




Cells 2022, 11(24), 3990; https://doi.org/10.3390/cells11243990 - 09 Dec 2022
Cited by 2 | Viewed by 1351
Abstract
►
Show Figures
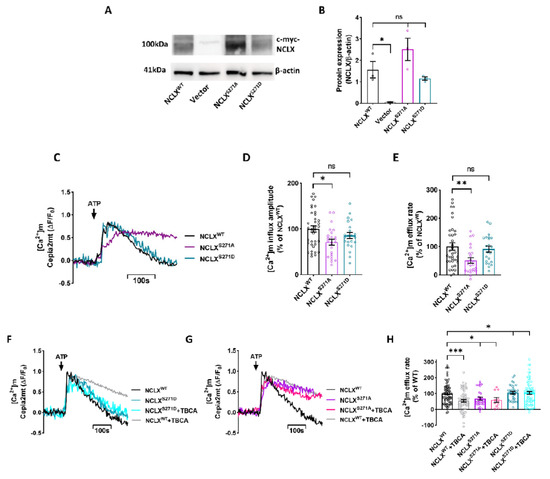
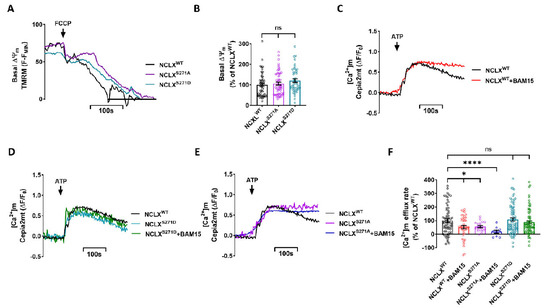
Mitochondrial Ca2+ efflux by NCLX is a critical rate-limiting step in mitochondria signaling. We previously showed that NCLX is phosphorylated at a putative Casein Kinase 2 (CKII) site, the serine 271 (S271). Here, we asked if NCLX is regulated by CKII and
[...] Read more.
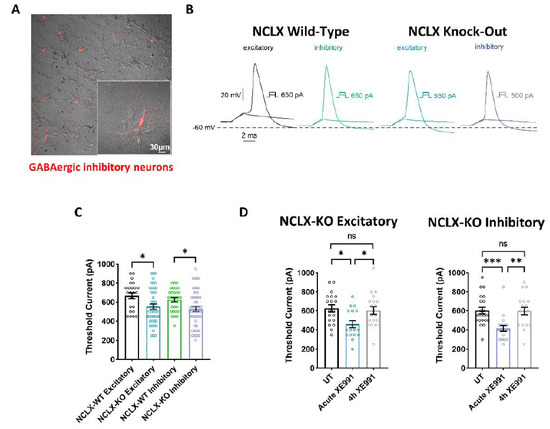
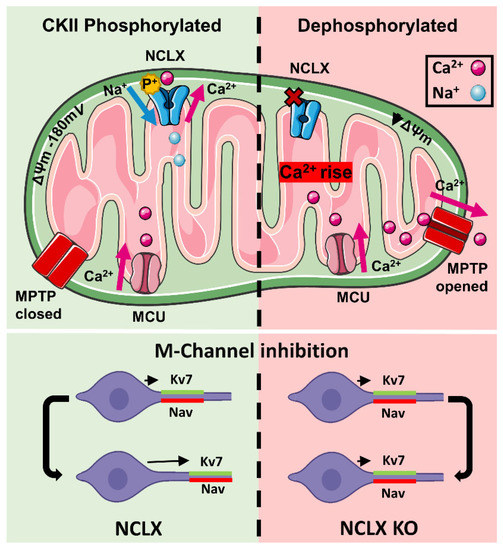
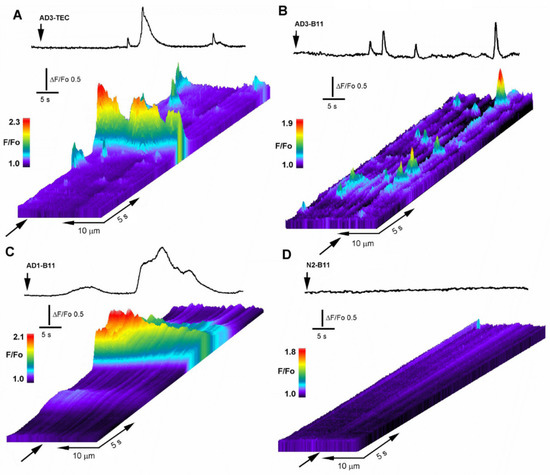
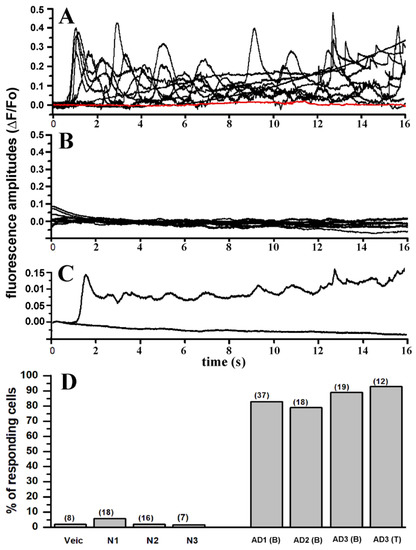
Mitochondrial Ca2+ efflux by NCLX is a critical rate-limiting step in mitochondria signaling. We previously showed that NCLX is phosphorylated at a putative Casein Kinase 2 (CKII) site, the serine 271 (S271). Here, we asked if NCLX is regulated by CKII and interrogated the physiological implications of this control. We found that CKII inhibitors down-regulated NCLX-dependent Ca2+ transport activity in SH-SY5Y neuronal cells and primary hippocampal neurons. Furthermore, we show that the CKII phosphomimetic mutants on NCLX inhibited (S271A) and constitutively activated (S271D) NCLX transport, respectively, rendering it insensitive to CKII inhibition. These phosphomimetic NCLX mutations also control the allosteric regulation of NCLX by mitochondrial membrane potential (ΔΨm). Since the omnipresent CKII is necessary for modulating the plasticity of the axon initial segment (AIS), we interrogated, in hippocampal neurons, if NCLX is required for this process. Similarly to WT neurons, NCLX-KO neurons can exhibit homeostatic plasticity following M-channel block. However, while WT neurons utilize a CKII-sensitive distal relocation of AIS Na+ and Kv7 channels to decrease their intrinsic excitability, we did not observe such translocation in NCLX-KO neurons. Thus, our results indicate that NCLX is regulated by CKII and is a crucial link between CKII signaling and fast neuronal plasticity.
Full article
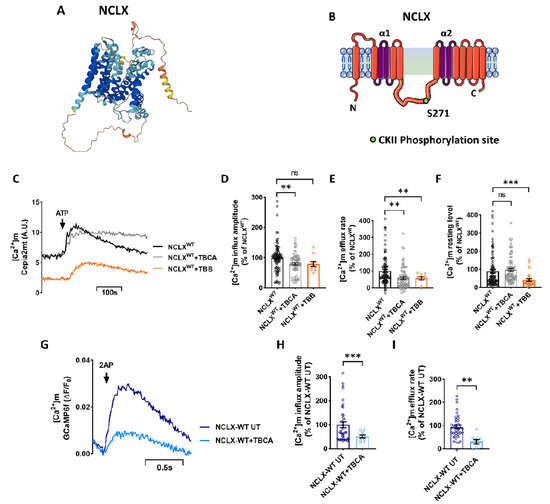
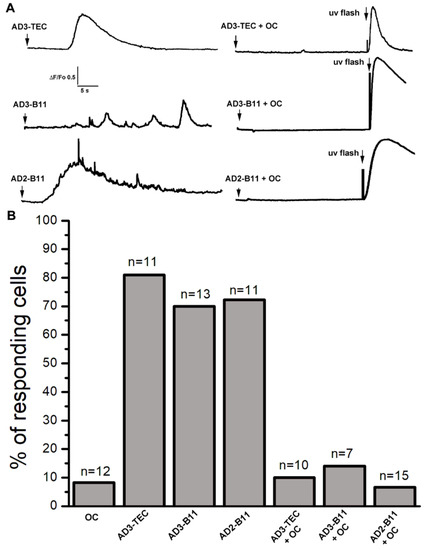
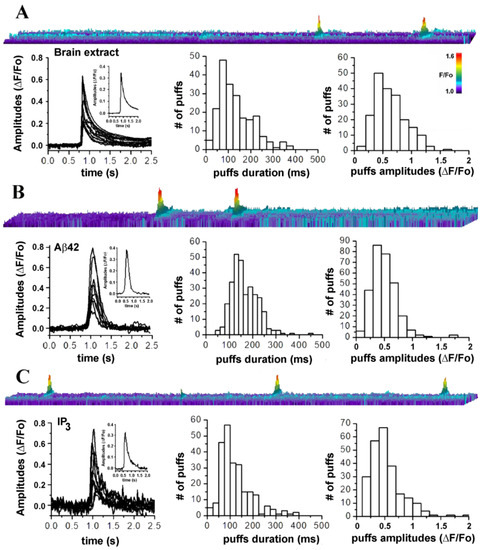
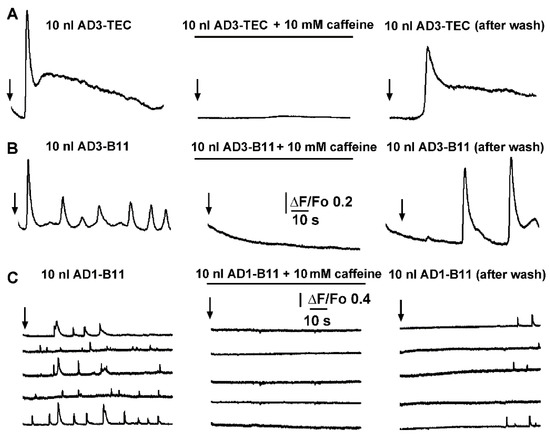
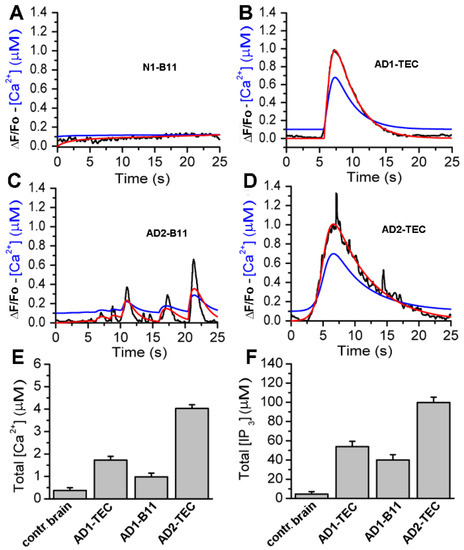
Figure 1
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}