



Membranes 2023, 13(8), 737; https://doi.org/10.3390/membranes13080737 - 17 Aug 2023
Cited by 1 | Viewed by 978
Abstract
►
Show Figures
PSD95-disc large-zonula occludens (PDZ) domains are globular modules of 80–90 amino acids that co-evolved with multicellularity. They commonly bind to carboxy-terminal sequences of a plethora of membrane-associated proteins and influence their trafficking and signaling. We previously built a PDZ resource (PDZome) allowing us
[...] Read more.
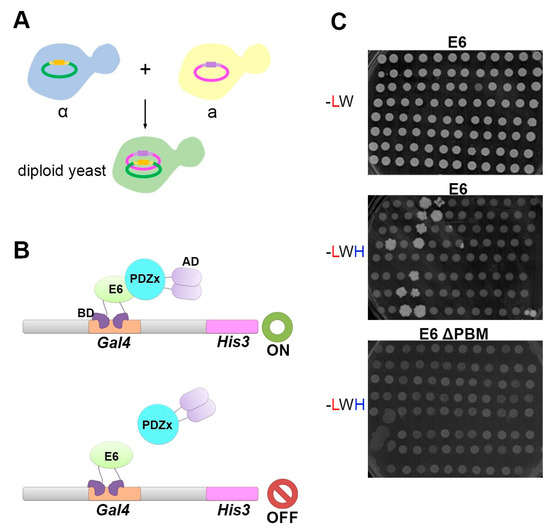
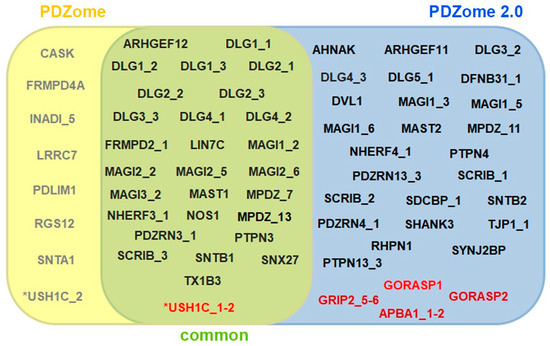
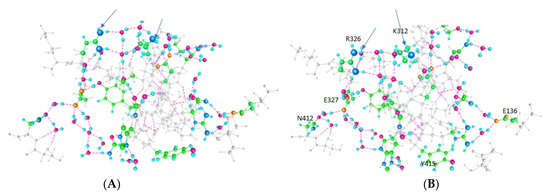
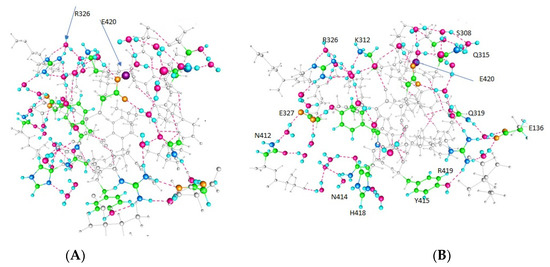
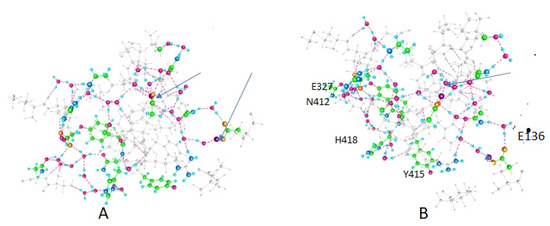
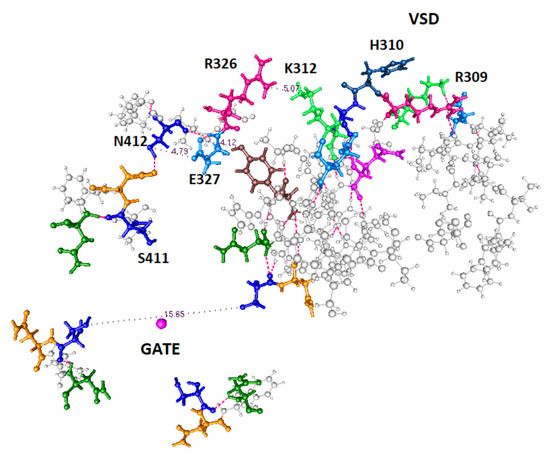
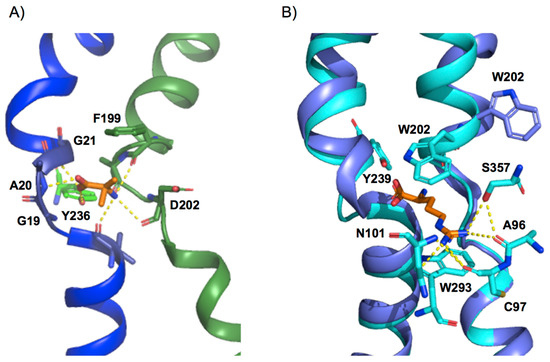
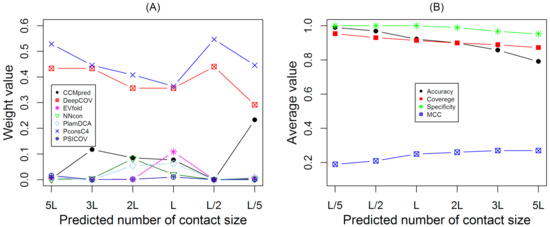
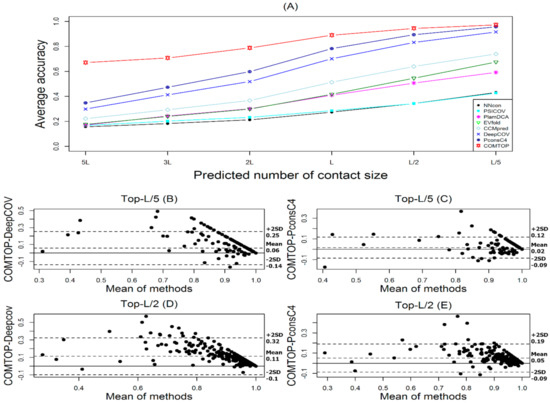
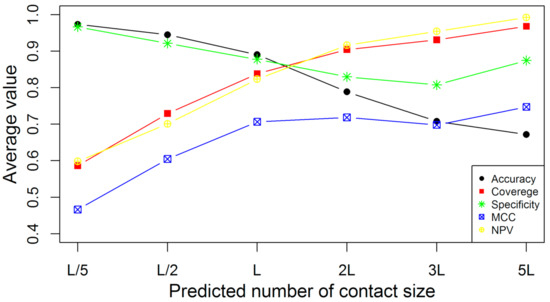
PSD95-disc large-zonula occludens (PDZ) domains are globular modules of 80–90 amino acids that co-evolved with multicellularity. They commonly bind to carboxy-terminal sequences of a plethora of membrane-associated proteins and influence their trafficking and signaling. We previously built a PDZ resource (PDZome) allowing us to unveil human PDZ interactions by Yeast two-hybrid. Yet, this resource is incomplete according to the current knowledge on the human PDZ proteome. Here we built the PDZome 2.0 library for Yeast two-hybrid, based on a PDZ library manually curated from online resources. The PDZome2.0 contains 305 individual clones (266 PDZ domains in isolation and 39 tandems), for which all boundaries were designed based on available PDZ structures. Using as bait the E6 oncoprotein from HPV16, a known promiscuous PDZ interactor, we show that PDZome 2.0 outperforms the previous resource.
Full article
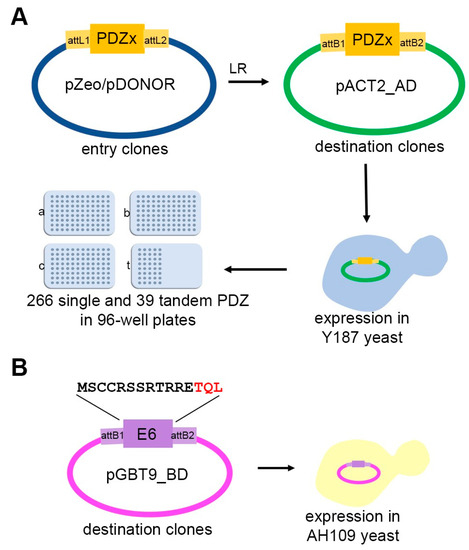
Figure 1
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}