
Isolated Myelopathy in Occult Breast Carcinoma with Negative Paraneoplastic Antibodies: A Case Report of a Rare Condition
 ,
,  , and
, and 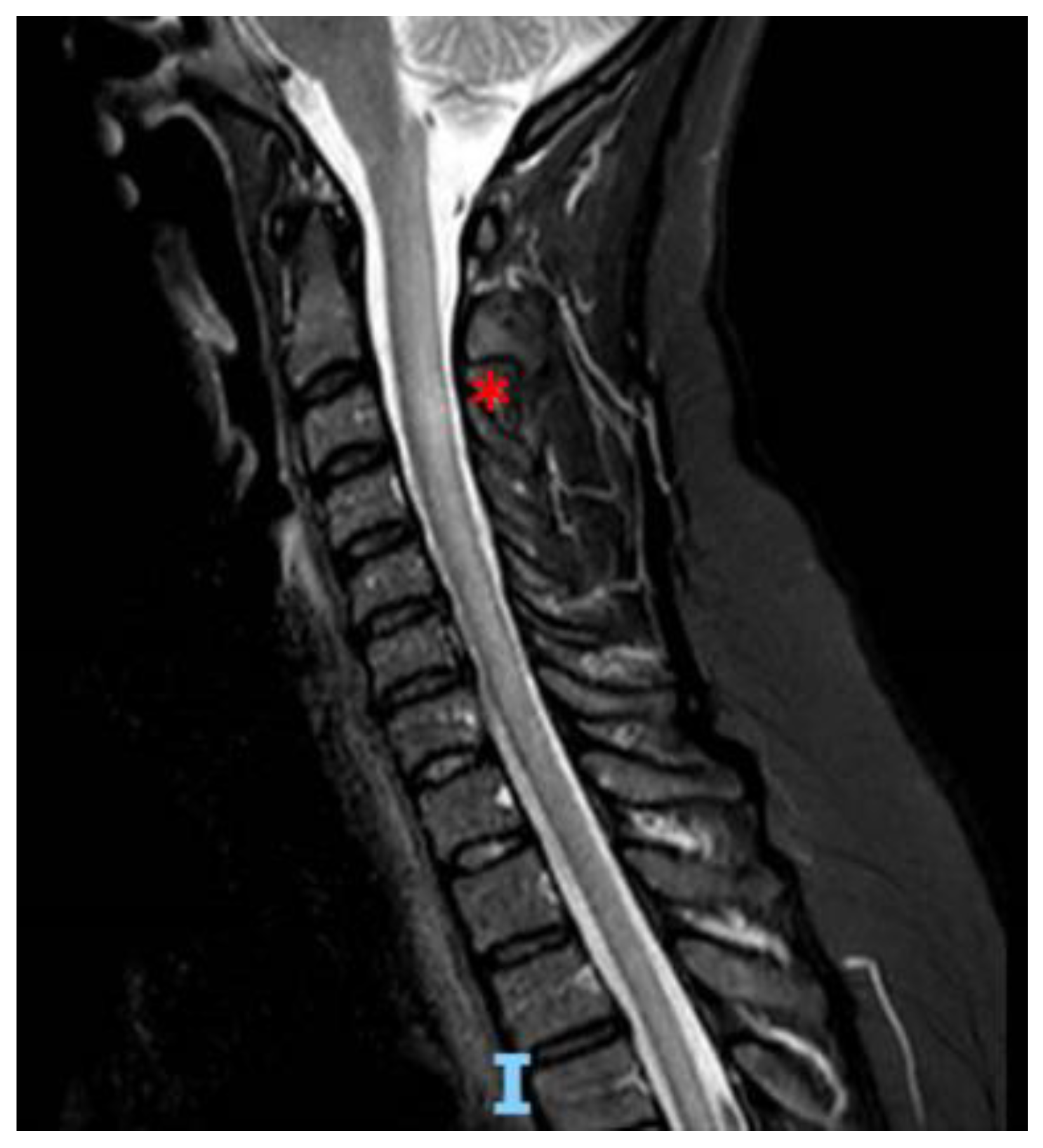{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
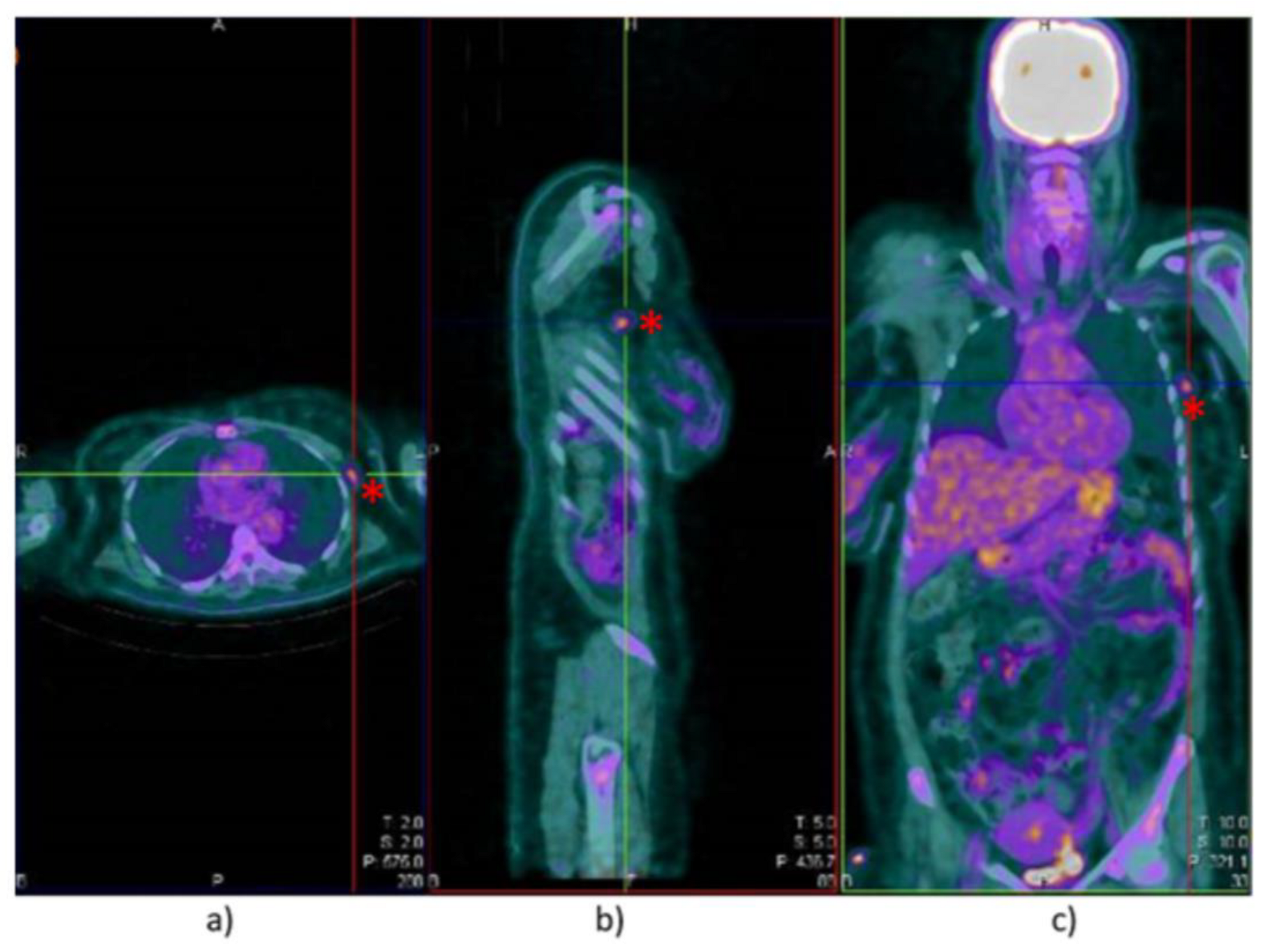
2. Case Report
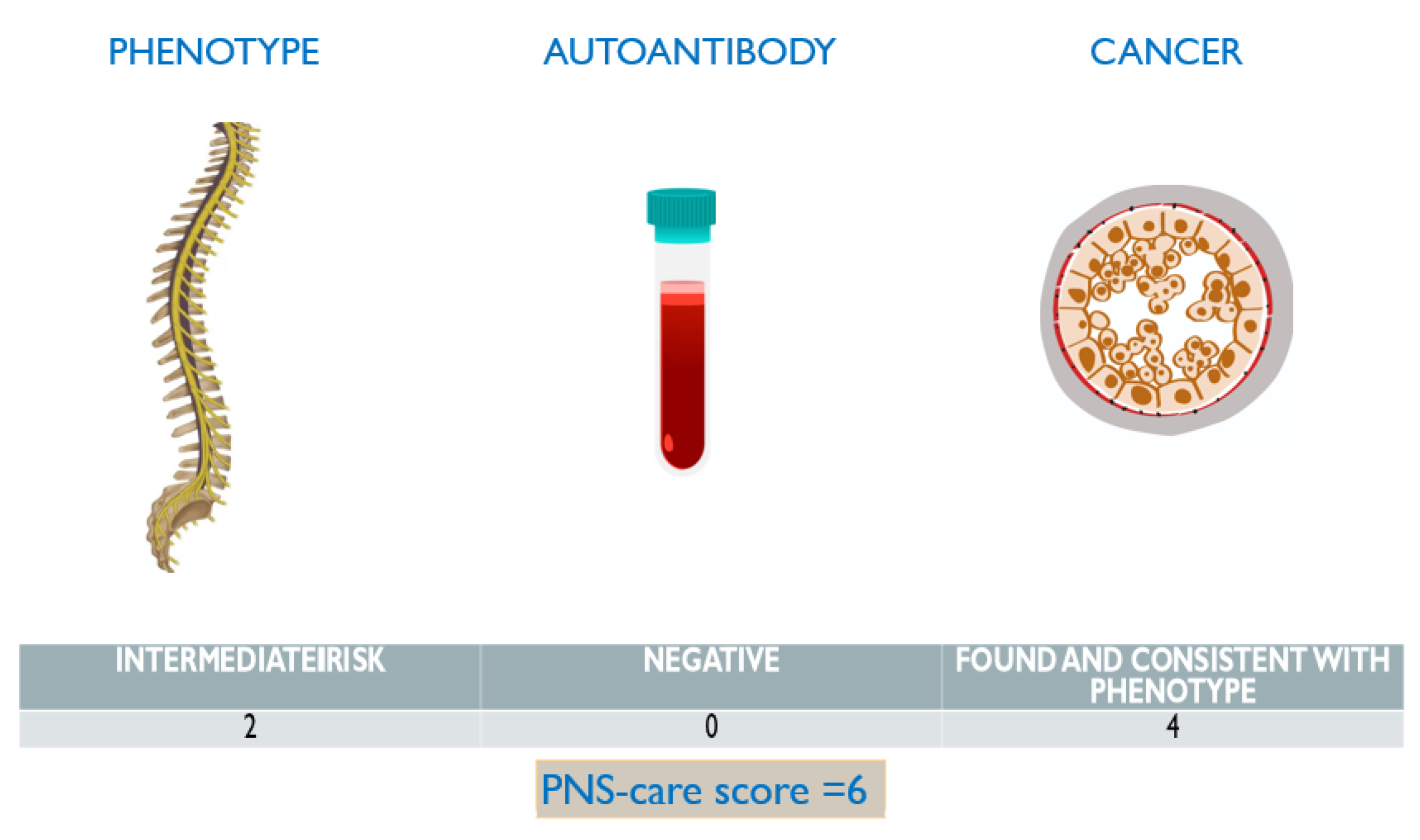
3. Discussion
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Vecchio, D.; Virgilio, E.; Naldi, P.; Comi, C.; Cantello, R. MOG-antibody demyelinating diseases: A case of post-partum severe rhombencephalitis and transverse myelitis. Mult. Scler. Relat. Disord. 2018, 21, 9–10. [Google Scholar] [CrossRef] [PubMed]
- Virgilio, E.; Vecchio, D.; Vercellino, M.; Naldi, P.; Tesser, F.; Cantello, R.; Cavalla, P.; Comi, C. Paraneoplastic neuromyelitis optica spectrum disorders: A case series. Neurol. Sci. 2021, 42, 2519–2522. [Google Scholar] [CrossRef] [PubMed]
- Cacciaguerra, L.; Sechi, E.; Rocca, M.A.; Filippi, M.; Pittock, S.J.; Flanagan, E.P. Neuroimaging features in inflammatory myelopathies: A review. Front. Neurol. 2022, 13, 993645. [Google Scholar] [CrossRef] [PubMed]
- Brinar, V.V.; Habek, M.; Brinar, M.; Malojcić, B.; Boban, M. The differential diagnosis of acute transverse myelitis. Clin. Neurol. Neurosurg. 2006, 108, 278–283. [Google Scholar] [CrossRef] [PubMed]
- McKeon, A.; Pittock, S.J.; Lennon, V.A. CSF complements serum for evaluating paraneoplastic antibodies and NMO-IgG. Neurology 2011, 76, 1108–1110. [Google Scholar] [CrossRef] [PubMed]
- Flanagan, E.P.; McKeon, A.; Lennon, V.A.; Kearns, J.; Weinshenker, B.G.; Krecke, K.N.; Matiello, M.; Keegan, B.M.; Mokri, B.; Aksamit, A.J.; et al. Paraneoplastic isolated myelopathy: Clinical course and neuroimaging clues. Neurology 2011, 76, 2089–2095. [Google Scholar] [CrossRef] [PubMed]
- Flanagan, E.P.; Keegan, B.M. Paraneoplastic myelopathy. Neurol. Clin. 2013, 31, 307–318. [Google Scholar] [CrossRef] [PubMed]
- Graus, F.; Vogrig, A.; Muñiz-Castrillo, S.; Antoine, J.G.; Desestret, V.; Dubey, D.; Giometto, B.; Irani, S.R.; Joubert, B.; Leypoldt, F.; et al. Updated Diagnostic Criteria for Paraneoplastic Neurologic Syndromes. Neurol. Neuroimmunol. Neuroinflamm 2021, 8. [Google Scholar] [CrossRef] [PubMed]
- Keegan, B.M.; Pittock, S.J.; Lennon, V.A. Autoimmune myelopathy associated with collapsin response-mediator protein-5 immunoglobulin G. Ann. Neurol. 2008, 63, 531–534. [Google Scholar] [CrossRef] [PubMed]
- Graus, F.; Dalmau, J. Paraneoplastic neurological syndromes. Curr. Opin. Neurol. 2012, 25, 795–801. [Google Scholar] [CrossRef] [PubMed]
- Tirthani, E.; Said, M.S.; Smith, R.G.; Jadhav, N.; Shanina, E. Paraneoplastic Encephalomyelitis. In StatPearls; StatPearls Publishing LLC.: Treasure Island, FL, USA, 2022. [Google Scholar]
- Flanagan, E.P. Autoimmune myelopathies. Handb. Clin. Neurol. 2016, 133, 327–351. [Google Scholar] [CrossRef] [PubMed]
- Graus, F.; Delattre, J.Y.; Antoine, J.C.; Dalmau, J.; Giometto, B.; Grisold, W.; Honnorat, J.; Smitt, P.S.; Vedeler, C.; Verschuuren, J.J.; et al. Recommended diagnostic criteria for paraneoplastic neurological syndromes. J. Neurol. Neurosurg. Psychiatry 2004, 75, 1135–1140. [Google Scholar] [CrossRef] [PubMed]
- Greenlee, J.E. Treatment of paraneoplastic neurologic disorders. Curr. Treat. Options Neurol. 2010, 12, 212–230. [Google Scholar] [CrossRef] [PubMed]
- Thapa, B.; Mahendraker, N.; Ramphul, K. Paraneoplastic Syndromes. In StatPearls; StatPearls Publishing LLC.: Treasure Island, FL, USA, 2022. [Google Scholar]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Paciolla, L.; Galli, G.; Vecchio, D.; Padelli, S.; Comi, C.; Cantello, R.; Virgilio, E. Isolated Myelopathy in Occult Breast Carcinoma with Negative Paraneoplastic Antibodies: A Case Report of a Rare Condition. Sclerosis 2023, 1, 60-66. https://doi.org/10.3390/sclerosis1010007
Paciolla L, Galli G, Vecchio D, Padelli S, Comi C, Cantello R, Virgilio E. Isolated Myelopathy in Occult Breast Carcinoma with Negative Paraneoplastic Antibodies: A Case Report of a Rare Condition. Sclerosis. 2023; 1(1):60-66. https://doi.org/10.3390/sclerosis1010007
Chicago/Turabian StylePaciolla, Loredana, Giulia Galli, Domizia Vecchio, Samuel Padelli, Cristoforo Comi, Roberto Cantello, and Eleonora Virgilio. 2023. "Isolated Myelopathy in Occult Breast Carcinoma with Negative Paraneoplastic Antibodies: A Case Report of a Rare Condition" Sclerosis 1, no. 1: 60-66. https://doi.org/10.3390/sclerosis1010007