
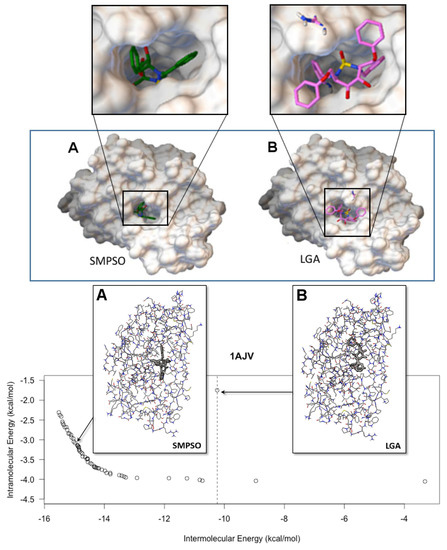
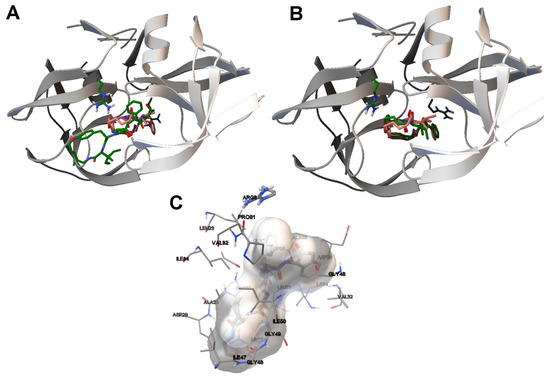
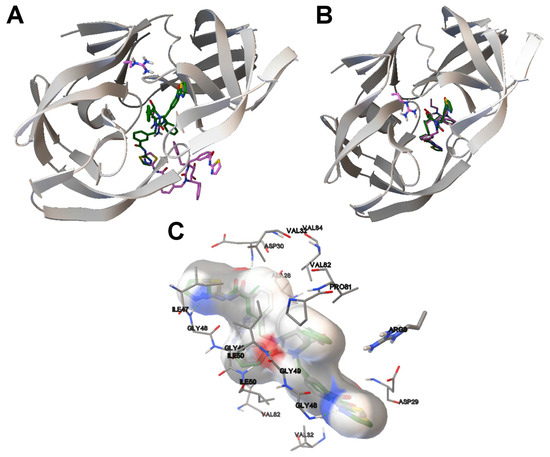
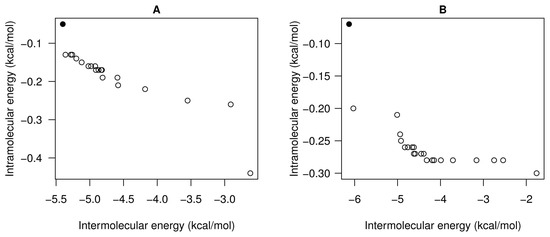
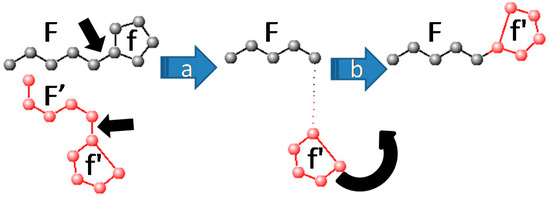
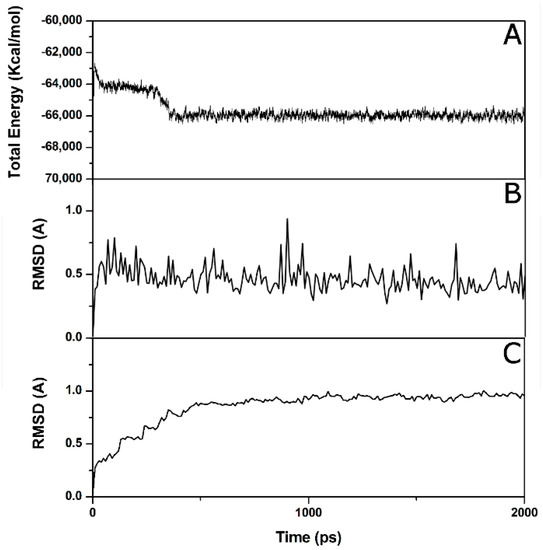
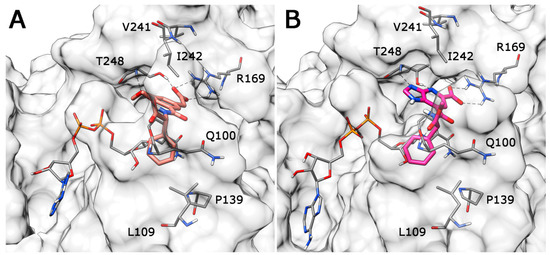
Collagen contains hydroxyproline (Hyp), which is a unique amino acid. Three collagen-derived small peptides (Gly-Pro-Hyp, Pro-Hyp, and Gly-Hyp) interacting across a lipid bilayer (POPC model membrane) for cellular uptakes of these collagen-derived small peptides were studied using accelerated molecular dynamics simulation. The ligands
[...] Read more.
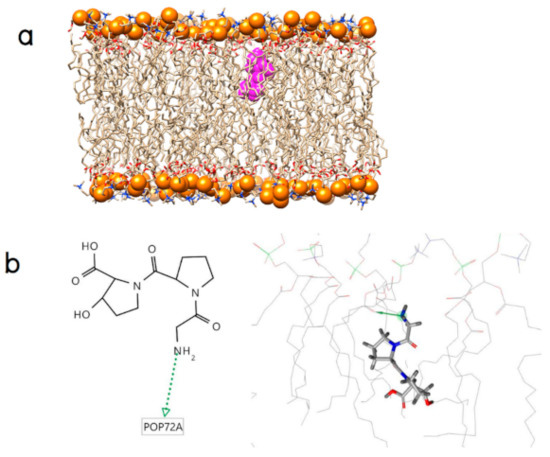
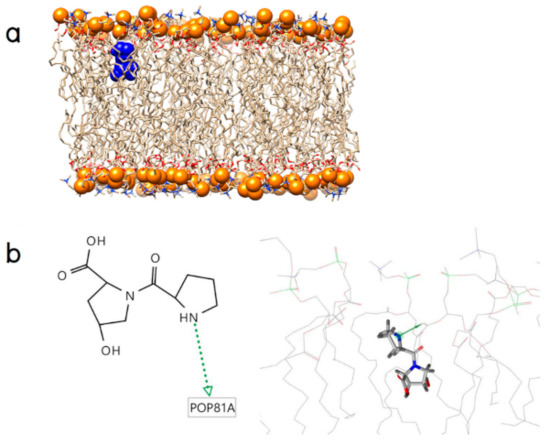
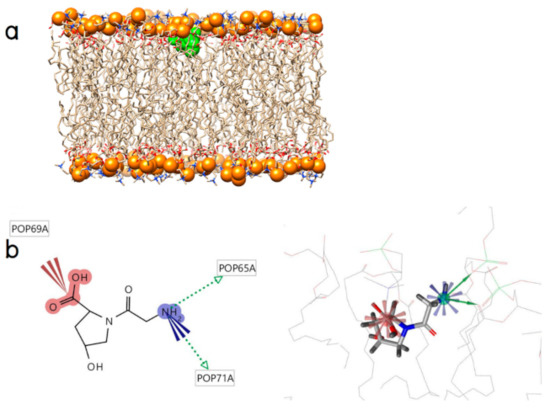
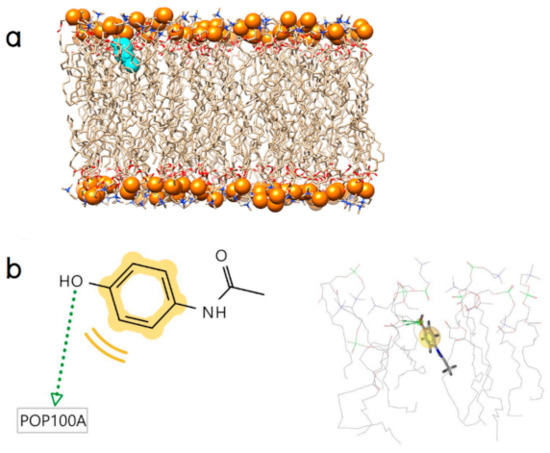
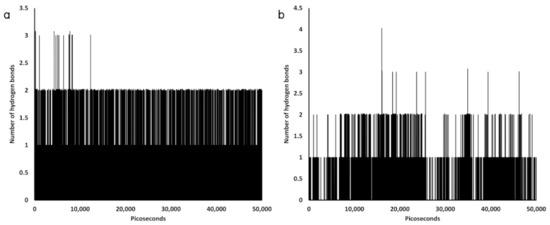
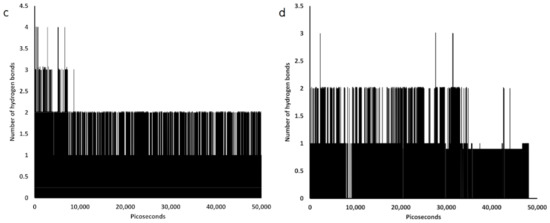
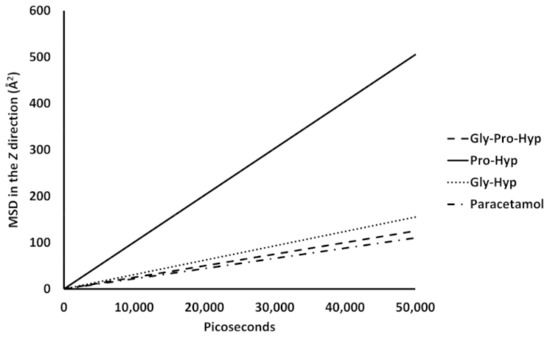
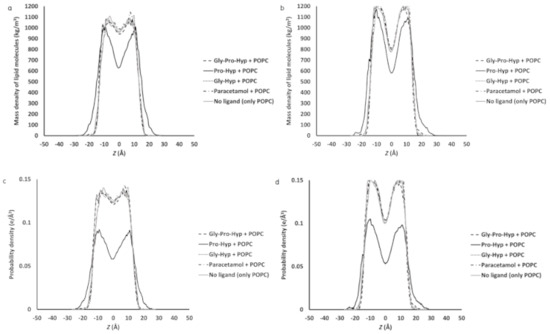
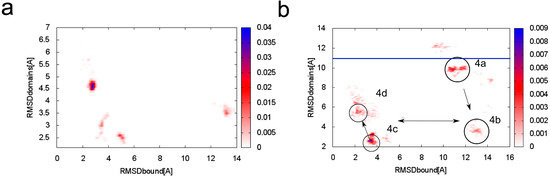
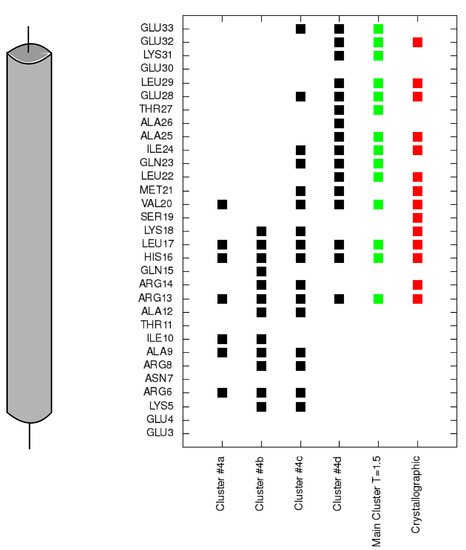
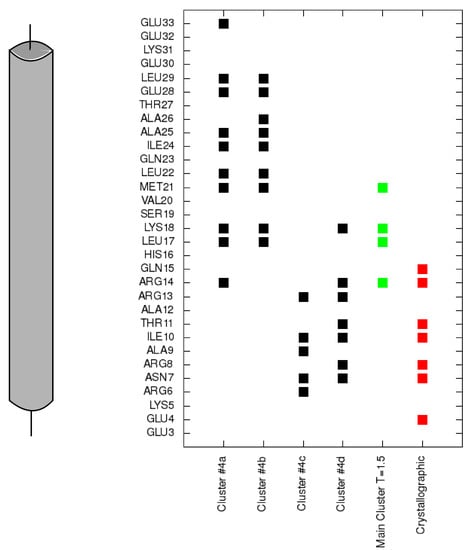
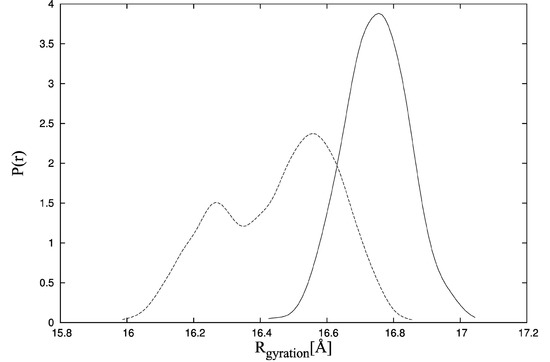
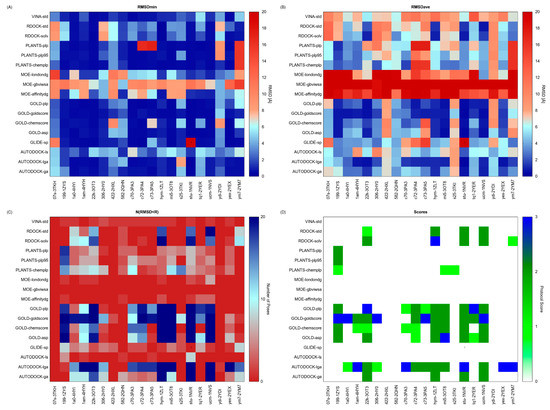
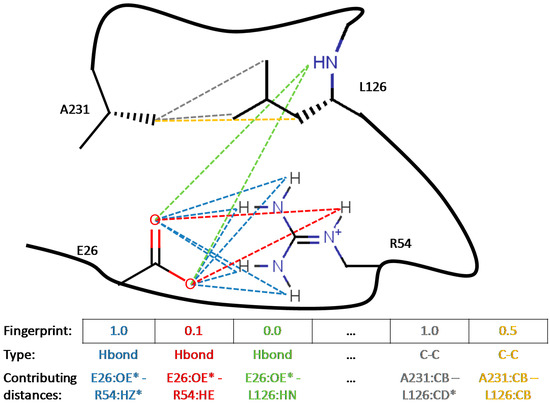
Collagen contains hydroxyproline (Hyp), which is a unique amino acid. Three collagen-derived small peptides (Gly-Pro-Hyp, Pro-Hyp, and Gly-Hyp) interacting across a lipid bilayer (POPC model membrane) for cellular uptakes of these collagen-derived small peptides were studied using accelerated molecular dynamics simulation. The ligands were investigated for their binding modes, hydrogen bonds in each coordinate frame, and mean square displacement (MSD) in the Z direction. The lipid bilayers were evaluated for mass and electron density profiles of the lipid molecules, surface area of the head groups, and root mean square deviation (RMSD). The simulation results show that hydrogen bonding between the small collagen peptides and plasma membrane plays a significant role in their internalization. The translocation of the small collagen peptides across the cell membranes was shown. Pro-Hyp laterally condensed the membrane, resulting in an increase in the bilayer thickness and rigidity. Perception regarding molecular behaviors of collagen-derived peptides within the cell membrane, including their interactions, provides the novel design of specific bioactive collagen peptides for their applications.
Full article
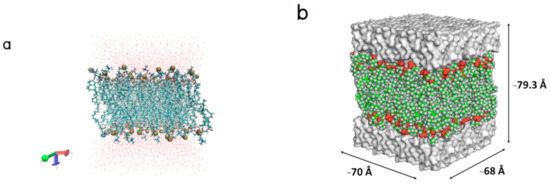
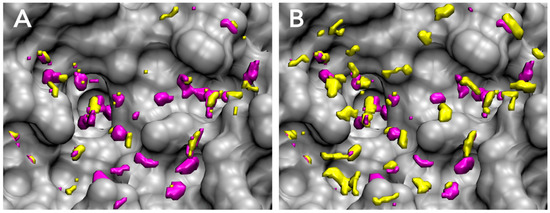
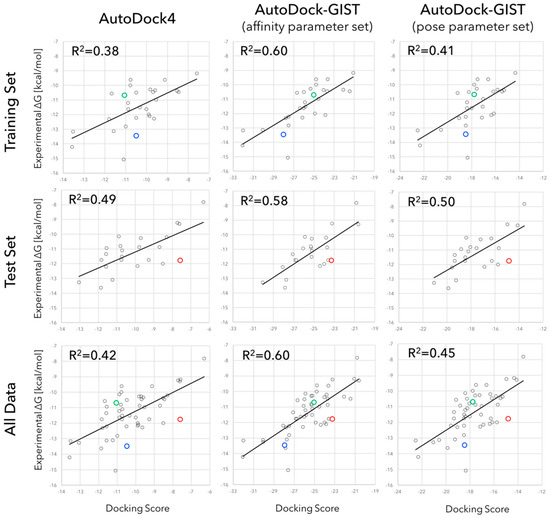
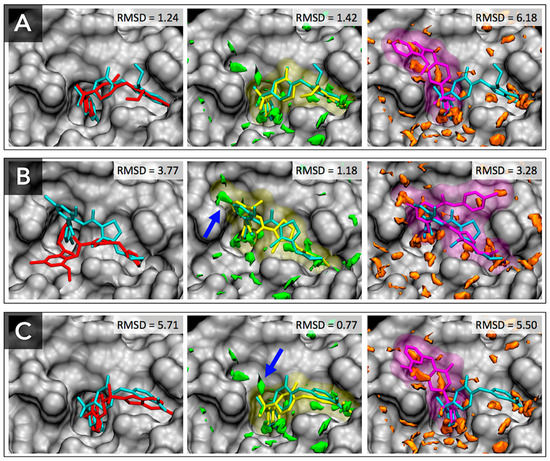
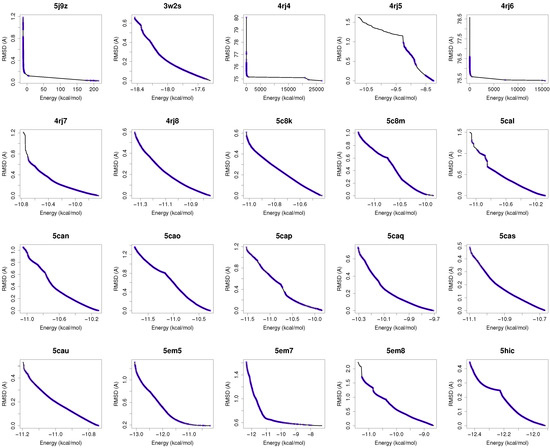
Figure 1







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
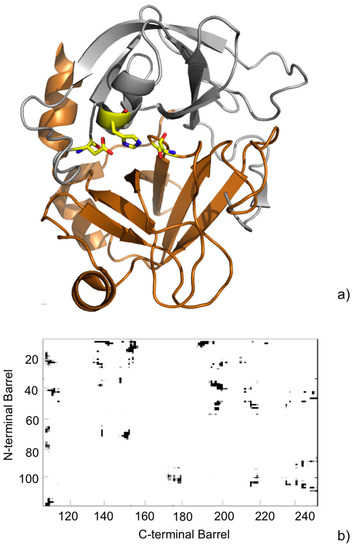{kind=link}
{kind=link}
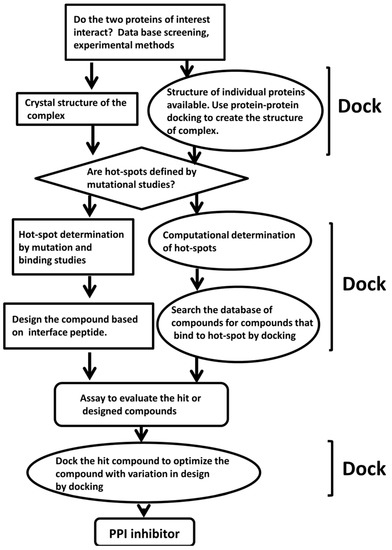{kind=link}
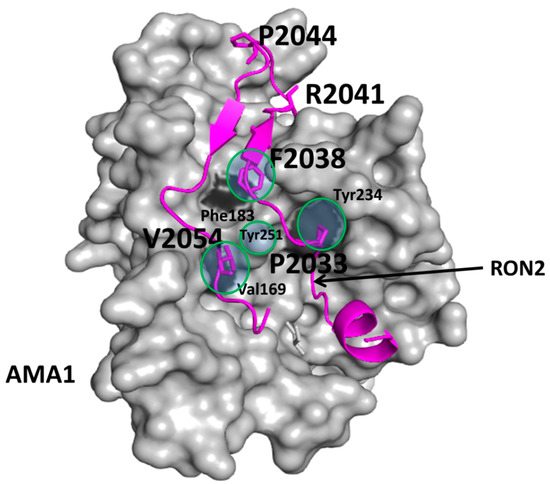{kind=link}
{kind=link}
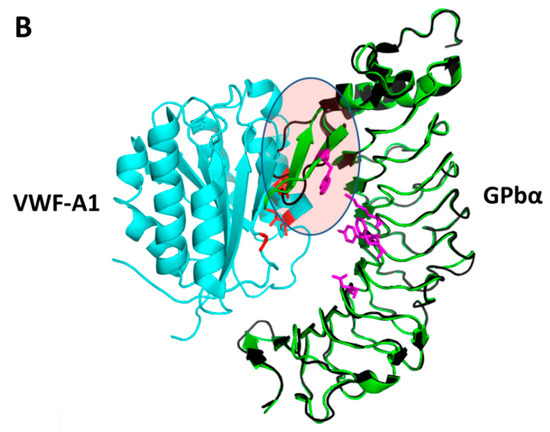{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}