Dear Colleagues,
Lysosomal storage diseases (LSDs) comprise a group of over 50 genetically inherited disorders that are characterized by accumulation of undigested material inside the lysosome, most often caused by a deficiency of the enzyme normally responsible for catabolism of various molecules derived from cellular turnover. This condition results in accumulation of the undegraded substrate in various tissues and organs of the body causing these organs to function less efficiently, resulting in progressive deterioration in physical and/or mental state, and eventually cell degeneration.
This Topical Collection provides an open access opportunity for investigators to contribute with original research articles as well as review articles that will allow a better understanding of the biochemical and molecular physiopathology underlying LSDs, potential biomarkers of disease progression and new therapeutic strategies to treat these diseases, and evaluation of treatment efficiency.
Potential topics include, but are not limited to:
- Recent progress in LSDs research
- Diagnosis of LSDs
- Technological developments in newborn and population screening
- New pathophysiological mechanisms involved in LSDs
- Biomarkers in disease staging, monitoring, and efficiency of treatment
- Recent advances in treatment and drug delivery
Prof. Dr. Jose A. Sanchez-Alcazar
Dr. Luis M. Jiménez Jiménez
Collection Editors
Manuscript Submission Information
Manuscripts should be submitted online at www.mdpi.com by registering and logging in to this website. Once you are registered, click here to go to the submission form. Manuscripts can be submitted until the deadline. All submissions that pass pre-check are peer-reviewed. Accepted papers will be published continuously in the journal (as soon as accepted) and will be listed together on the collection website. Research articles, review articles as well as short communications are invited. For planned papers, a title and short abstract (about 100 words) can be sent to the Editorial Office for announcement on this website.
Submitted manuscripts should not have been published previously, nor be under consideration for publication elsewhere (except conference proceedings papers). All manuscripts are thoroughly refereed through a single-blind peer-review process. A guide for authors and other relevant information for submission of manuscripts is available on the Instructions for Authors page. Diseases is an international peer-reviewed open access monthly journal published by MDPI.
Please visit the Instructions for Authors page before submitting a manuscript.
The Article Processing Charge (APC) for publication in this open access journal is 1800 CHF (Swiss Francs).
Submitted papers should be well formatted and use good English. Authors may use MDPI's
English editing service prior to publication or during author revisions.
The below list represents only planned manuscripts. Some of these
manuscripts have not been received by the Editorial Office yet. Papers
submitted to MDPI journals are subject to peer-review.
Type of the paper: Review
Tentative title: Nonsense Suppression as an Approach to Treat Lysosomal Storage Diseases
Author: Kim M. Keeling
Affiliations: University of Alabama at Birmingham, Department of Biochemistry and Molecular Genetics, UAB Gregory Fleming Cystic Fibrosis Research Center, UAB Comprehensive Arthritis, Musculoskeletal, Bone, and Autoimmunity Center Birmingham, AL 35294, USA
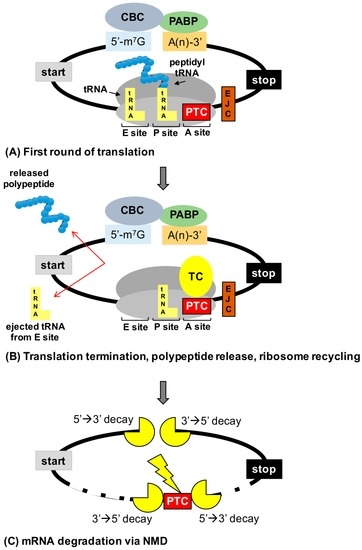
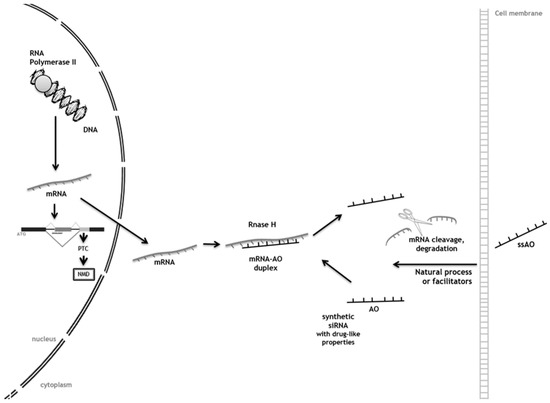
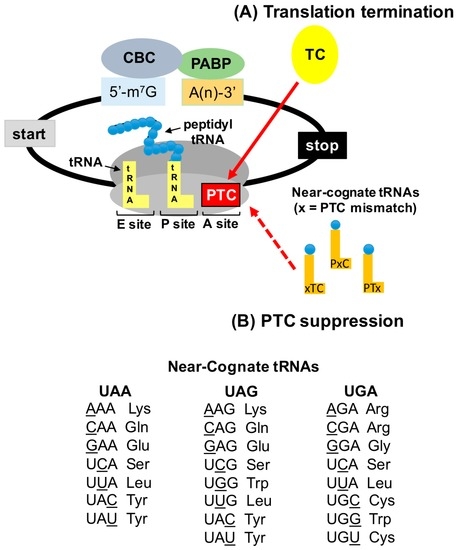
Abstract: In-frame premature termination codons (PTCs), also known as nonsense mutations, comprise 11% of all disease-associated gene lesions. PTCs reduce gene expression in two ways. First, PTCs mediate premature translation termination of an mRNA, leading to the production of a truncated polypeptide that often is unstable and/or lacks normal function. Second, PTCs trigger degradation of an mRNA by activating nonsense-mediated mRNA decay (NMD), a cellular pathway that recognizes and degrades mRNAs containing a PTC. Thus, translation termination and NMD serve as putative therapeutic targets to develop treatments for genetic diseases caused by PTCs. Over the past decade, significant progress has been made in the identification of drugs with the ability to suppress translation termination (also called readthrough) of PTCs. More recently, NMD inhibitors have also been explored as a way to enhance the efficiency of PTC suppression. Lysosomal storage diseases are a particularly interesting group of diseases to investigate the effectiveness of PTC suppression drugs due to their relativity low threshold for correction compared to other genetic diseases such as cystic fibrosis and Duchenne muscular dystrophy. In this review, the current status of PTC suppression and NMD inhibition as a potential treatment for lysosomal storage diseases will be discussed.
Type of Paper: Short communication
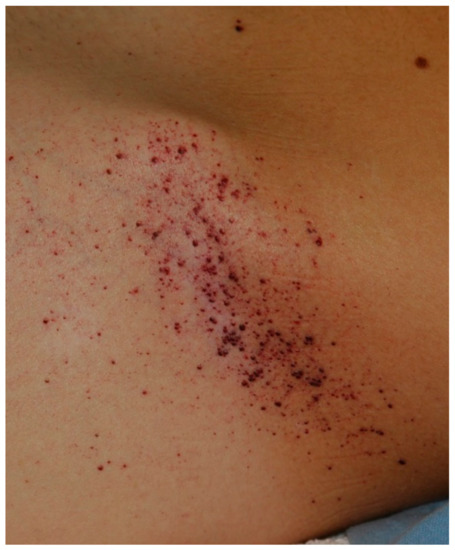
Tentative Title: Sweat gland biopsy: A possible early diagnostic tool in the Anderson-Fabry disease
Authors: Pistone Giuseppe, Caputo Valentina, Fattore Davide, Tilotta Giovanna, Bongiorno Maria Rita
Affiliations: Department of Dermatology, University of Palermo, via del Vespro 131, 90127, Palermo, Italy.
Abstract: Anderson-Fabry disease is a rare X-linked lysosomal storage disorder caused by deficient or absent activity of the enzyme alfa-galactosidase A. This defect enzyme leads to accumulation of glycolipids, primarily globotriaosylceramide (Gb3), in the vascular endothelium of several organs, including the skin, kidneys, nervous system, and heart. The characteristic early clinical features of Fabry disease include acroparaesthesia, angiokeratoma, heat intolerance, hypohidrosis, cornea verticillata and gastrointestinal symptoms. Later complications occur with the disease progression and include progressive renal failure, hypertrofic cardiomyopathy, cerebrovascular disease and reduced life expectancy. Anderson Fabry disease is therefore a disabling and systemic disease which requires a timely diagnosis. The purpose of our study is to define sweat glands morphological abnormalities in children and adolescents with Fabry disease with minimal symptoms and in patients affected by variants of Fabry disease in which biopsy is essential, to establish a baseline morphological diagnosis of the disease before to undergo to kidney or endomyocardial biopsy or when the classical approach is not possible because of some complications, with minimal discomfort for patients.
Type of the paper: Review
Tentative title: LSD and malignancy
Authors: Gregory M. Pastores, MD and Derralynn A. Hughes, MD, PhD
Affiliations: Department of Medicine (Genetics), University College Dublin, Ireland and University College London, Royal Free London NHS Foundation Trust, London NW3 2QG, UK
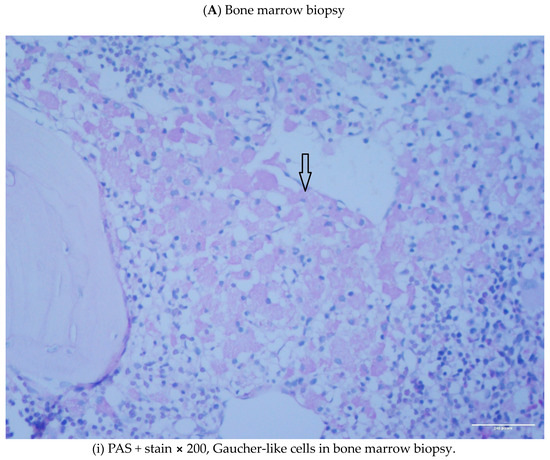
Abstract: Lysosomal storage disorders (LSD) are infrequent to rare conditions caused by mutations that lead to a disruption in the usual sequential degradation of macromolecules or their transit within the cell. Gaucher disease (GD), a lipidosis, is among the most common LSD, with an estimated incidence of 1 in 40,000 among the Caucasian, non-Jewish population. Studies have indicated an increased frequency of polyclonal and monoclonal gammopathy. As lipid storage in GD is restricted to cells of monocyte-/macrophage lineage, it had been speculated the occurrence of gammopathies reflect 'chronic macrophage activation'. It has been shown, β-glucosylceramide 22: 0 (βGL1-22) and glucosylsphingosine (LGL1), two major sphingolipids that accumulate in GD, can be recognized by a distinct subset of CD1d-restricted human and murine type II natural killer T (NKT) cells. Investigations undertaken in the affected mouse model revealed βGL1-22- and LGL1-specific NKT cells were present and constitutively expressed a T-follicular helper (TFH) phenotype; injection of these lipids led to downstream induction of germinal center B cells, hypergammaglobulinemia, and the production of antilipid antibodies. Subsequent studies have found clonal immunoglobulin in 33% of sporadic human monoclonal gammopathies is also specific for the lysolipids LGL1 and lysophosphatidylcholine (LPC). Furthermore, substrate reduction ameliorated GD-associated gammopathy in mice. These findings illustrate the value of investigations into rare diseases, which as 'experiments of nature' may provide insights into conditions found in the general population that continue to remain incompletely understood.
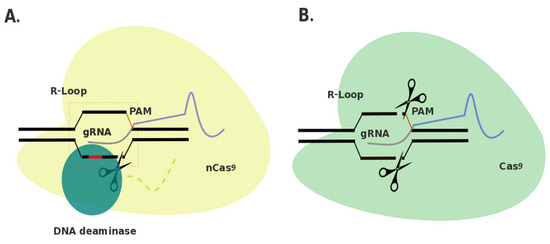
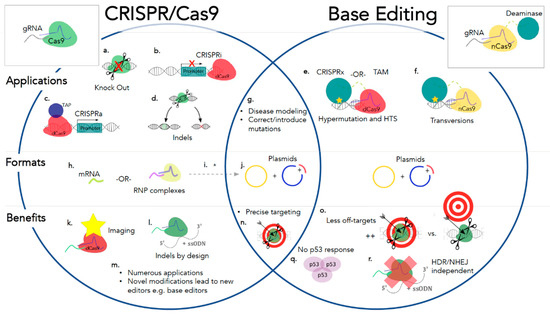
Tentative Title: A Prospective Treatment Option for Lysosomal Storage Diseases: CRISPR/Cas9 Gene editing Technology for Mutation Correction in Induced Pluripotent Stem Cells
Authors: Chloe L. Christensen and Francis Y.M. Choy
Affiliations: Department of Biology, Centre for Biomedical Research, University of Victoria, B.C., Canada
Abstract: Ease of design, relatively low cost, and a multitude of gene-altering capabilities have all led to the adoption of the sophisticated, yet simple gene-editing system: clustered regularly interspaced short palindromic repeats/CRISPR-associated protein 9 (CRISPR/Cas9). The CRISPR/Cas9 system holds promise for the correction of deleterious mutations by taking advantage of the homology directed repair pathway and by supplying a correction template to the affected patient’s cells. Currently this technique is being applied /in vitro/in human induced pluripotent stem cells (iPSCs) to correct a variety of severe genetic diseases, but has not, as of yet, been used in iPSCs derived from patients affected with a lysosomal storage disease (LSD). If adopted into clinical practice, corrected iPSCs derived from cells that originate from the patient themselves, could be used for therapeutic amelioration of LSD symptoms without the risks associated with allogeneic stem cell transplantation, such as graft versus host disease. CRISPR/Cas9 editing in patient’s cells could also overcome the costly, lifelong process associated with substrate reduction therapy, pharmacological chaperone therapy, or enzyme replacement therapy in order to reverse LSD symptoms. In this review, the overall impact of the CRISPR/Cas9 gene-editing technique for treatment of LSDs, as well as methods currently employed to increase the efficiency of this re-engineered biological system will be discussed.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}