




Genes 2024, 15(3), 349; https://doi.org/10.3390/genes15030349 - 10 Mar 2024
Viewed by 918
Abstract
►
Show Figures
The production of milk by dairy cows far exceeds the nutritional needs of the calf and is vital for the economical use of dairy cattle. High milk yield is a unique production trait that can be effectively enhanced through traditional selection methods. The
[...] Read more.
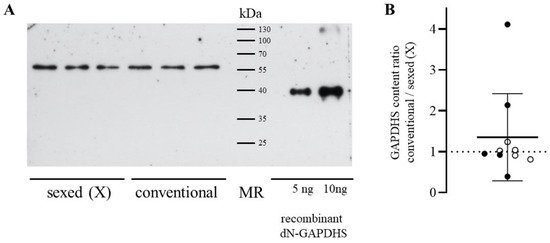
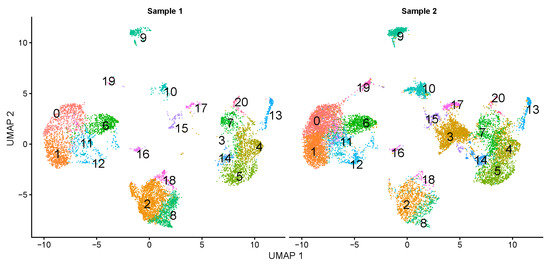
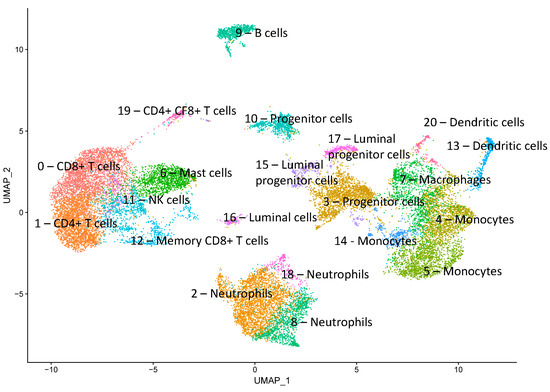
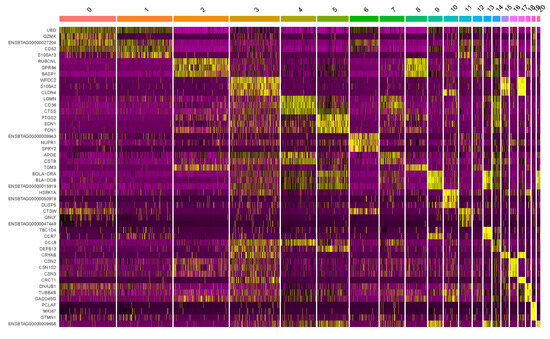
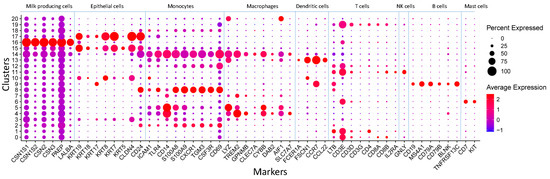
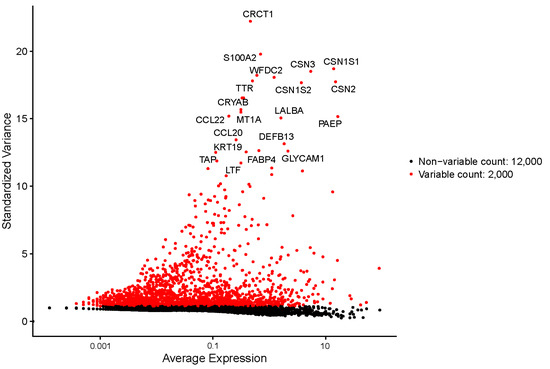
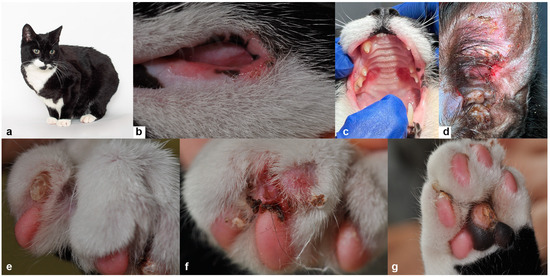
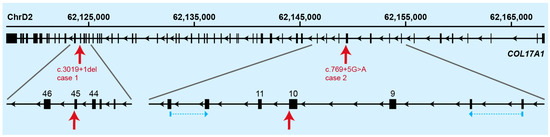
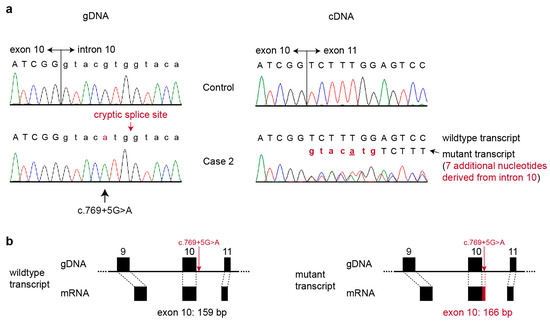
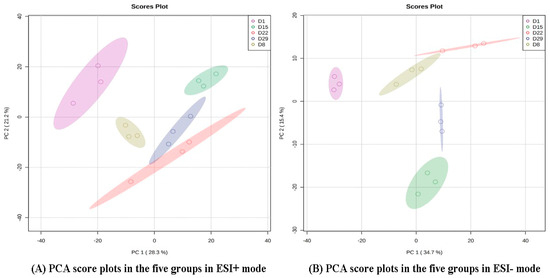
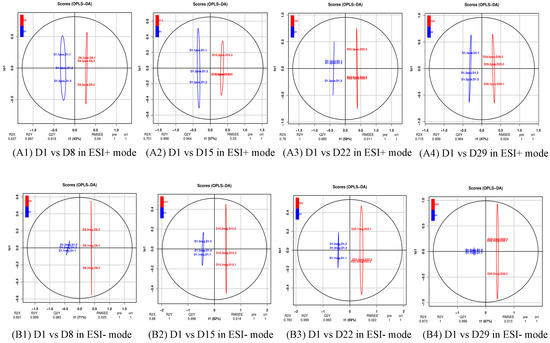
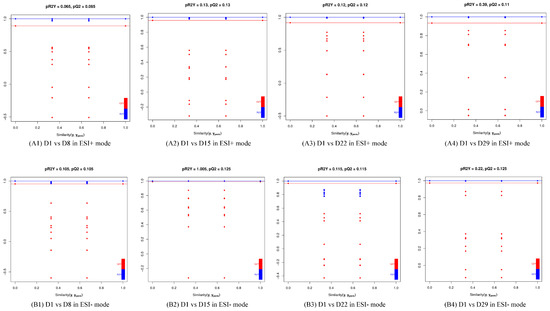
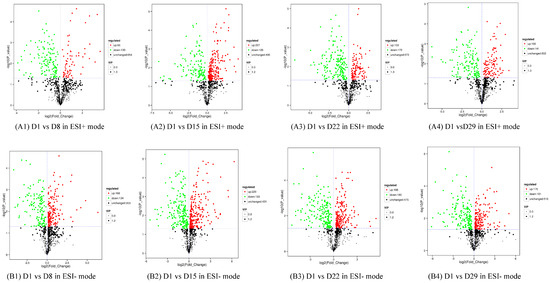
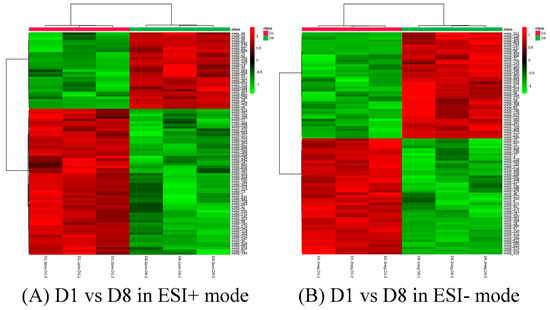
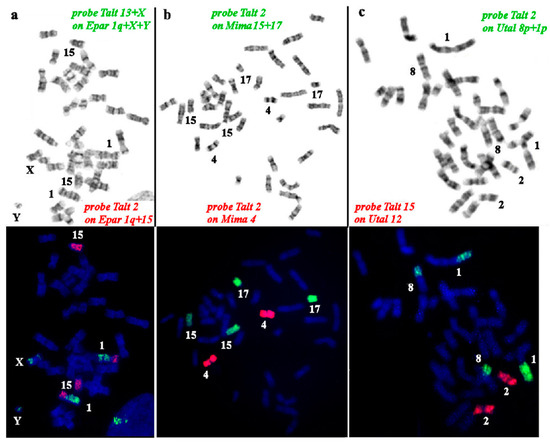
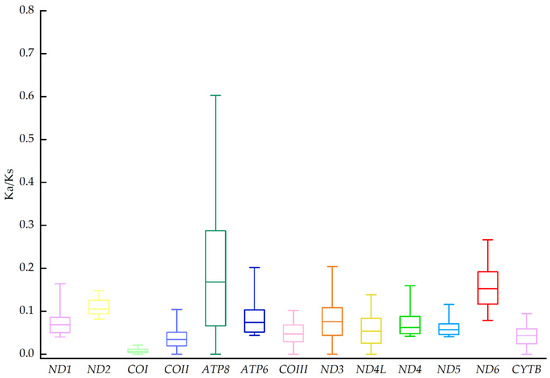
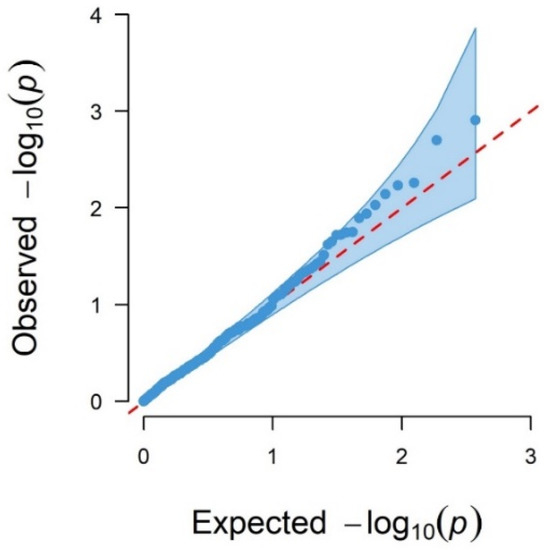
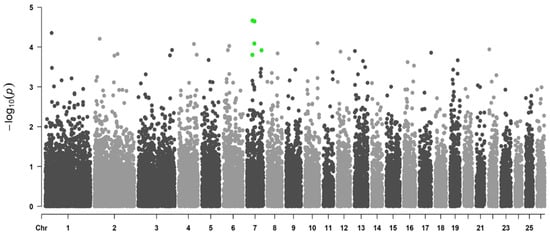
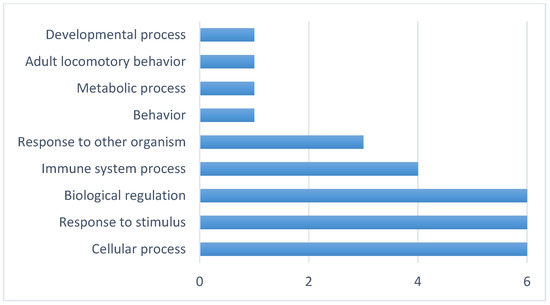
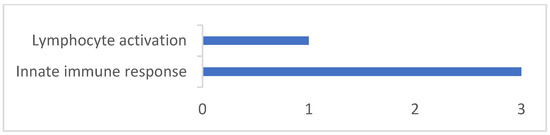
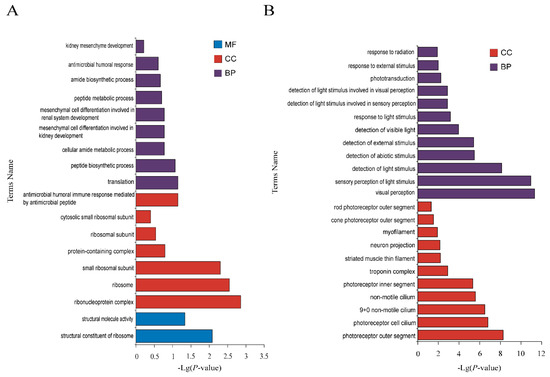
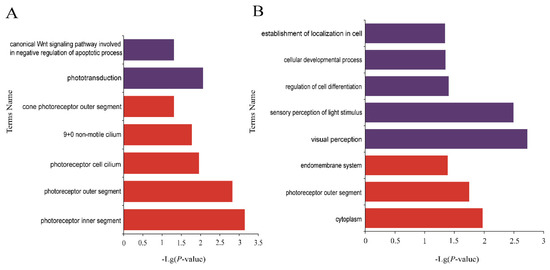
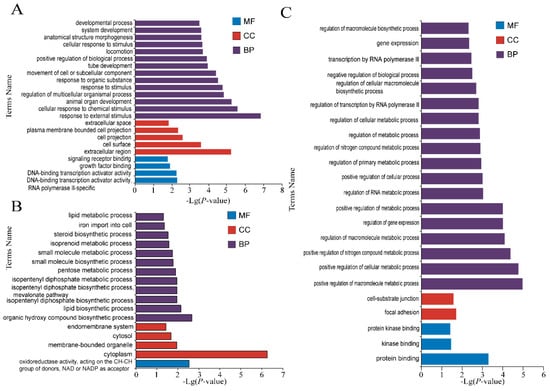
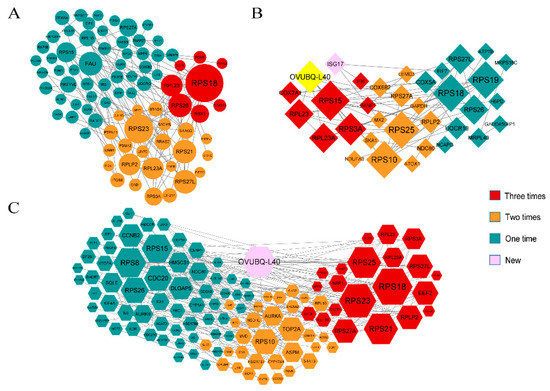
The production of milk by dairy cows far exceeds the nutritional needs of the calf and is vital for the economical use of dairy cattle. High milk yield is a unique production trait that can be effectively enhanced through traditional selection methods. The process of lactation in cows serves as an excellent model for studying the biological aspects of lactation with the aim of exploring the mechanistic base of this complex trait at the cellular level. In this study, we analyzed the milk transcriptome at the single-cell level by conducting scRNA-seq analysis on milk samples from two Holstein Friesian cows at mid-lactation (75 and 93 days) using the 10× Chromium platform. Cells were pelleted and fat was removed from milk by centrifugation. The cell suspension from each cow was loaded on separate channels, resulting in the recovery of 9313 and 14,544 cells. Library samples were loaded onto two lanes of the NovaSeq 6000 (Illumina) instrument. After filtering at the cell and gene levels, a total of 7988 and 13,973 cells remained, respectively. We were able to reconstruct different cell types (milk-producing cells, progenitor cells, macrophages, monocytes, dendritic cells, T cells, B cells, mast cells, and neutrophils) in bovine milk. Our findings provide a valuable resource for identifying regulatory elements associated with various functions of the mammary gland such as lactation, tissue renewal, native immunity, protein and fat synthesis, and hormonal response.
Full article
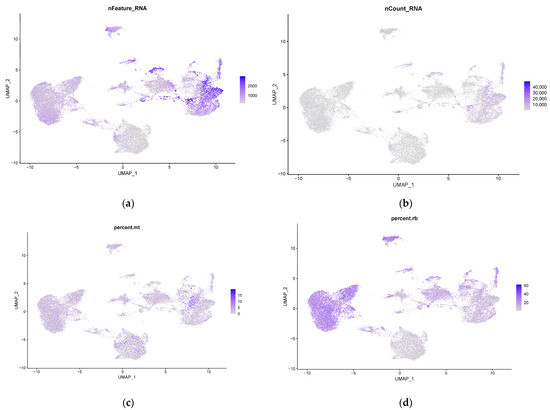
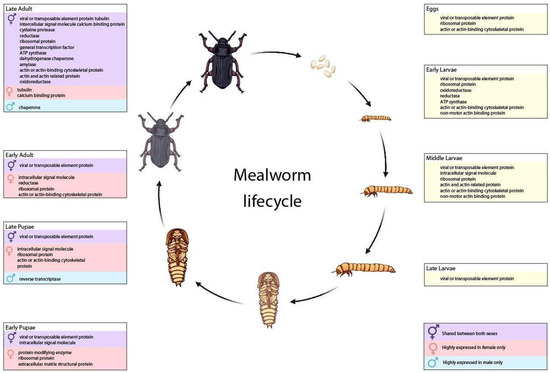
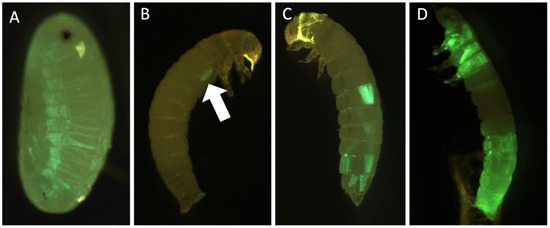
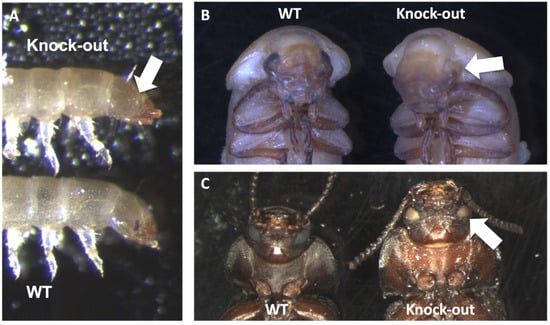
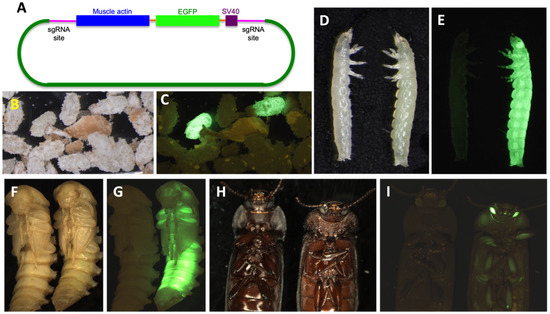
Figure 1






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}