



Animals 2024, 14(8), 1145; https://doi.org/10.3390/ani14081145 - 09 Apr 2024
Viewed by 365
Abstract
►
Show Figures
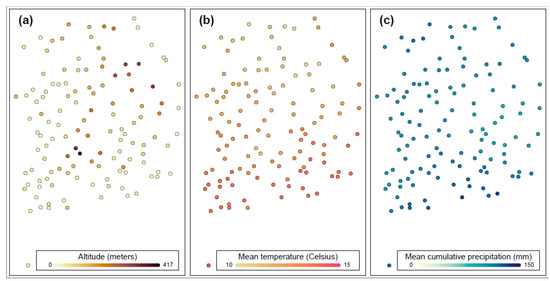
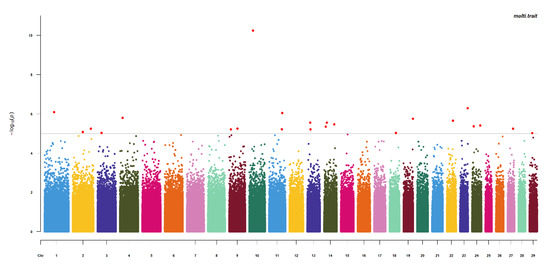
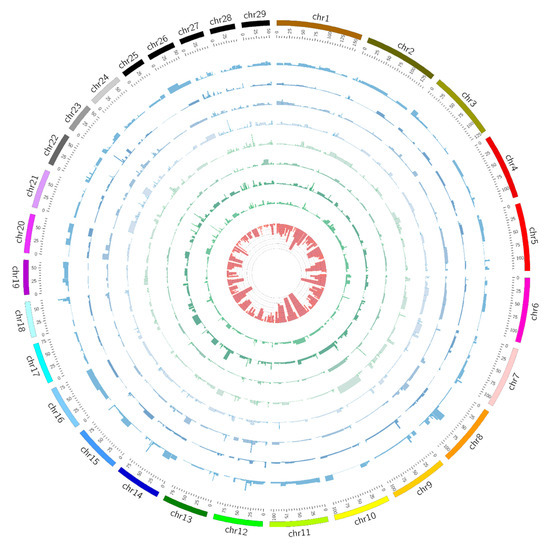
Identifying genetic markers of economically valuable traits has practical benefits for the meat goat industry. To better understand the genomic variations influencing body conformation traits, a genome-wide association study was performed on Tashi goats, an indigenous Chinese goat breed. A total of 155
[...] Read more.
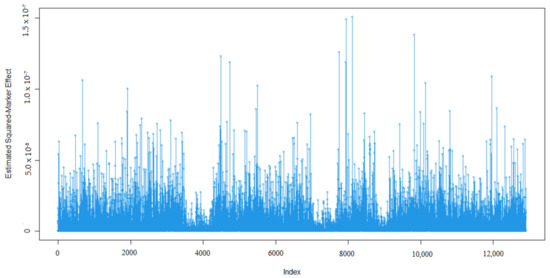
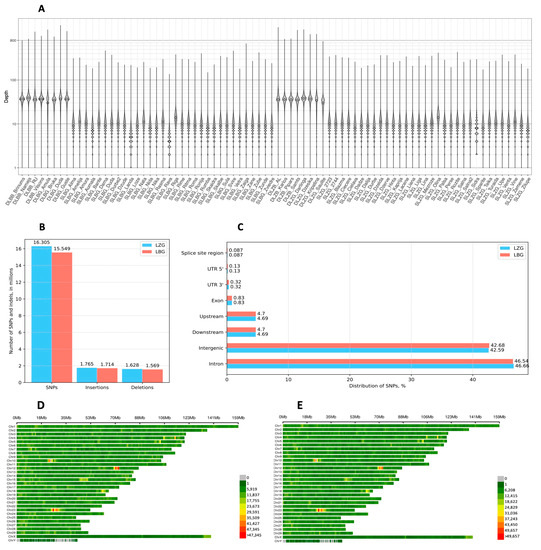
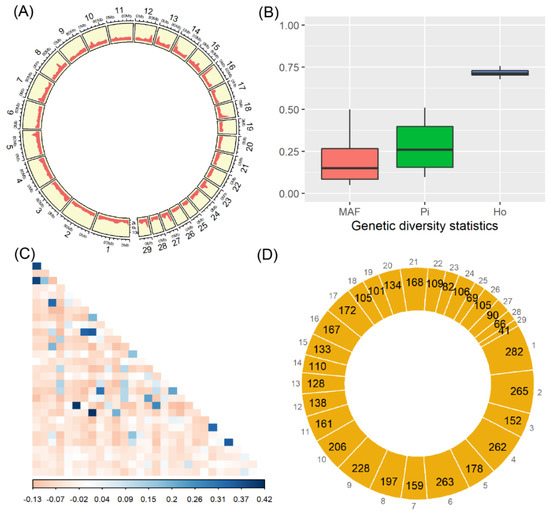
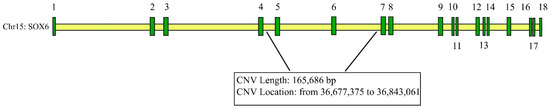
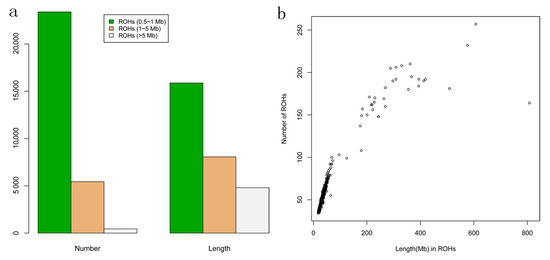
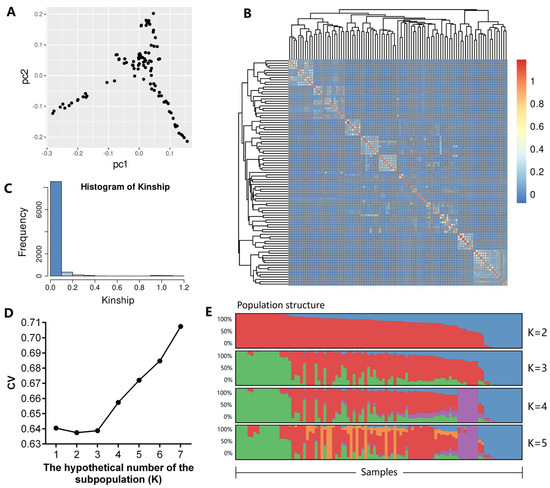
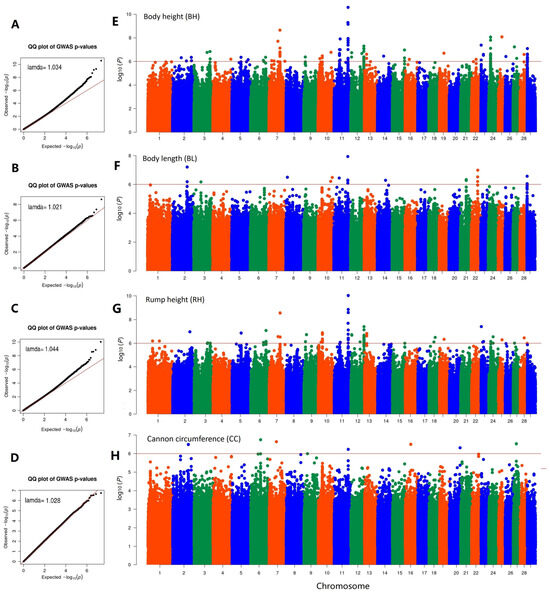
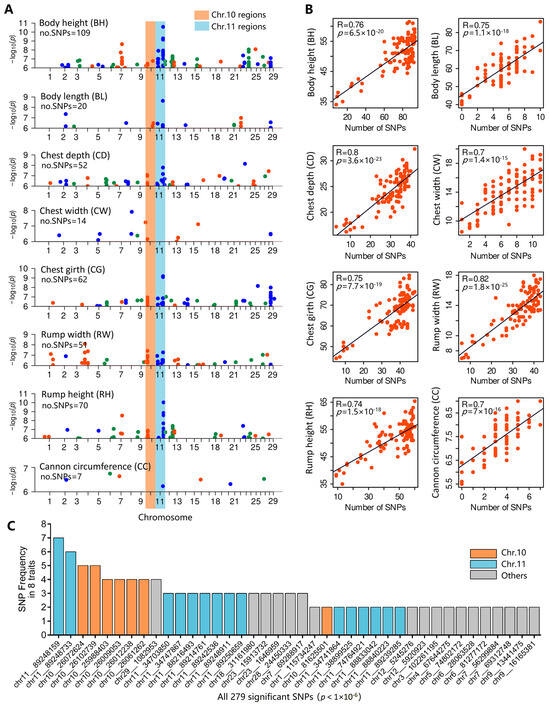
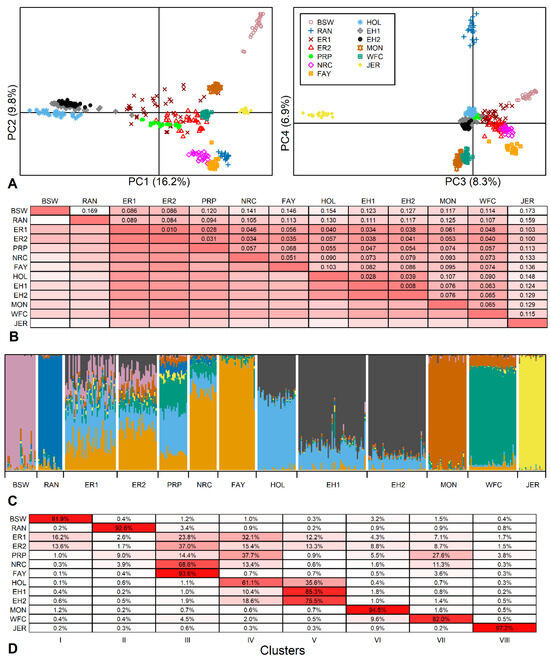
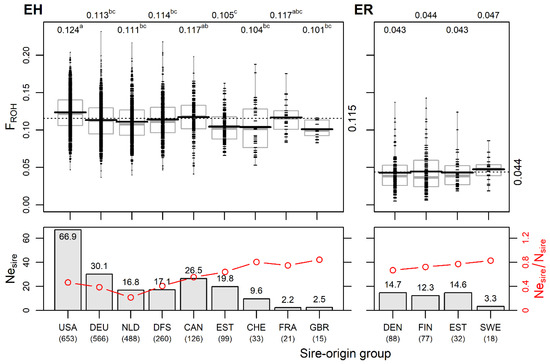
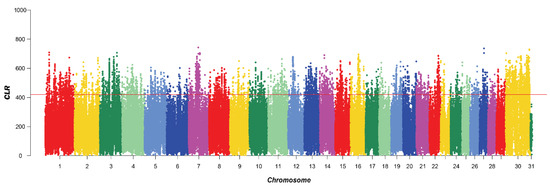
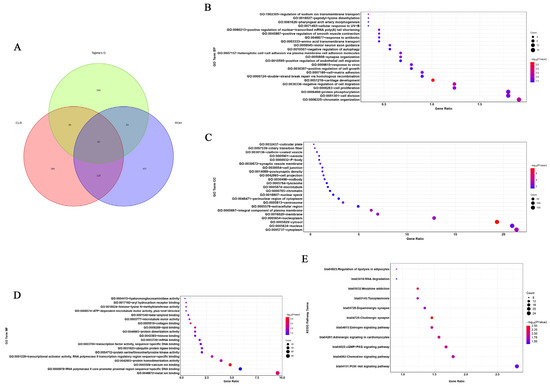
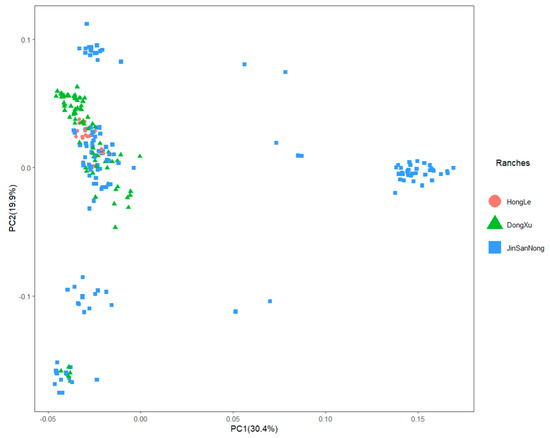
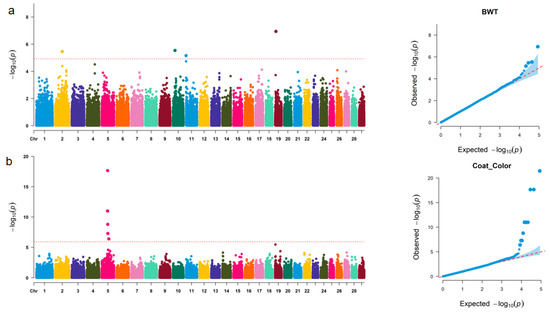
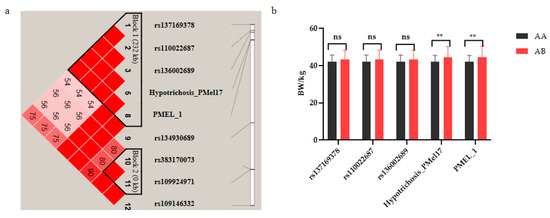
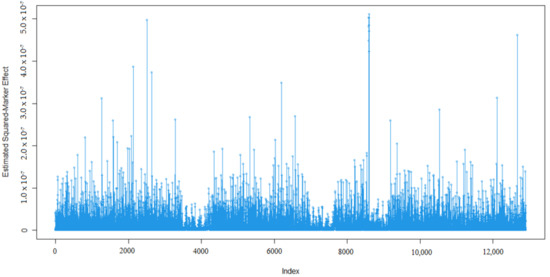
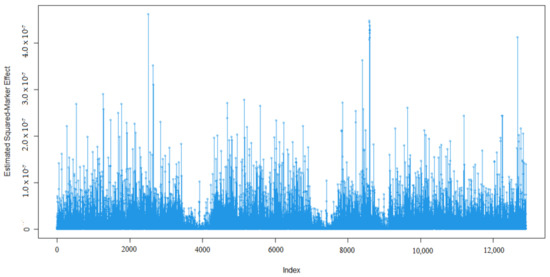
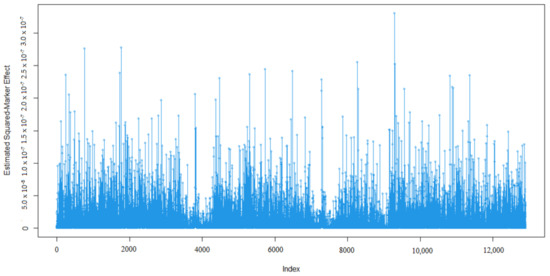
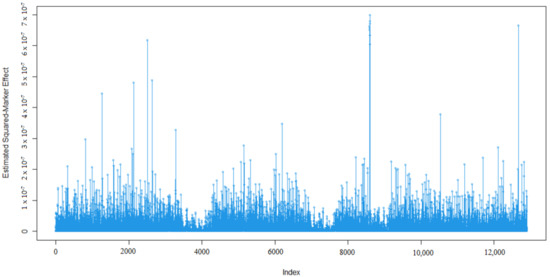
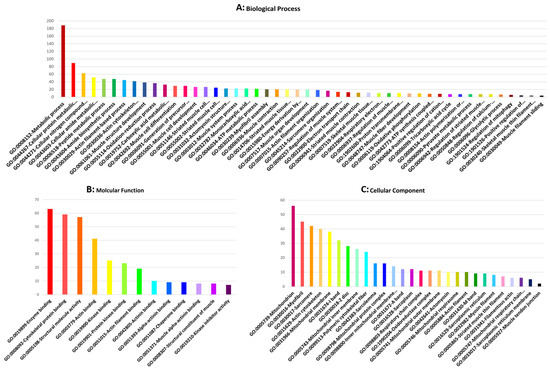
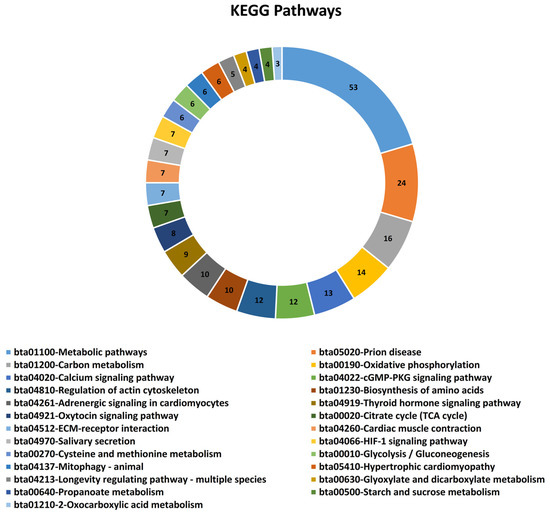
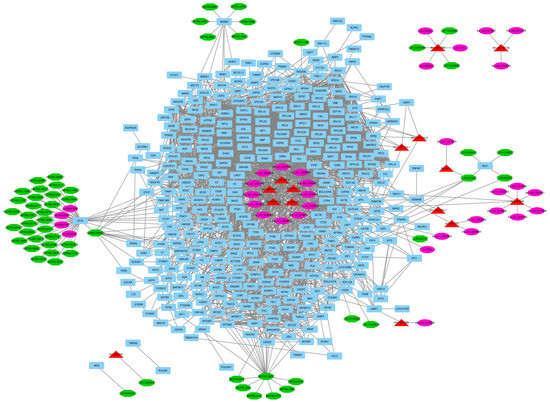
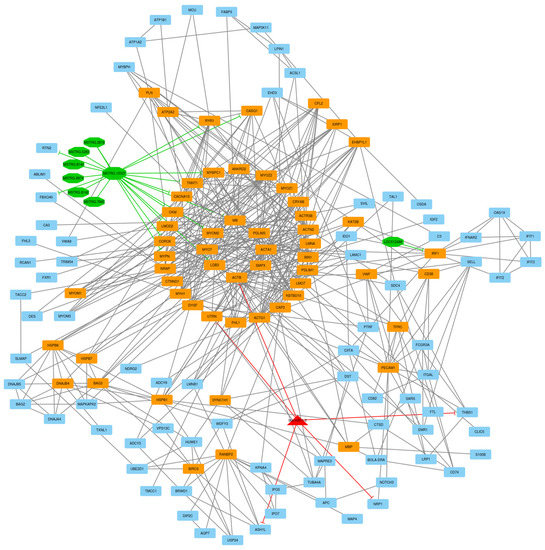
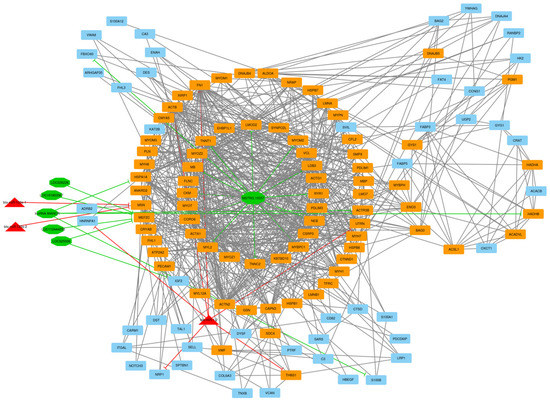
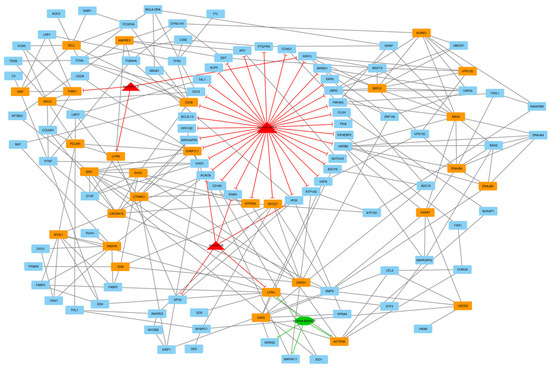
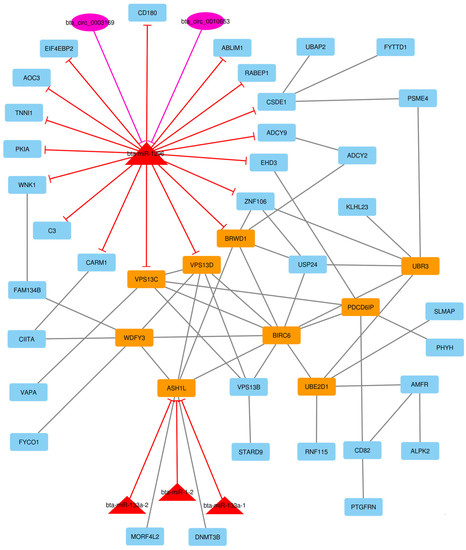
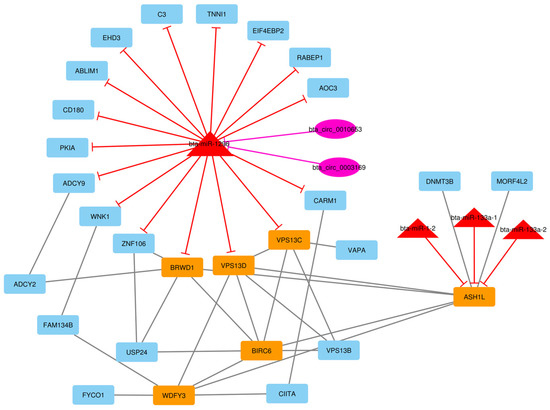
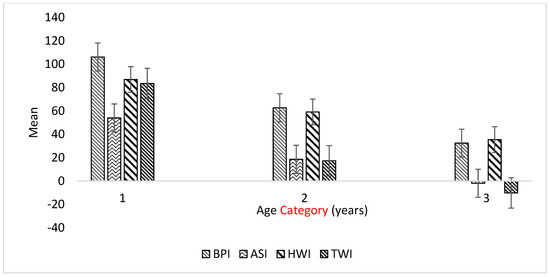
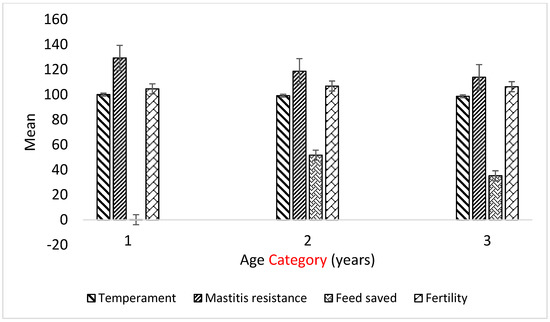
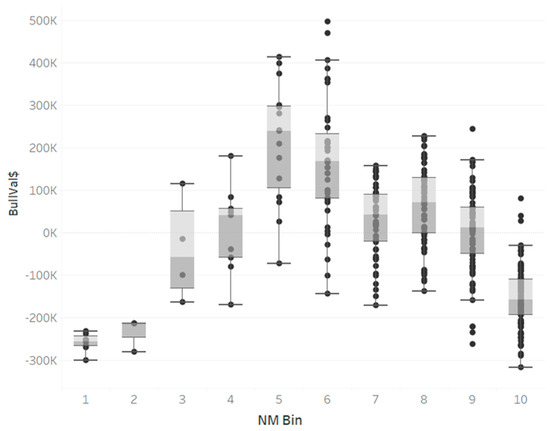
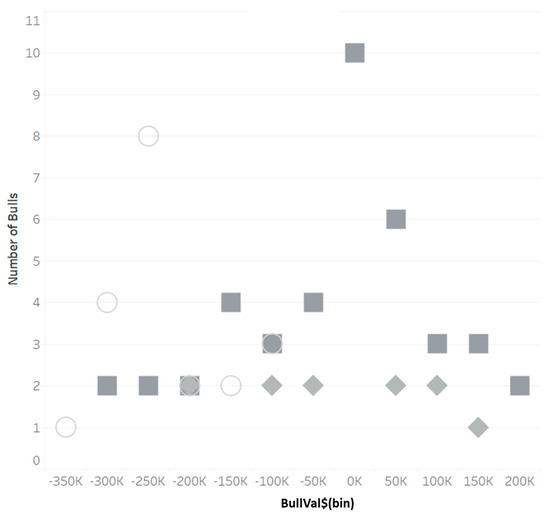
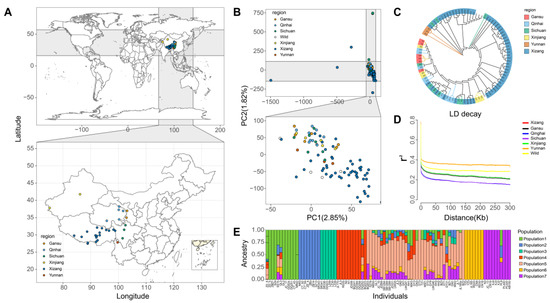
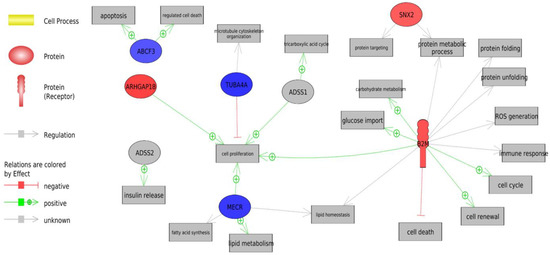
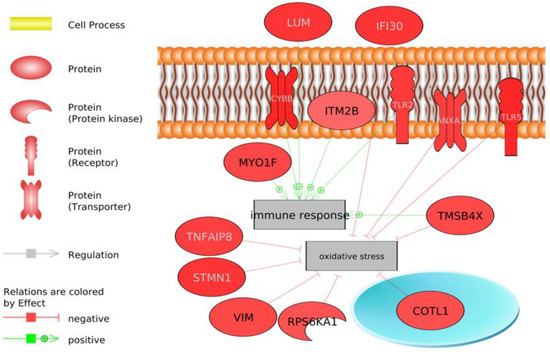
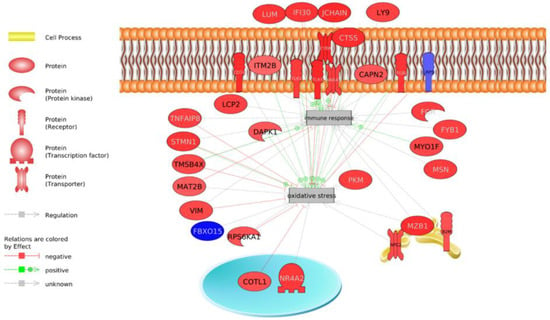
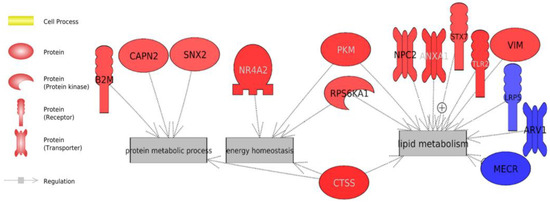
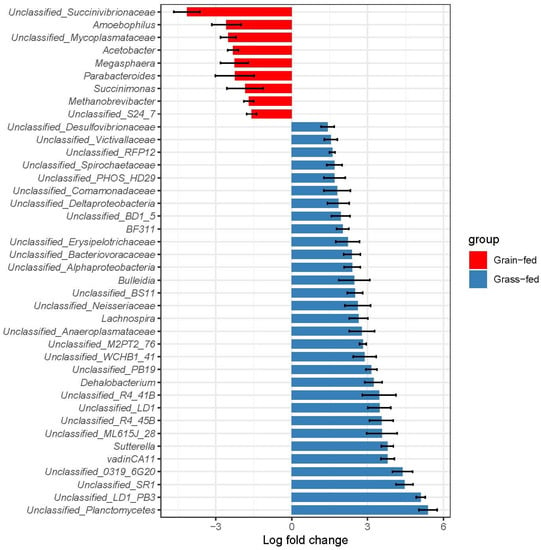
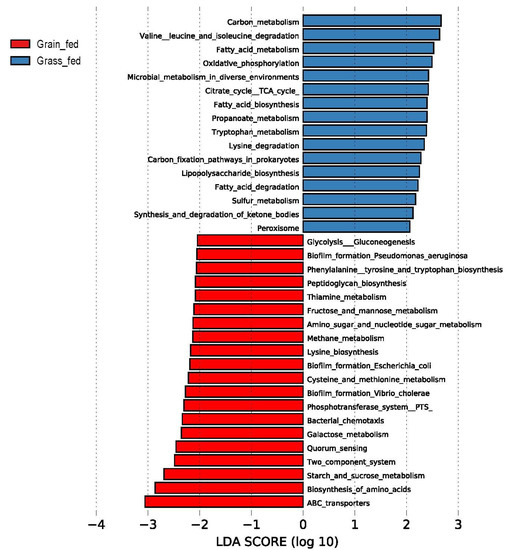
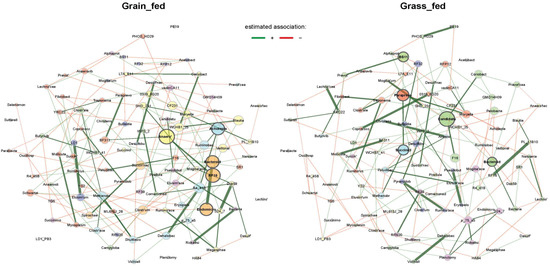
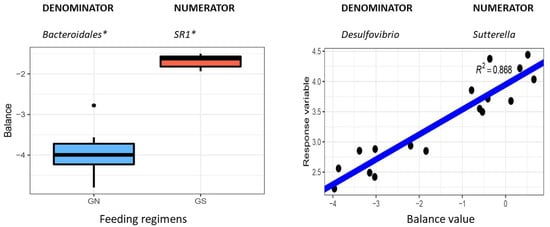
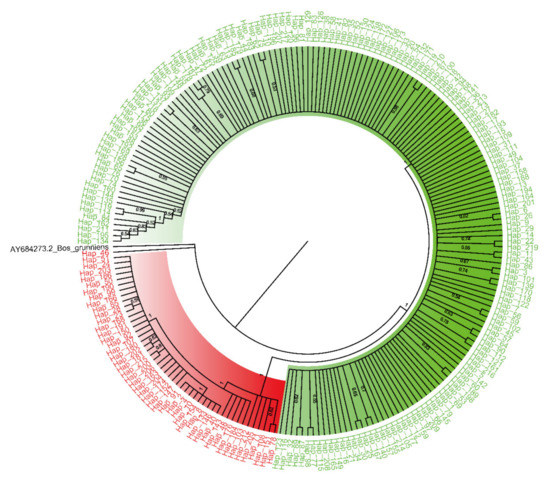
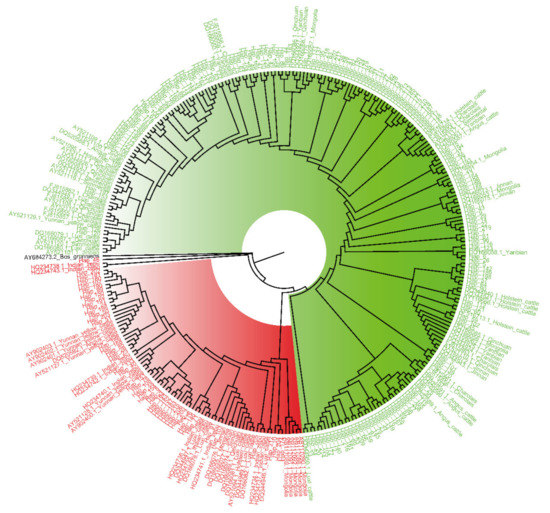
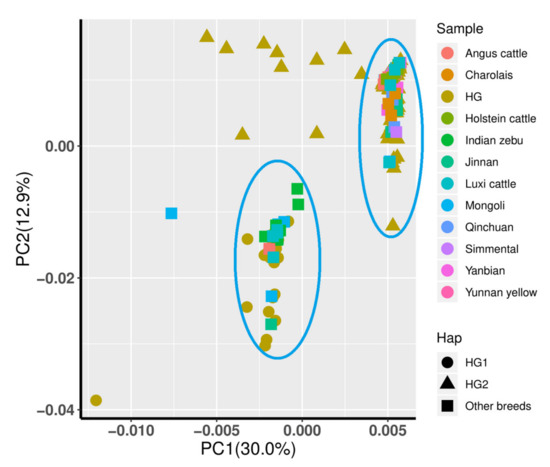
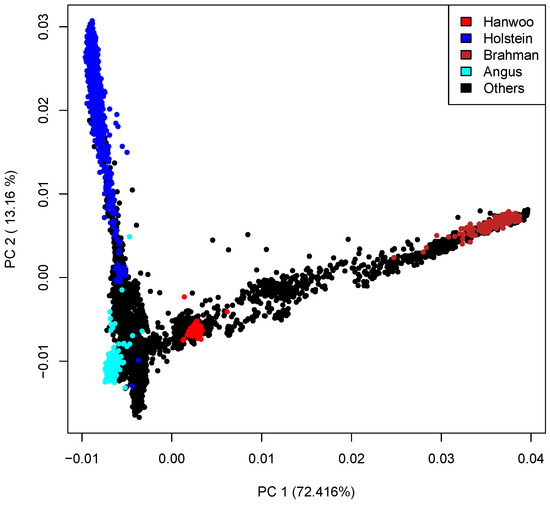
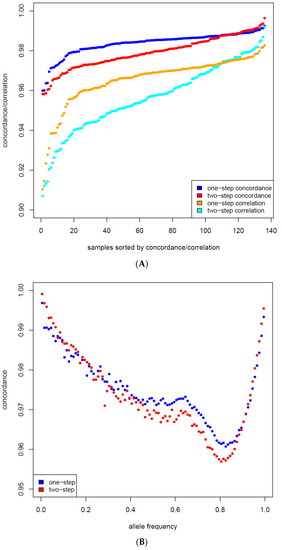
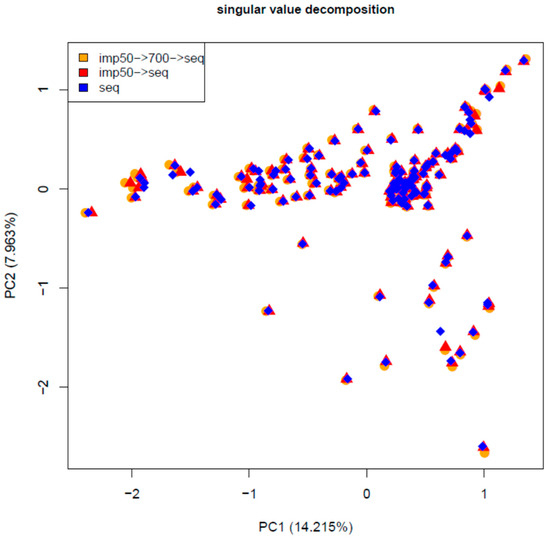
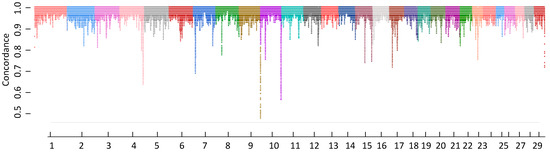
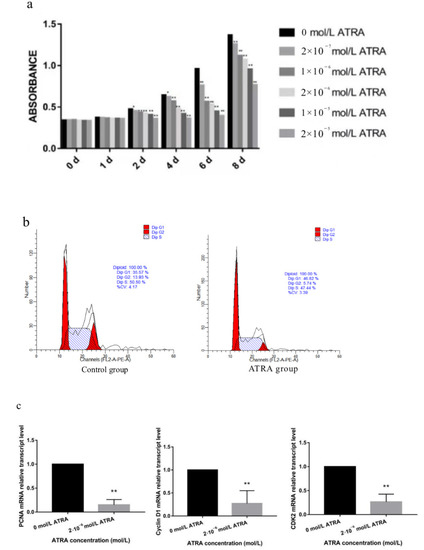
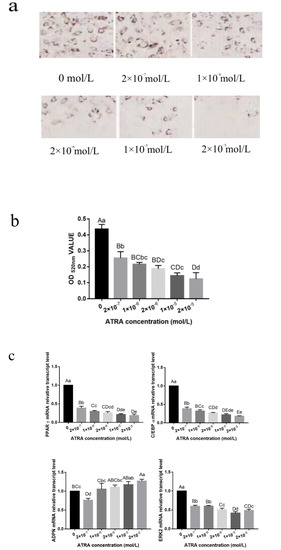
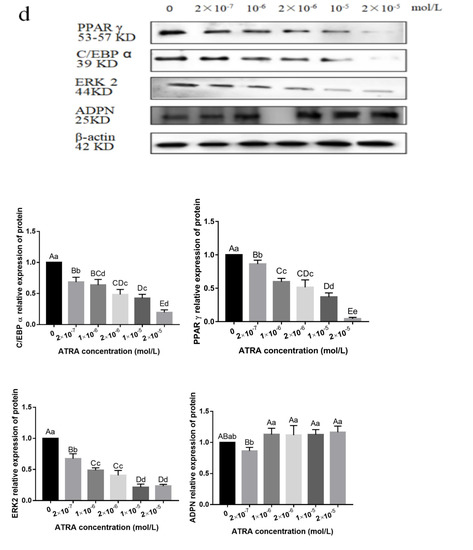
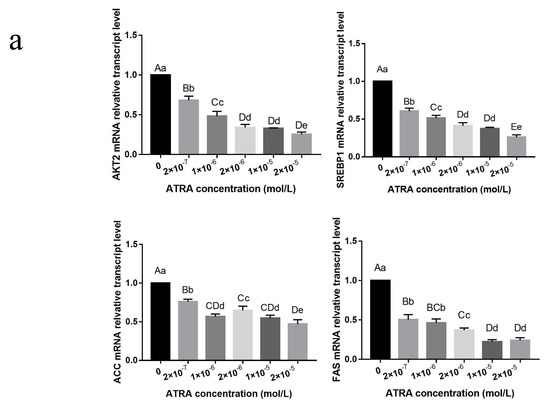
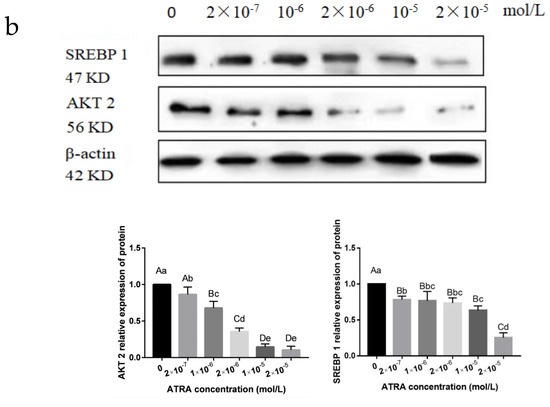
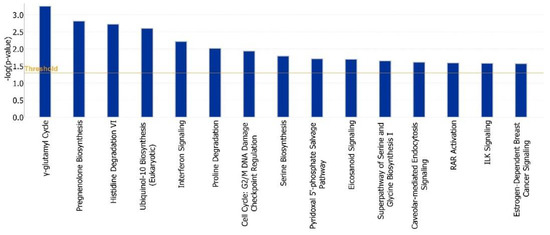
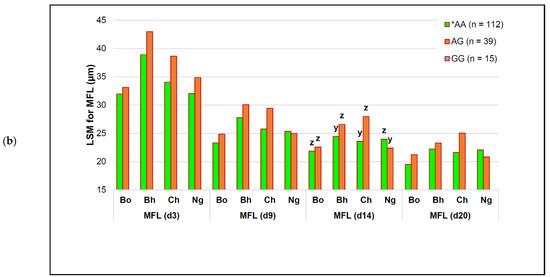
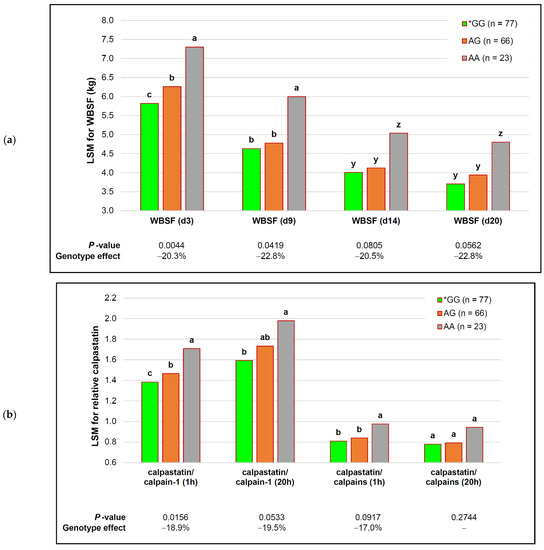
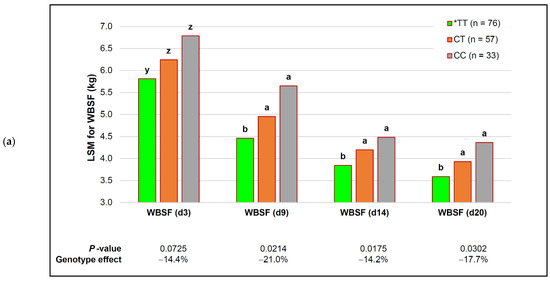
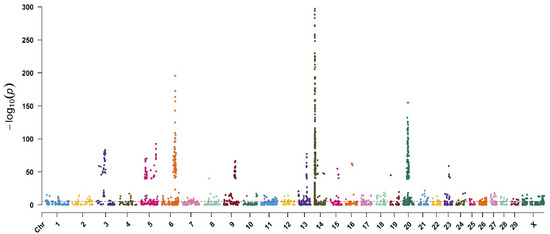
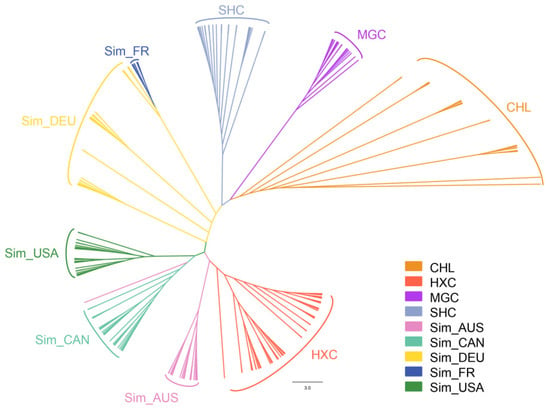
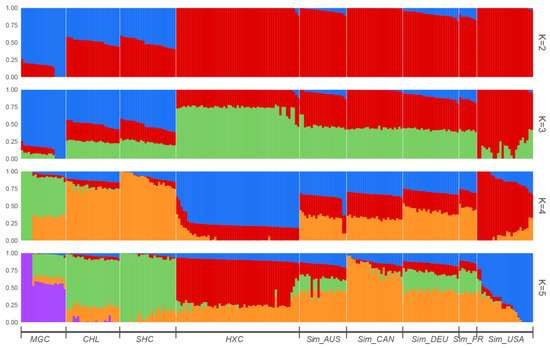
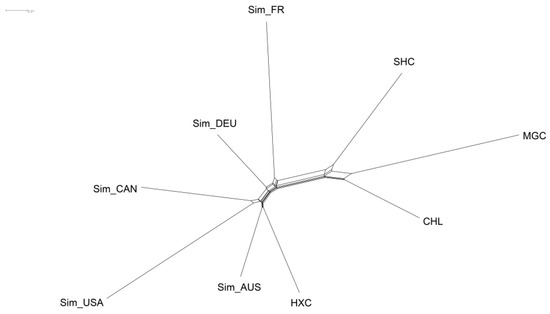
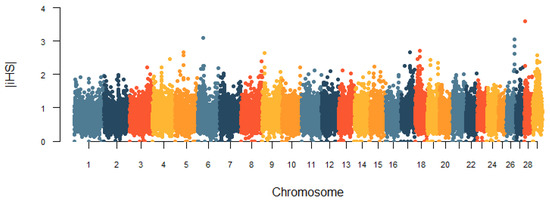
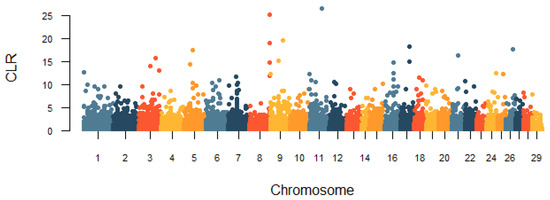
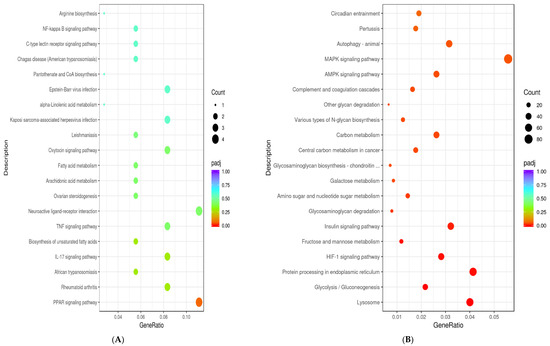
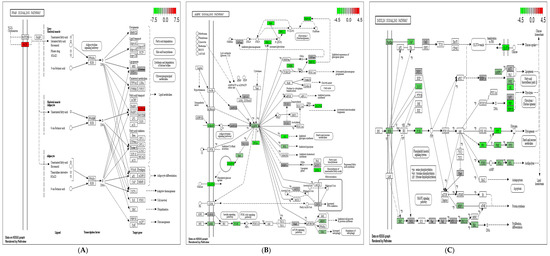
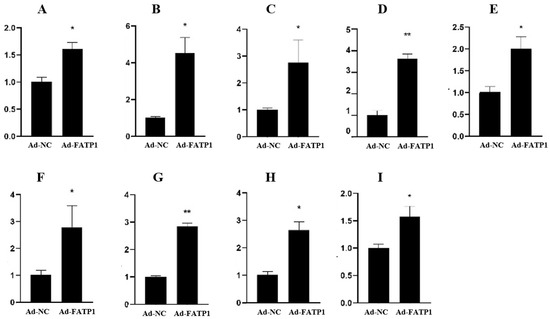
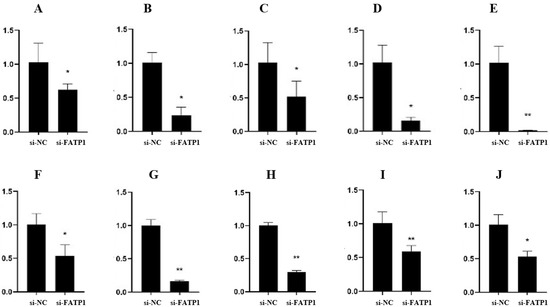
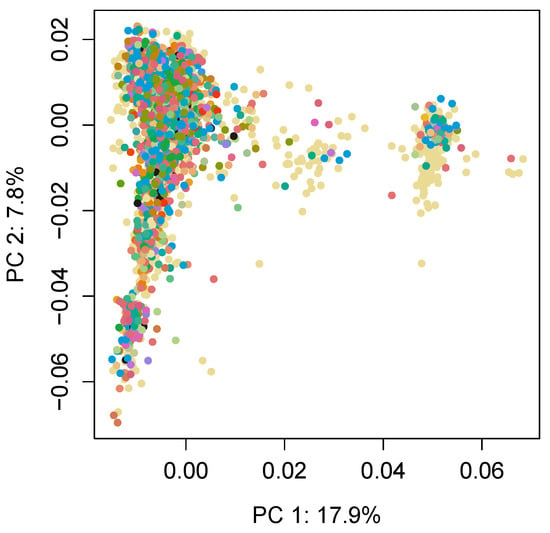
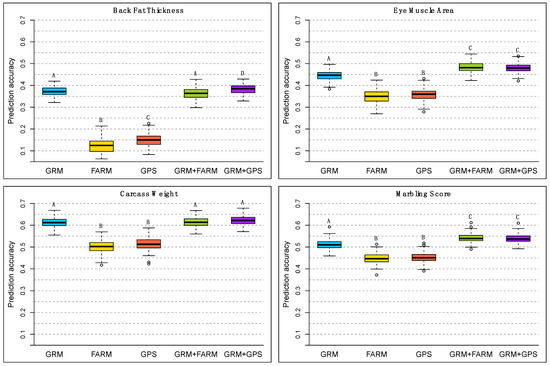
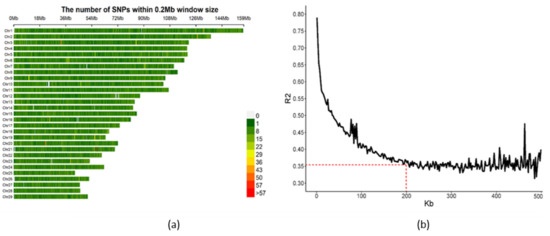
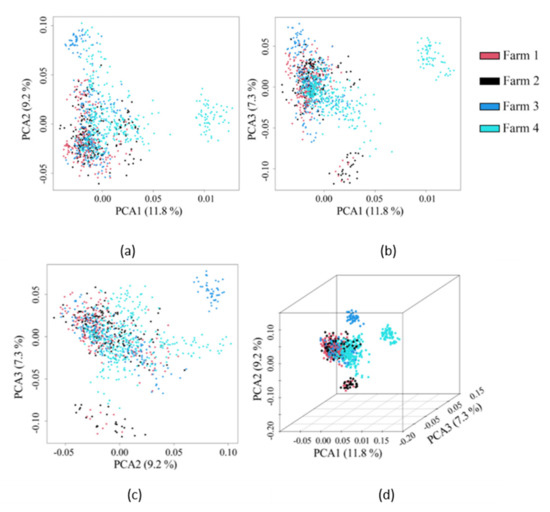
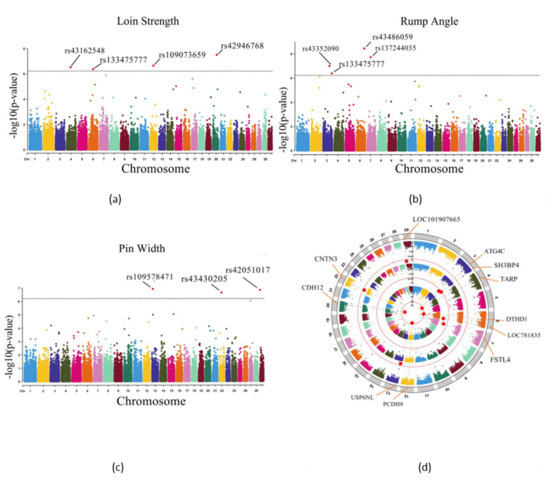
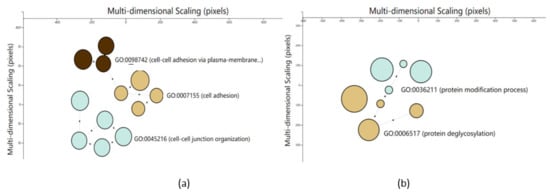
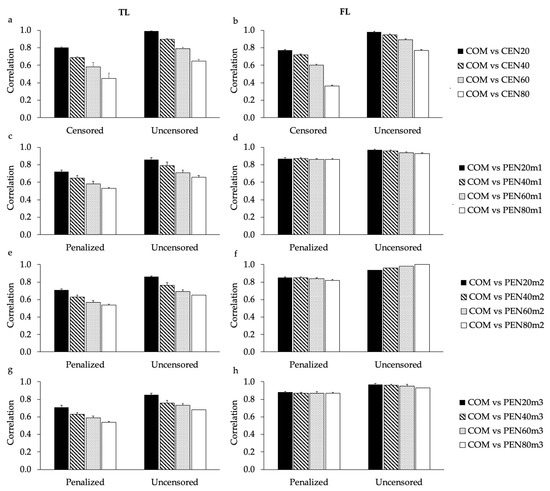
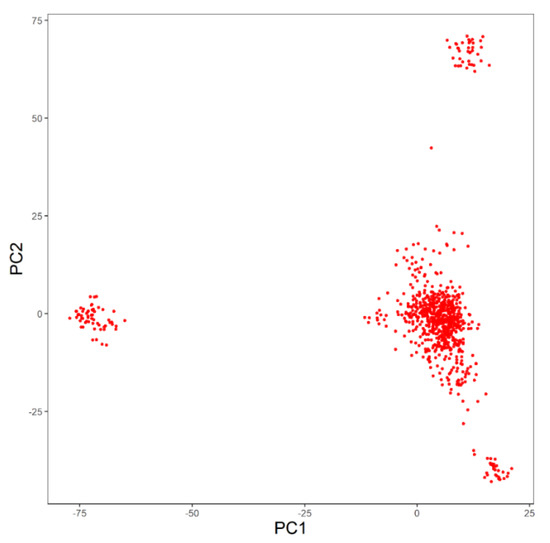
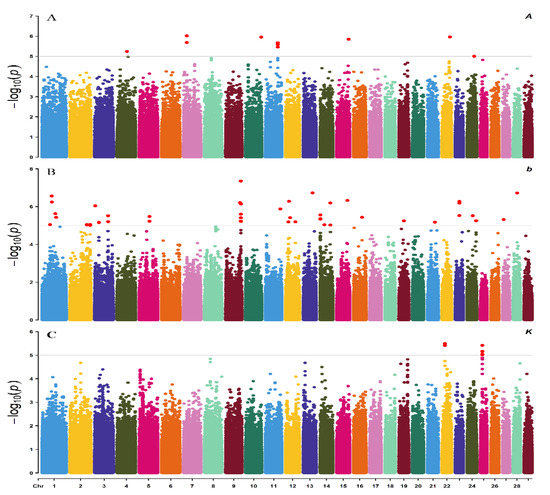
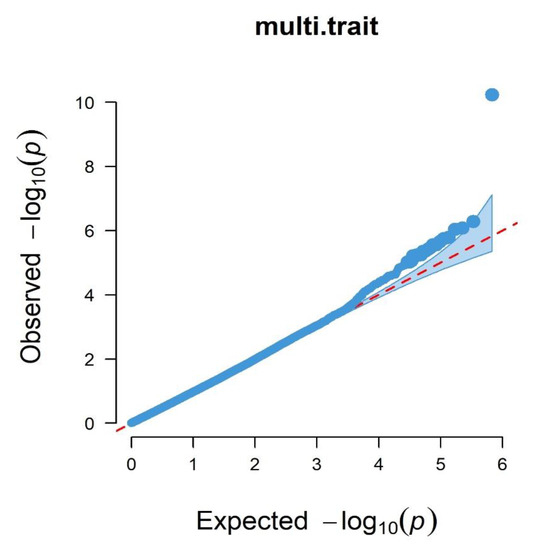
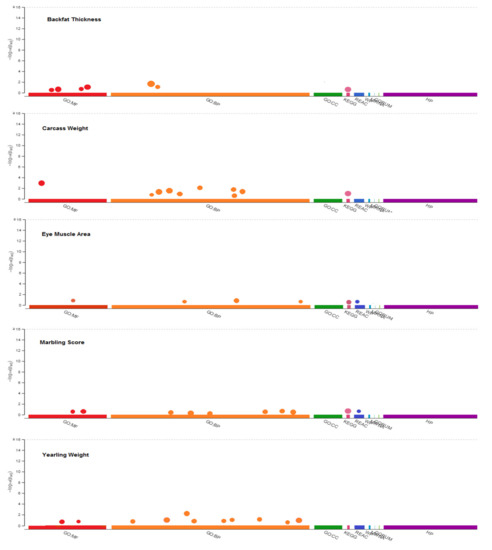
Identifying genetic markers of economically valuable traits has practical benefits for the meat goat industry. To better understand the genomic variations influencing body conformation traits, a genome-wide association study was performed on Tashi goats, an indigenous Chinese goat breed. A total of 155 Tashi goats were phenotyped for eight body conformation traits: body height, body length, chest depth, chest width, chest girth, rump width, rump height, and cannon bone circumference. Then, 100 Tashi goats were randomly selected for whole-genome sequencing and genotyped. We obtained 1676.4 Gb of raw data with an average sequencing depth of 6.2X. Clean reads were aligned to the ARS1.2 reference genome, and 11,257,923 single nucleotide polymorphisms (SNPs) were identified. The structure analysis showed that these Tashi goats were almost not genetically related. The 109, 20, 52, 14, 62, 51, 70, and 7 SNPs were significantly associated with body height, body length, chest depth, chest width, chest girth, rump width, rump height, and cannon bone circumference. Within the ±500 kb region of significant SNPs, 183 genes were annotated. The most significantly enriched KEGG pathway was “olfactory transduction”, and the most significantly enriched gene ontology (GO) terms were “cellular process”, “cellular anatomical entity”, and “molecular transducer activity”. Interestingly, we found several SNPs on chromosomes 10 and 11 that have been identified multiple times for all eight body conformation traits located in two fragments (114 kb and 1.03 Mb). In chr.10:25988403-26102739, the six SNPs were tightly linked, the TACTAG genotype was the highest at 91.8%, and the FNTB (Farnesyltransferase, CAAX Box Beta) and CHURC1 (Churchill Domain Containing 1) genes were located. In chr.11:88216493-89250659, ten SNPs were identified with several dependent linkage disequilibrium (LD) blocks, and seven related genes were annotated, but no significant SNP was located in them. Our results provide valuable biological information for improving growth performance with practical applications for genomic selection in goats.
Full article
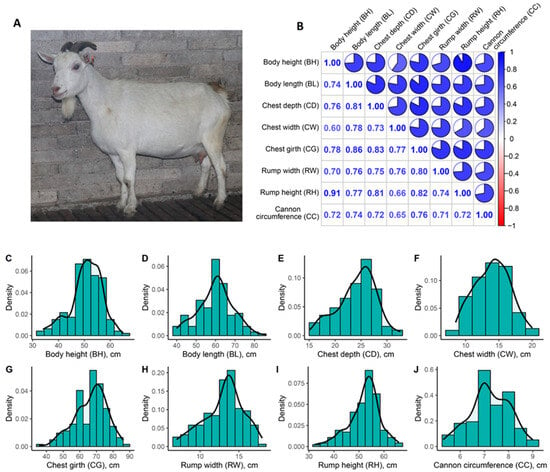
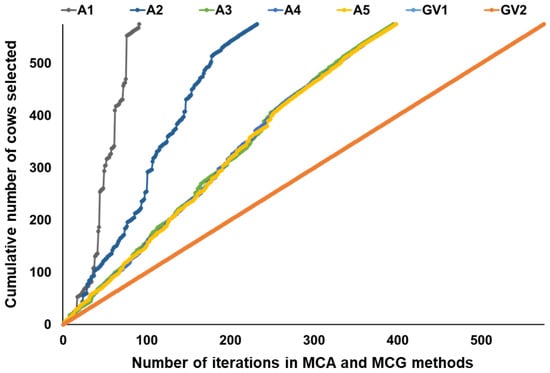
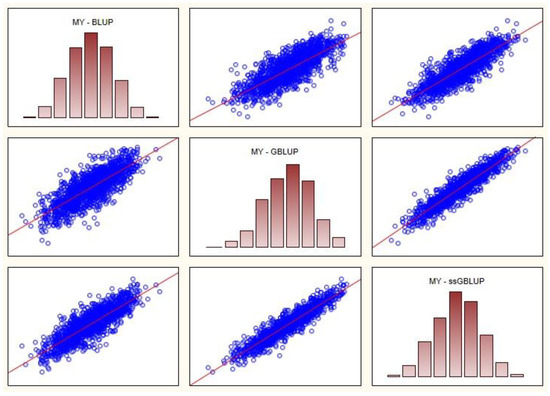
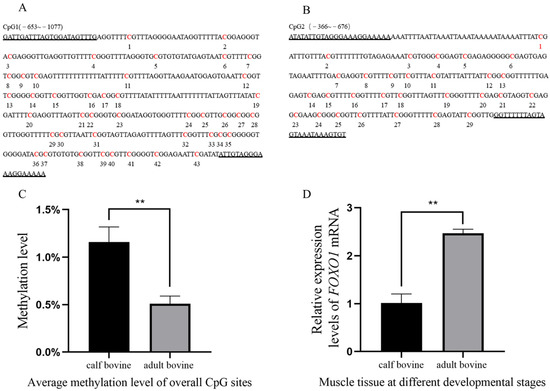
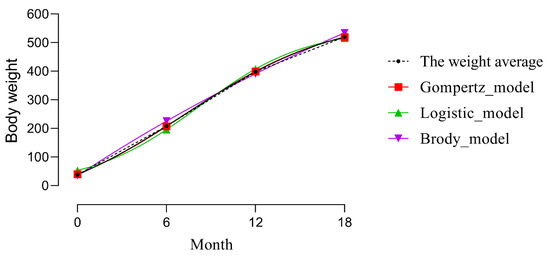
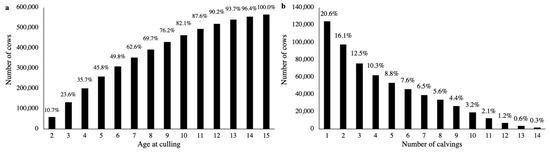
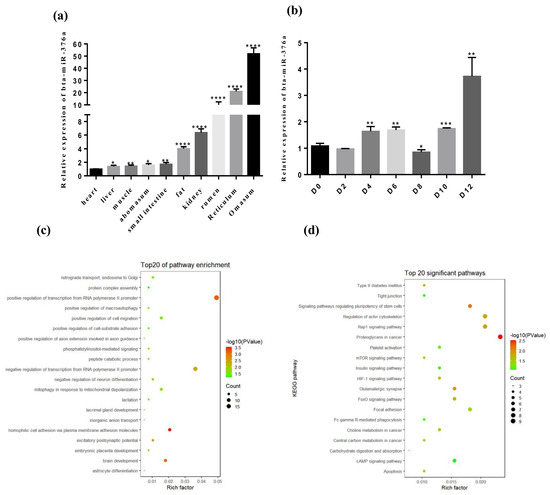
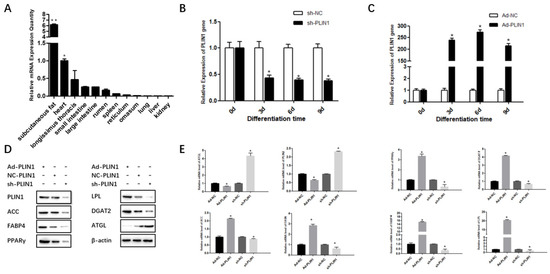
Figure 1













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}