



Cells 2022, 11(22), 3538; https://doi.org/10.3390/cells11223538 - 09 Nov 2022
Cited by 3 | Viewed by 1530
Abstract
►
Show Figures
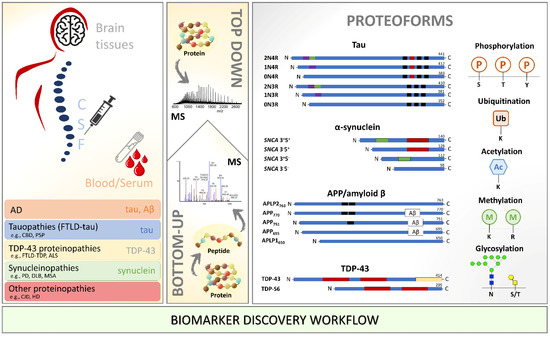
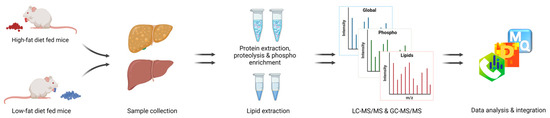
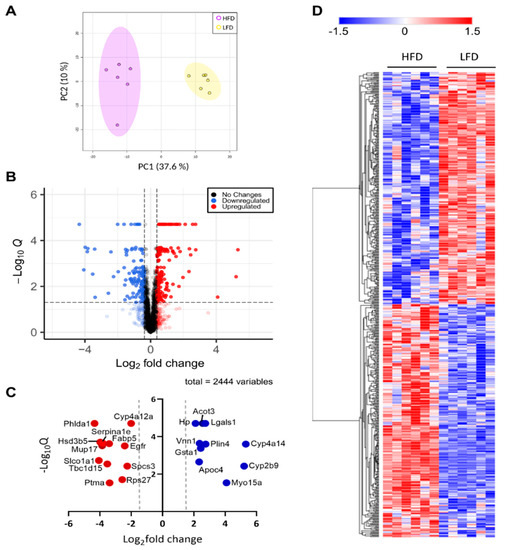
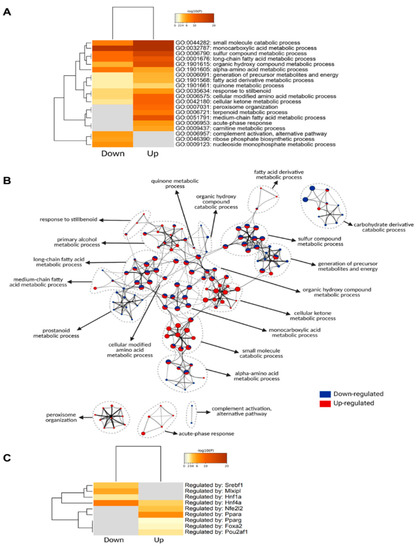
Neuromelanin granules (NMGs) are organelle-like structures present in the human substantia nigra pars compacta. In addition to neuromelanin, NMGs contain proteins, lipids and metals. As NMG-containing dopaminergic neurons are preferentially lost in Parkinson’s disease and dementia with Lewy bodies (DLB), it is
[...] Read more.
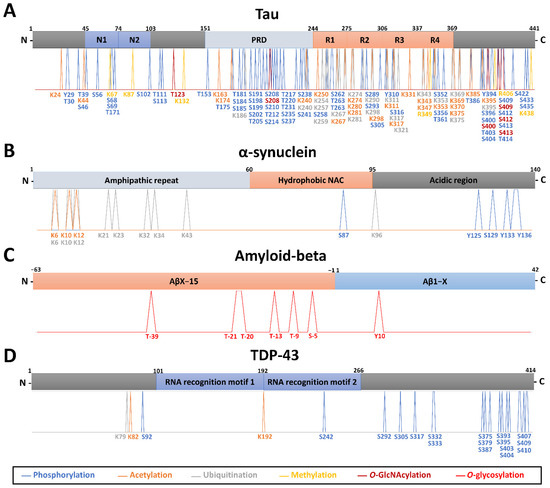
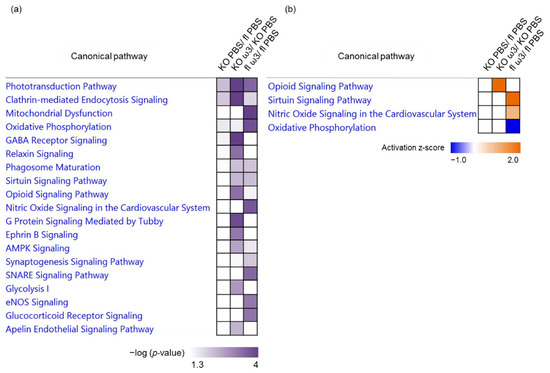
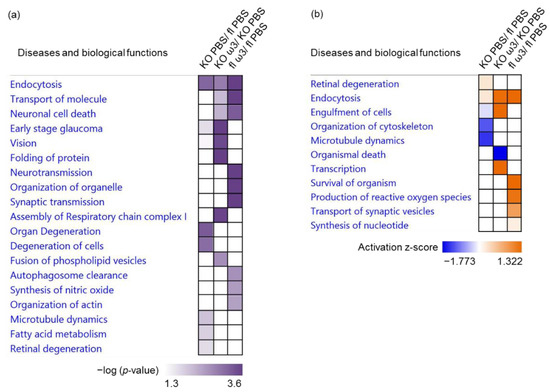
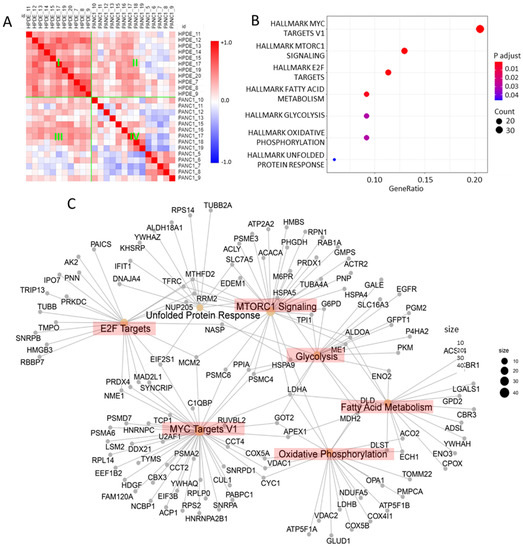
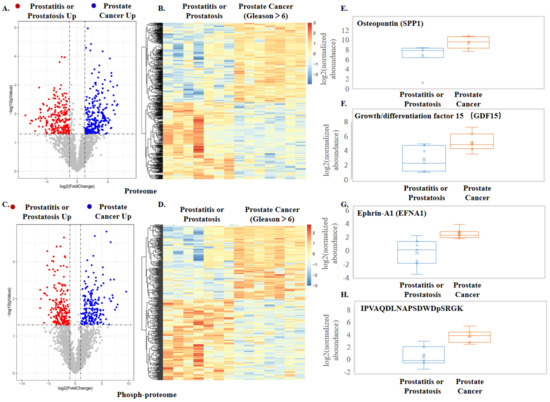
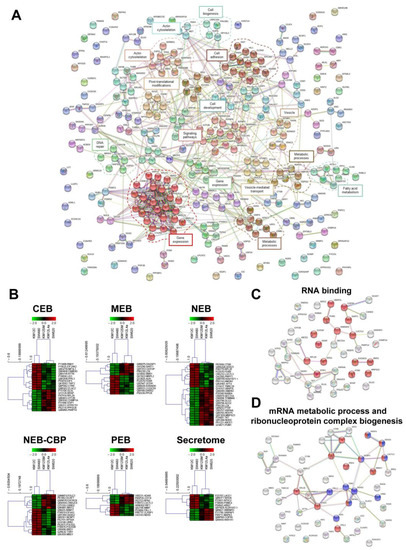
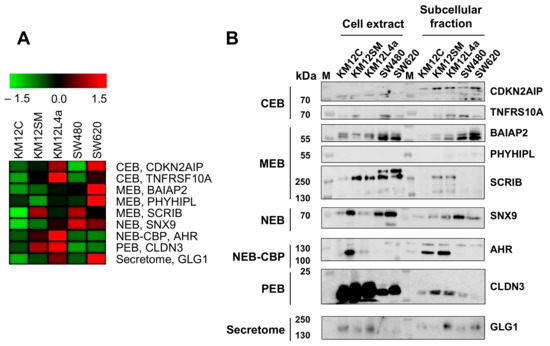
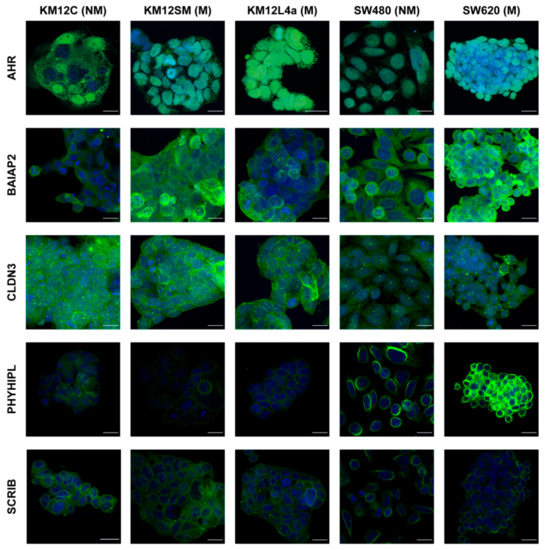
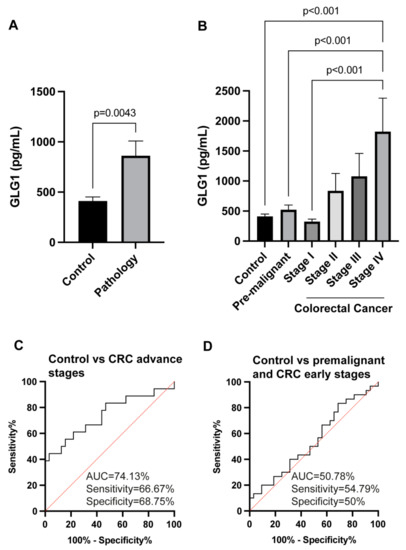
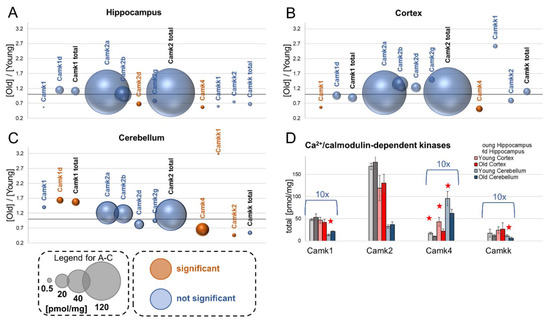
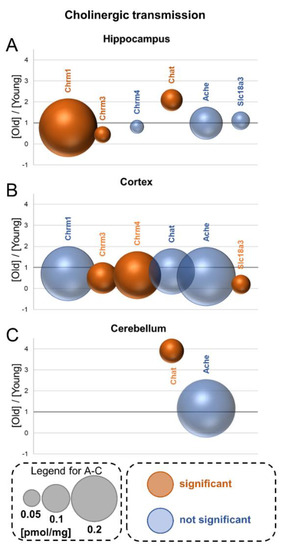
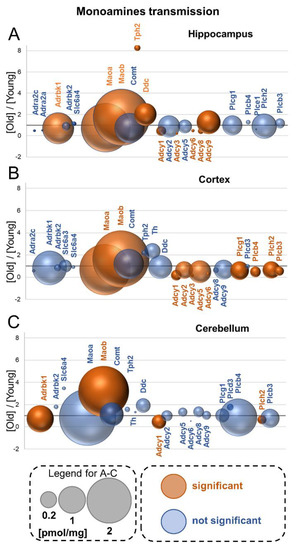
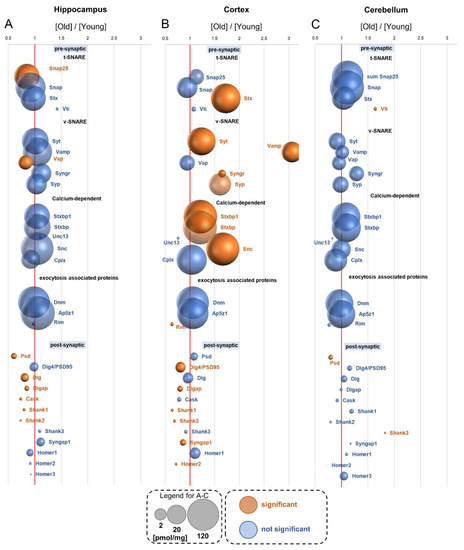
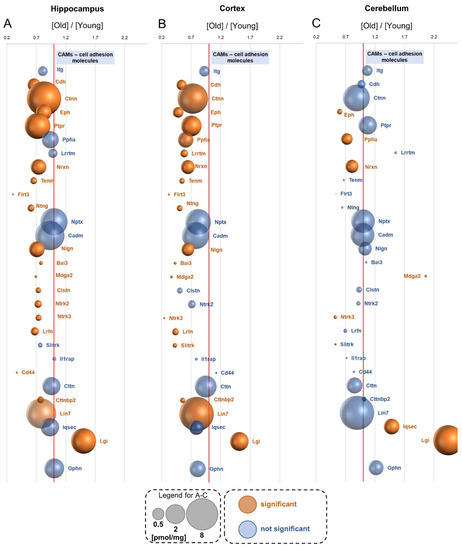
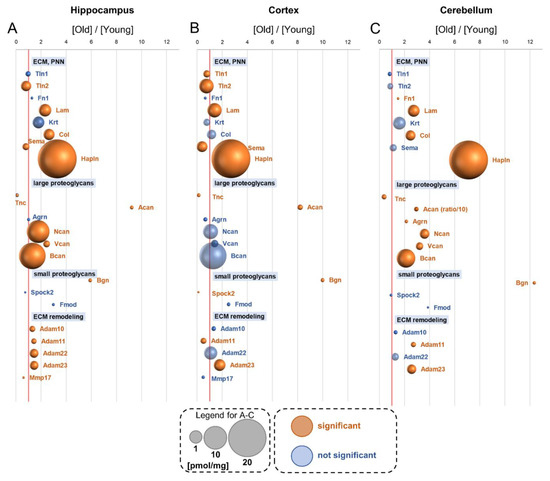
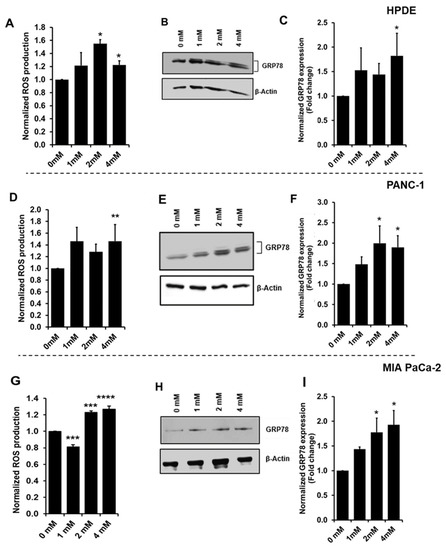
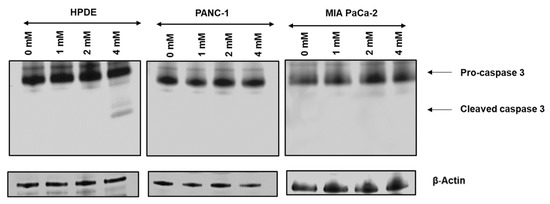
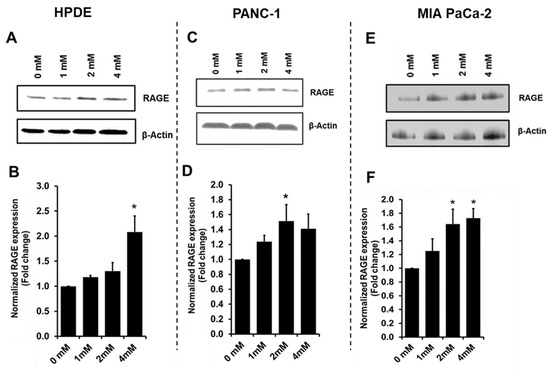
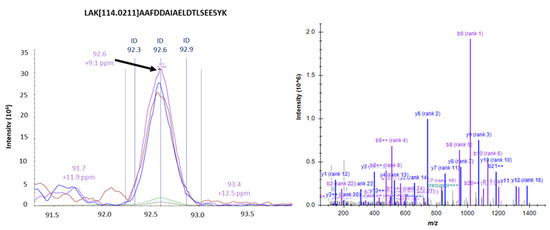
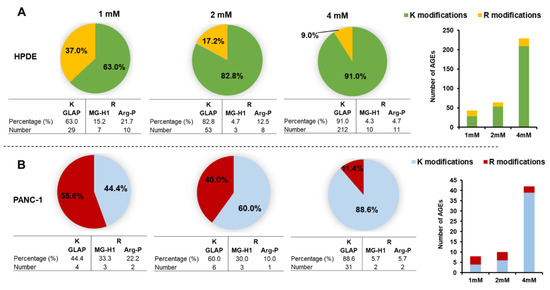
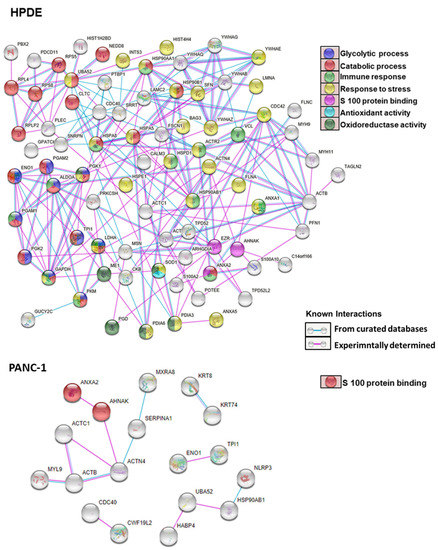
Neuromelanin granules (NMGs) are organelle-like structures present in the human substantia nigra pars compacta. In addition to neuromelanin, NMGs contain proteins, lipids and metals. As NMG-containing dopaminergic neurons are preferentially lost in Parkinson’s disease and dementia with Lewy bodies (DLB), it is assumed that NMGs may play a role in neurodegenerative processes. Until now, this role is not completely understood and needs further investigation. We therefore set up an exploratory proteomic study to identify differences in the proteomic profile of NMGs from DLB patients (n = 5) compared to healthy controls (CTRL, n = 5). We applied a laser microdissection and mass-spectrometry-based approach, in which we used targeted mass spectrometric experiments for validation. In NMG-surrounding (SNSurr.) tissue of DLB patients, we found evidence for ongoing oxidative damage and an impairment of protein degradation. As a potentially disease-related mechanism, we found α-synuclein and protein S100A9 to be enriched in NMGs of DLB cases, while the abundance of several ribosomal proteins was significantly decreased. As S100A9 is known to be able to enhance the formation of toxic α-synuclein fibrils, this finding points towards an involvement of NMGs in pathogenesis, however the exact role of NMGs as either neuroprotective or neurotoxic needs to be further investigated. Nevertheless, our study provides evidence for an impairment of protein degradation, ongoing oxidative damage and accumulation of potentially neurotoxic protein aggregates to be central mechanisms of neurodegeneration in DLB.
Full article
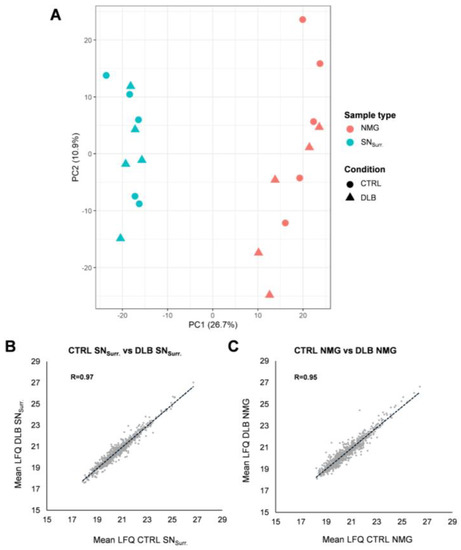
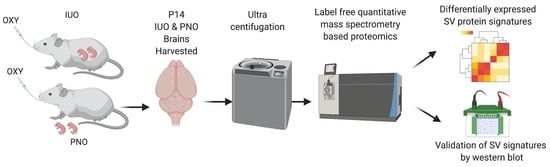
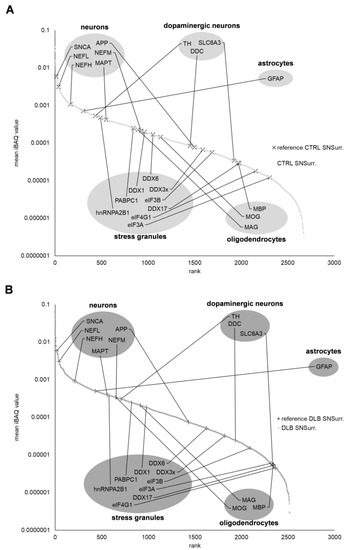
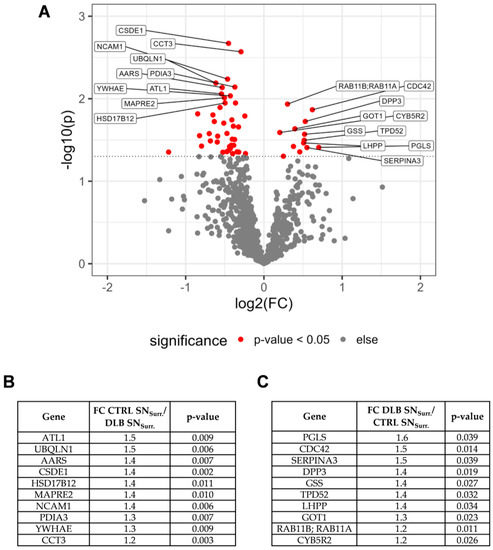
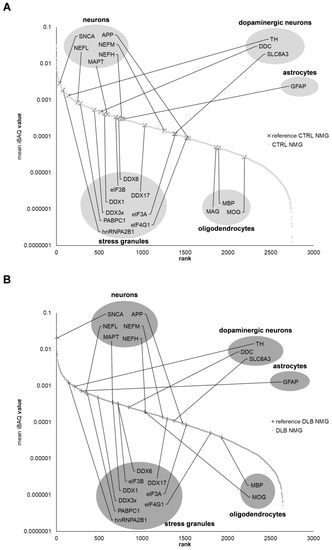
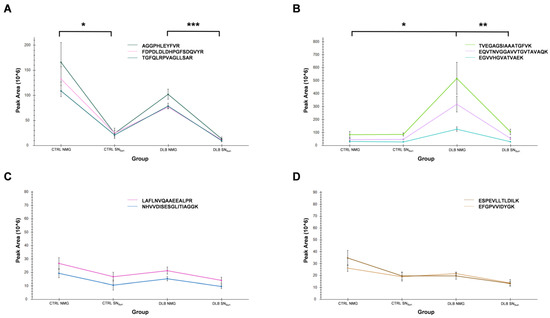
Figure 1
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}