The Complete Nucleotide Sequence and Gene Organization of the Mitochondrial Genome of Triatoma boliviana (Hemiptera, Reduviidae, Triatominae) and Phylogenetic Comparisons

 , , , and
, , , and
Abstract
:1. Introduction
2. Materials and Methods
3. Results and Discussion
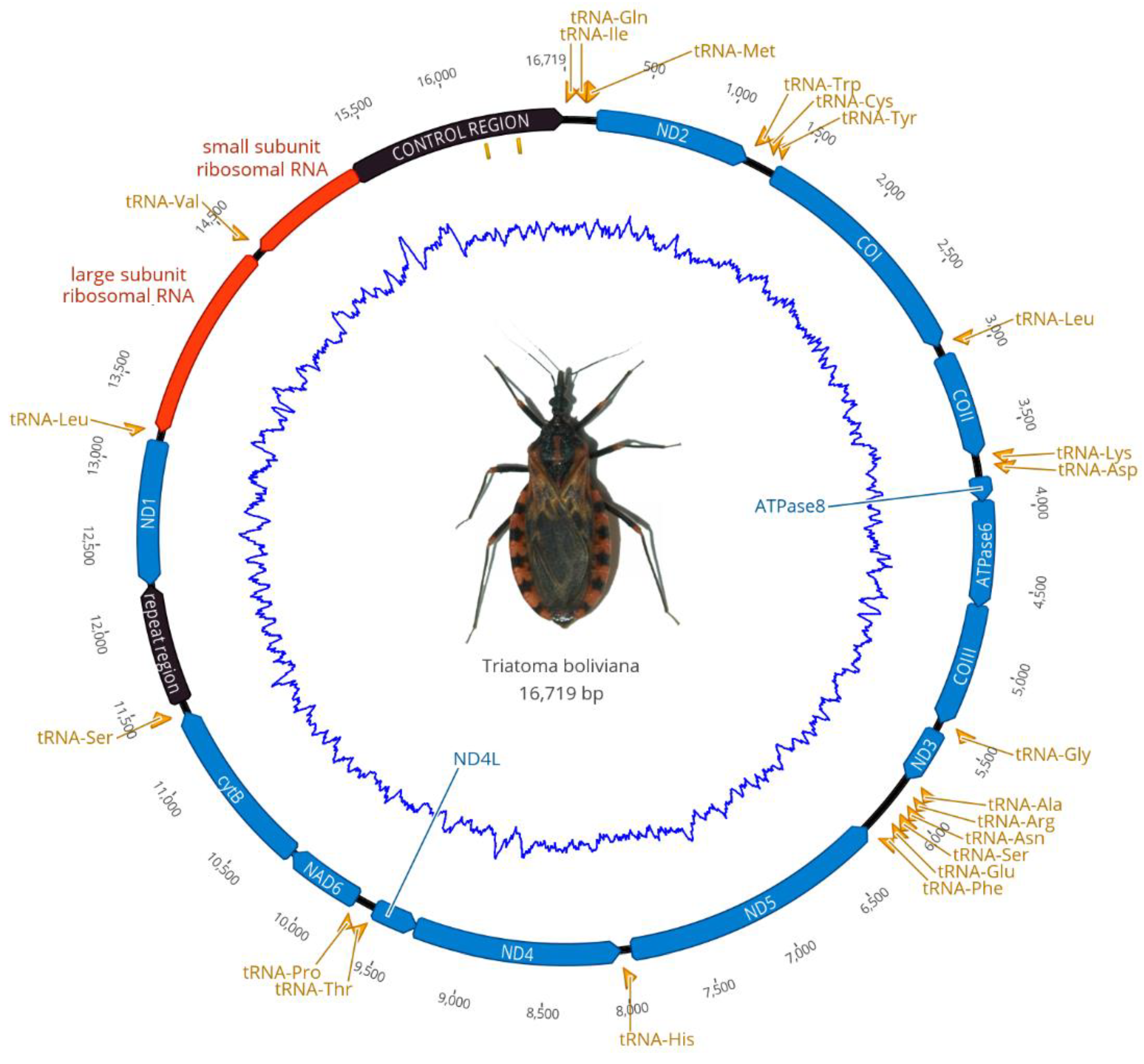
3.1. Mitogenome of Triatoma boliviana
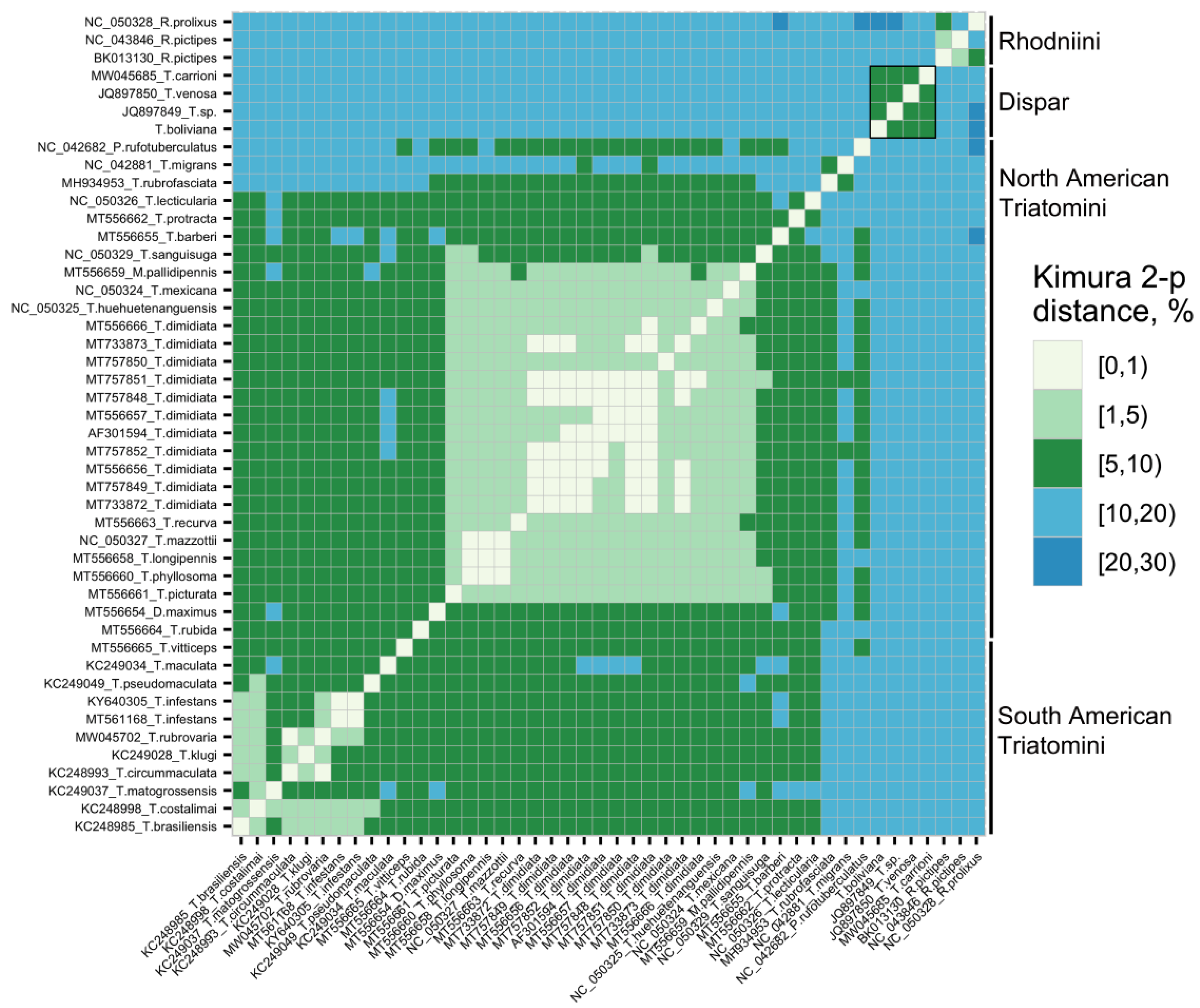
3.2. Phylogenetic Analyses
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Lent, H.; Wygodzinsky, P. Revision of the Triatominae (Hemiptera: Reduviidae), and their significance as vectors of Chagas disease. Bull. Am. Mus. Nat. Hist. 1979, 163, 123–520. [Google Scholar]
- Alevi, K.C.C.; de Oliveira, J.; da Silva Rocha, D.; Galvão, C. Trends in taxonomy of Chagas disease vectors (Hemiptera, Reduviidae, Triatominae): From Linnaean to integrative taxonomy. Pathogens 2021, 10, 1627. [Google Scholar] [CrossRef] [PubMed]
- Monteiro, F.A.; Weirauch, C.; Felix, M.; Lazoski, C.; Abad-Franch, F. Evolution, systematics, and biogeography of the Triatominae, vectors of Chagas disease. Adv. Parasitol. 2018, 99, 265–344. [Google Scholar] [CrossRef] [PubMed]
- Kieran, T.J.; Gordon, E.R.; Zaldívar-Riverón, A.; Ibarra-Cerdeña, C.N.; Glenn, T.C.; Weirauch, C. Ultraconserved elements reconstruct the evolution of Chagas disease-vectoring kissing bugs (Reduviidae: Triatominae). Syst. Entomol. 2021, 46, 725–740. [Google Scholar] [CrossRef]
- Dujardin, J.P.; Schofield, C.J.; Panzera, F. Los vectores de la enfermedad de Chagas. In Classe des Sciences Naturelles et Médicales; Mémoire in-8, Nouvelle Série, Tome 25, fasc. 3; Académie Royale des Sciences D’Outre-Mer: Brussels, Belgium, 2002; p. 198. [Google Scholar]
- Schofield, C.J.; Galvão, C. Classification, evolution, and species groups within the Triatominae. Acta Trop. 2009, 110, 88–100. [Google Scholar] [CrossRef]
- Justi, S.A.; Galvão, C.; Schrago, C.G. Geological changes of the Americas and their influence on the diversification of the Neotropical kissing bugs (Hemiptera: Reduviidae: Triatominae). PLoS Negl. Trop. Dis. 2016, 10, e0004527. [Google Scholar] [CrossRef]
- Martínez Avendaño, E.; Chávez Espada, T.; Sossa Gil, D.; Aranda Asturizaga, R.; Vargas Mamani, B.; Vidaurre Prieto, P. Triatoma boliviana sp. n. (Hemiptera: Reduviidae: Triatominae) de los valles subandinos de La Paz-Bolivia, similar a Triatoma nigromaculata Stål, 1859. Cuad. Hosp. Clín. 2007, 52, 9–16. [Google Scholar]
- Rojas de Arias, A.; Monroy, C.; Guhl, F.; Sosa-Estani, S.; Santos, W.S.; Abad-Franch, F. Chagas disease control-surveillance in the Americas: The multinational initiatives and the practical impossibility of interrupting vector-borne Trypanosoma cruzi transmission. Mem. Inst. Oswaldo Cruz 2021, 116, e210130. [Google Scholar] [CrossRef]
- Santillán-Guayasamín, S.; Barnabé, C.; Magallón-Gastelum, E.; Waleckx, E.; Yumiseva, C.A.; Grijalva, M.J.; Villacís, A.G.; Brenière, S.F. Molecular data supports monophyly of Triatoma dispar complex within genus Triatoma. Infect. Genet. Evol. 2020, 85, 104429. [Google Scholar] [CrossRef]
- Hwang, W.S.; Weirauch, C. Evolutionary history of assassin bugs (Insecta: Hemiptera: Reduviidae): Insights from divergence dating and ancestral state reconstruction. PLoS ONE 2012, 7, e45523. [Google Scholar] [CrossRef]
- Dierckxsens, N.; Mardulyn, P.; Smits, G. NOVOPlasty: De novo assembly of organelle genomes from whole genome data. Nucleic Acids Res. 2017, 45, e18. [Google Scholar] [CrossRef] [PubMed]
- Pita, S.; Panzera, F.; Vela, J.; Mora, P.; Palomeque, T.; Lorite, P. Complete mitochondrial genome of Triatoma infestans (Hemiptera, Reduviidae, Triatominae), main vector of Chagas disease. Infect. Genet. Evol. 2017, 54, 158–163. [Google Scholar] [CrossRef] [PubMed]
- Bernt, M.; Donath, A.; Jühling, F.; Externbrink, F.; Florentz, C.; Fritzsch, G.; Pütz, J.; Middendorf, M.; Stadler, P.F. MITOS: Improved de novo metazoan mitochondrial genome annotation. Mol. Phylogen. Evol. 2013, 69, 313–319. [Google Scholar] [CrossRef] [PubMed]
- Dotson, E.M.; Beard, C.B. Sequence and organization of the mitochondrial genome of the Chagas disease vector, Triatoma dimidiata. Insect Mol. Biol. 2001, 10, 205–215. [Google Scholar] [CrossRef]
- Clary, D.O.; Wolstenholme, D.R. The mitochondrial DNA molecule of Drosophila yakuba: Nucleotide sequence, gene organization, and genetic code. J. Mol. Evol. 1985, 22, 252–271. [Google Scholar] [CrossRef]
- Beard, C.B.; Hamm, D.M.; Collins, F.H. The mitochondrial genome of the mosquito Anopheles gambiae: DNA sequence, genome organization, and comparisons with mitochondrial sequences of other insects. Insect Mol. Biol. 1993, 2, 103–124. [Google Scholar] [CrossRef]
- Ojala, D.; Montoya, J.; Attardi, G. tRNA punctuation model of RNA processing in human mitochondria. Nature 1981, 290, 470–474. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef]
- Capella-Gutiérrez, S.; Silla-Martínez, J.M.; Gabaldón, T. trimAl: A tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 2009, 25, 1972–1973. [Google Scholar] [CrossRef]
- Paradis, E.; Schliep, K. ape 5.0: An environment for modern phylogenetics and evolutionary analyses in R. Bioinformatics 2019, 35, 526–528. [Google Scholar] [CrossRef] [PubMed]
- Schliep, K.P. phangorn: Phylogenetic analysis in R. Bioinformatics 2011, 27, 592–593. [Google Scholar] [CrossRef] [PubMed]
- Schliep, K.P. Estimating Phylogenetic Trees with Phangorn. Available online: https://mran.microsoft.com/snapshot/2015-12-12/web/packages/phangorn/vignettes/Trees.pdf (accessed on 2 February 2022).
- Yu, G.; Smith, D.K.; Zhu, H.; Guan, Y.; Lam, T.T.-Y. GGTREE: An R package for visualization and annotation of phylogenetic trees with their covariates and other associated data. Methods Ecol. Evol. 2017, 8, 28–36. [Google Scholar] [CrossRef]
- Aguilera-Uribe, M.; Meza-Lázaro, R.N.; Kieran, T.J.; Ibarra-Cerdeña, C.N.; Zaldívar-Riverón, A. Phylogeny of the North-Central American clade of blood-sucking reduviid bugs of the tribe Triatomini (Hemiptera: Triatominae) based on the mitochondrial genome. Infect. Genet. Evol. 2020, 84, 104373. [Google Scholar] [CrossRef] [PubMed]
- Wickham, H. ggplot2: Elegant Graphics for Data Analysis; Springer: New York, NY, USA, 2016; Available online: https://ggplot2.tidyverse.org (accessed on 2 February 2022).
- Dong, L.; Ma, X.; Wang, M.; Zhu, D.; Feng, Y.; Zhang, Y.; Wang, J. Complete mitochondrial genome of the Chagas disease vector, Triatoma rubrofasciata. Korean J. Parasitol. 2018, 56, 515–519. [Google Scholar] [CrossRef]
- Zhao, Y.; Jiang, M.; Wu, Y.; Song, F.; Cai, W.; Li, H. Mitochondrial genomes of three kissing bugs (Reduviidae: Triatominae) and their phylogenetic implications. Int. J. Biol. Macromol. 2019, 134, 36–42. [Google Scholar] [CrossRef]
- Cameron, S.L.; Lo, N.; Bourguignon, T.; Svenson, G.J.; Evans, T.A. A mitochondrial genome phylogeny of termites (Blattodea: Termitoidae): Robust support for interfamilial relationships and molecular synapomorphies define major clades. Mol. Phylogenet. Evol. 2012, 65, 163–173. [Google Scholar] [CrossRef]
- Hua, J.; Li, M.; Dong, P.; Cui, Y.; Xie, Q.; Bu, W. Phylogenetic analysis of the true water bugs (Insecta: Hemiptera: Heteroptera: Nepomorpha): Evidence from mitochondrial genomes. BMC Evol. Biol. 2009, 9, 134. [Google Scholar] [CrossRef]
- Kocher, A.; Kamilari, M.; Lhuillier, E.; Coissa, E.; Peneau, J.; Chave, J.; Murienne, J. Shotgun assembly of the assassin bug Brontostoma colossus mitochondrial genome (Heteroptera, Reduviidae). Gene 2014, 552, 184–194. [Google Scholar] [CrossRef]
- Zhang, D.X.; Szymura, J.M.; Hewitt, G.M. Evolution and structural conservation of the control region of insect mitochondrial DNA. J. Mol. Evol. 1995, 40, 382–391. [Google Scholar] [CrossRef]
- Weirauch, C. Cladistic analysis of Reduviidae (Heteroptera: Cimicomorpha) based on morphological characters. Syst. Entomol. 2008, 33, 229–274. [Google Scholar] [CrossRef]
- Weirauch, C.; Munro, J.B. Molecular phylogeny of the assassin bugs (Hemiptera: Reduviidae), based on mitochondrial and nuclear ribosomal genes. Mol. Phylogenet. Evol. 2009, 53, 287–299. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Gordon, E.R.L.; Forthman, M.; Hwang, W.S.; Walden, K.; Swanson, D.R.; Johnson, K.P.; Meier, R.; Weirauch, C. Evolution of the assassin’s arms: Insights from a phylogeny of combined transcriptomic and ribosomal DNA data (Heteroptera: Reduvioidea). Sci. Rep. 2016, 6, 22177. [Google Scholar] [CrossRef] [PubMed]
- Panzera, Y.; Pita, S.; Ferreiro, M.J.; Ferrandis, I.; Lages, C.; Pérez, R.; Silva, A.E.; Guerra, M.; Panzera, F. High dynamics of rDNA cluster location in kissing bug holocentric chromosomes (Triatominae, Heteroptera). Cytogenet. Genome Res. 2012, 138, 56–67. [Google Scholar] [CrossRef] [PubMed]
- Bardella, V.B.; Pita, S.; Vanzela, A.L.L.; Galvão, C.; Panzera, F. Heterochromatin base pair composition and diversification in holocentric chromosomes of kissing bugs (Hemiptera, Reduviidae). Mem. Inst. Oswaldo Cruz 2016, 111, 614–624. [Google Scholar] [CrossRef] [PubMed]
- Panzera, F.; Pita, S.; Lorite, P. Chromosome structure and evolution of Triatominae: A review. In Triatominae—The Biology of Chagas Disease Vectors; Entomology in Focus; Guarneri, A., Lorenzo, M., Eds.; Springer Nature: Cham, Switzerland, 2021; Volume 5, pp. 65–99. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| T. boliviana | Triatoma sp. JQ897849 | T. venosa JQ897850 | South Am. Triatomini | North Am. Triatomini | Rhodniini Tribe | |
|---|---|---|---|---|---|---|
| T. boliviana | 0.000 | 15.289 | 14.396 | 18.747 | ||
| Triatoma sp. JQ897849 | 7.549 | 0.000 | 14.811 | 13.515 | 19.301 | |
| T. venosa JQ897850 | 7.862 | 7.185 | 0.000 | 14.061 | 13.095 | 17.401 |
| T. carrioni MW045685 | 7.629 | 7.646 | 7.294 | 13.556 | 12.355 | 16.293 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pita, S.; Mora, P.; Rojas-Cortez, M.; Palomeque, T.; Lorite, P.; Panzera, F. The Complete Nucleotide Sequence and Gene Organization of the Mitochondrial Genome of Triatoma boliviana (Hemiptera, Reduviidae, Triatominae) and Phylogenetic Comparisons. Arthropoda 2023, 1, 3-11. https://doi.org/10.3390/entomology1010002
Pita S, Mora P, Rojas-Cortez M, Palomeque T, Lorite P, Panzera F. The Complete Nucleotide Sequence and Gene Organization of the Mitochondrial Genome of Triatoma boliviana (Hemiptera, Reduviidae, Triatominae) and Phylogenetic Comparisons. Arthropoda. 2023; 1(1):3-11. https://doi.org/10.3390/entomology1010002
Chicago/Turabian StylePita, Sebastián, Pablo Mora, Mirko Rojas-Cortez, Teresa Palomeque, Pedro Lorite, and Francisco Panzera. 2023. "The Complete Nucleotide Sequence and Gene Organization of the Mitochondrial Genome of Triatoma boliviana (Hemiptera, Reduviidae, Triatominae) and Phylogenetic Comparisons" Arthropoda 1, no. 1: 3-11. https://doi.org/10.3390/entomology1010002