
Is It Lupus? Is It Neuromyelitis Optica Spectrum Disorder (NMOSD)?—Why Not Both?
by
 , , and
, , and
Niklas Alexander Kaempfer
1,
Mathias Fousse
1,*,
Michael Kettner
2,
Klaus Fassbender
1 and
Daniel Janitschke
1,* 1
Department of Neurology, Saarland University Medical Center, 66421 Homburg, Germany
2
Department of Neuroradiology, Saarland University Medical Center, 66421 Homburg, Germany
*
Authors to whom correspondence should be addressed.
Sclerosis 2023, 1(1), 51-59; https://doi.org/10.3390/sclerosis1010006
Submission received: 31 March 2023
/
Revised: 28 April 2023
/
Accepted: 8 May 2023
/
Published: 12 May 2023
Abstract
:Multiple sclerosis (MS) and neuromyelitis optica spectrum disorders (NMOSD) are among the commonly considered differential diagnoses in patients with inflammatory central nervous system (CNS)-diseases. Formerly diagnosed competing autoimmune diseases might impair diagnostics and treatment. Here, we report on a 41-year-old woman admitted to our hospital with primary manifestation of NMOSD (paresthesia, paralysis of the lower extremities, and urinary incontinence) while undergoing treatment for a diagnosed systemic lupus erythematosus (SLE) with hydroxychloroquine. CNS manifestation of the disease was considered. Magnetic resonance imaging (MRI) of the cranium and spinal cord showed multiple supratentorial lesions of the white matter and massive intramedullary lesions with contrast enhancement. Cerebrospinal fluid (CSF) showed pleocytosis (20/µL), positive antinuclear antibodies (ANA), antiphospholipid antibodies, and SSA/Ro antibodies, while formerly positive dsDNA antibodies were negative. Further diagnostics revealed a 1:10,240 serum titer of Aquaporine-4 antibodies. The patient received intravenous methylprednisolone for three days (2 g per day), which led to an escalation to plasmapheresis and to an improved EDSS from 8.0 to 4.0. Because of the comorbidity, a combined relapse prophylaxis with satralizumab and mycophenolate mofetil was established. Rehabilitation and continued treatment improved EDSS to 1.0 with no impairment of mobilization. Although formerly diagnosed SLE could have explained the symptoms, it is important to reconsider competitive diseases in order to establish adequate immunotherapy.
1. Introduction
Multiple sclerosis (MS) and neuromyelitis optica spectrum disorders (NMOSD) are among the commonly considered differential diagnoses in patients with inflammatory central nervous system (CNS)-diseases [1]. Formerly diagnosed competing autoimmune diseases might impair diagnostics and treatment. NMOSD is an autoimmune inflammatory disease affecting the central nervous system. Compared to multiple sclerosis, NMOSD is less common; its prevalence ranges up to 4.4 per 100,000 people, and females are considered more susceptible [2]. Clinically, patients most commonly present with symptoms associated with damage of the optic nerve, spinal cord, area postrema, brainstem, diencephalon, and cerebrum. The mentioned parts of the CNS and their respective clinical features are also taken into account in the diagnostic criteria (as conceptualized by Wingerchuk et al.) [3]. Considered the most specific clinical feature of NMOSD is longitudinally extensive transverse myelitis (LETM), which is less common in MS patients. Antibodies directed against aquaporine-4 (AQP-4), which is a protein located in cerebral regions with spatial reference to cerebral spinal fluid (CSF), might induce an increase in interleukin-6 expression in astrocytes interfering with blood–brain barrier function. Additionally, via complement activation, the process ultimately results in a loss of function in astrocytes by interfering with their supportive function for oligodendrocytes and neurons and leading to neutrophil infiltration and, subsequently, secondary demyelination [4]. NMO patients who are seronegative for AQP-4 antibodies might show a more heterogeneous clinical manifestation. In 30–70% of seronegative NMO patients antibodies directed against myelin oligodendrocyte glycoprotein (MOG) can be detected. The detection of MOG antibodies is not considered in the diagnostic criteria for NMOSD. It indicates the presence of myelin oligodendrocyte glycoprotein antibody-associated disease (MOGAD) despite the possibility of overlapping clinical features and MRI findings, thus complicating the distinction between MS and NMO, which are considered separate entities of disease [5]. An important differential diagnosis regarding inflammatory CNS diseases (especially for patients with non-neurological co-manifestations) is systemic lupus erythematosus (SLE). SLE is a chronic autoimmune disease with a wide range of possible clinical manifestations that predominantly affects females. The complex pathogenesis involves a reduced apoptotic clearance, the loss of immunological self-tolerance, and the deposition of immune complexes and autoantibodies, causing complement activation and tissue infiltration with neutrophilic granulocytes, monocytes, and lymphocytes. Key organ manifestations include skin, the musculoskeletal system, kidneys, and the CNS. While several classification criteria have been established in the past (e.g., SLICC and ACR/EULAR), a diagnosis is generally based on the combination of clinical and serological findings (e.g., the presence of antinuclear antibodies) [6]. Clinical manifestations regarding the central nervous system are summarized in the term ‘neuropsychiatric SLE’ (NPSLE), which is estimated to occur in 12–95% of SLE patients, presenting with a variety of symptoms. Clinical manifestation ranges from non-specific complaints (such as headache or cognitive deficits) to severe clinical manifestations (e.g., seizures or ischemic strokes). In 0.9–2.7% of all cases, a demyelinating syndrome can be observed; in 0.9–3.9% of cases, SLE-related myelopathy is described [7]. Although the pathophysiology regarding the damage of the spinal cord is not entirely known, the literature suggests small vessel vasculitis and thrombotic events as possible causes. A connection between the presence of antiphospholipid antibodies (aPL) and ischemic events in patient with NPSLE-related myelopathy has been discussed [8].
In this case report, we are reporting on a patient with a severe primary manifestation of seropositive NMOSD while undergoing treatment for systemic lupus erythematosus. Despite the already diagnosed SLE, extensive differential diagnostics were initiated as a competitive disease was suspected.
2. Materials and Methods
2.1. Laboratory Analysis
CSF parameters and routine serum laboratory diagnostics, as well as antinuclear antibodies (ANA), double-strand-DNA antibodies (dsDNA), antiphospholipid (aPL), complement factors, and SSA/Ro antibodies were analyzed in the central laboratory of the Saarland Medical Center. The department of Virology of the Saarland Medical Center carried out virus analysis. Bacterial analysis was performed in the department of Microbiology of the Saarland Medical Center. Aquaporin-4 antibody and MOG antibody analysis were conducted in an external laboratory (MVZ Labor PD Dr. Volkmann und Kollegen GbR, Karlsruhe, Germany). The Department of Internal Medicine I at Saarland Medical Center carried out fluorescence-activated cell sorting (FACS) analysis of the CSF. The Department of Neuropathology at the Saarland Medical Center conducted the neuropathological analysis of the CSF.
2.2. MR-Imaging
MRI images were acquired in the Department for Neuroradiology. Cranial MRI images were acquired in a 3-tesla MRI scanner, whereas spinal images were obtained in a 1.5-tesla MRI scanner. Further description of the shown image is given in the figure legend.
2.3. Neurophysiological Diagnostic
Visual sensory evoked potentials (VEP), sensory evoked potentials (SEP), and motoric evoked potentials were performed in our sub-department of Neurophysiology.
2.4. Diagnostic Flow
Serum and CSF were acquired at the beginning of the inpatient stay for all of the parameters mentioned in Section 2.1. MRI and evoked potentials were initiated simultaneously. All findings were received on the day of initiation, except the test results for AQP-4 antibodies, anti-MOG antibodies (conducted by an external laboratory), and neuropathological analysis (delay of nine days).
3. Case Presentation
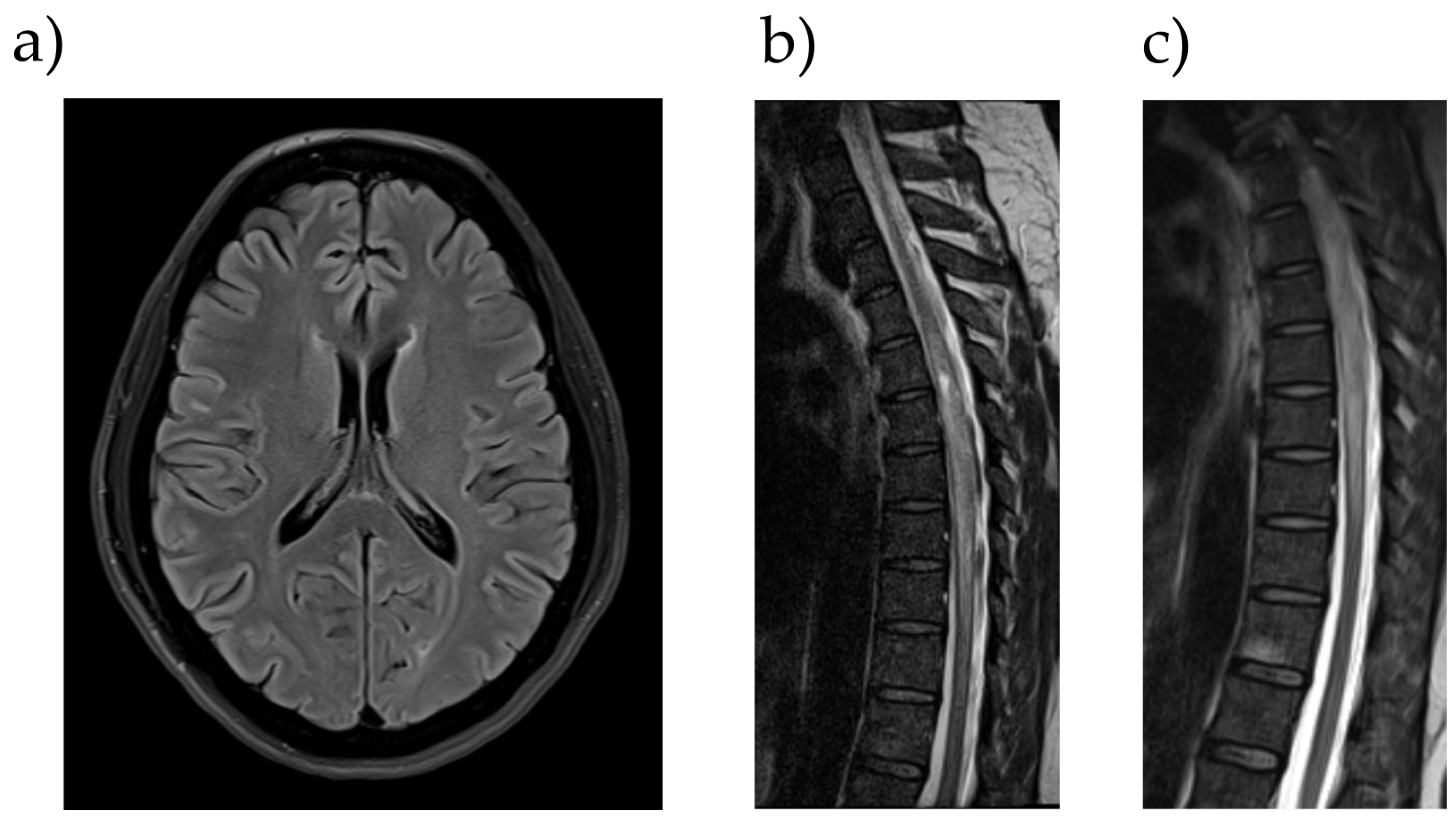
A 41-year-old woman was admitted to our hospital in March 2022 with paralysis of the lower extremities, as well as urinary incontinence. According to the patient, the first symptoms had occurred a month before with paresthesia of the lower extremities, while first signs of paralysis were noticed about a week before hospitalization. After this, the patient suffered from urinary incontinence. The patient had no history of neurological disorders, but since the patient was diagnosed with systemic lupus erythematosus in 2018, CNS manifestation of the disease was considered. When first diagnosed, the patient presented relapsing episodes of thrombocytopenia and hemolysis, and she tested positive for antinuclear antibodies (ANA), double-strand-DNA antibodies (dsDNA), and antiphospholipid (aPL) antibodies in serum probes (Table 1). The patient initially was being treated exclusively with prednisolone; later, a treatment with hydroxychloroquine was established, which had appeared to be a sufficient relapse prophylaxis. At the day of admission to our Department of Neurology, the clinical examination saw a vigilant and fully oriented patient, with no impairment of the cranial nerves and no aphasia, dysarthria, or dysphagia. Cognitive impairment or psychiatric symptoms were not observed. While the upper limbs showed no signs of paresis or sensory disturbance, severe paresis of the legs (1/5 on the British Medical Council research scale) as well as hypoesthesia and bimalleolar pallhypesthesia (0/8) were detected. The lower limbs also showed weakened reflexes, while Babinski’s sign was bilaterally positive. MRIs of the cranium and spinal cord showed multiple supratentorial white matter lesions as a sign of past inflammation and massive intramedullary lesions (at the level of thoracic vertebrae 4–11) with signs of contrast enhancement in T1-weighted images. Because of the extent of the intramedullary lesions with diffuse signal alteration, our colleagues from the Department of Neuroradiology discussed a possible intramedullary tumorous mass (e.g., ependymoma or glioma). Spinal ischemia was also discussed but considered less likely as the symptoms did not occur abruptly (Figure 1).
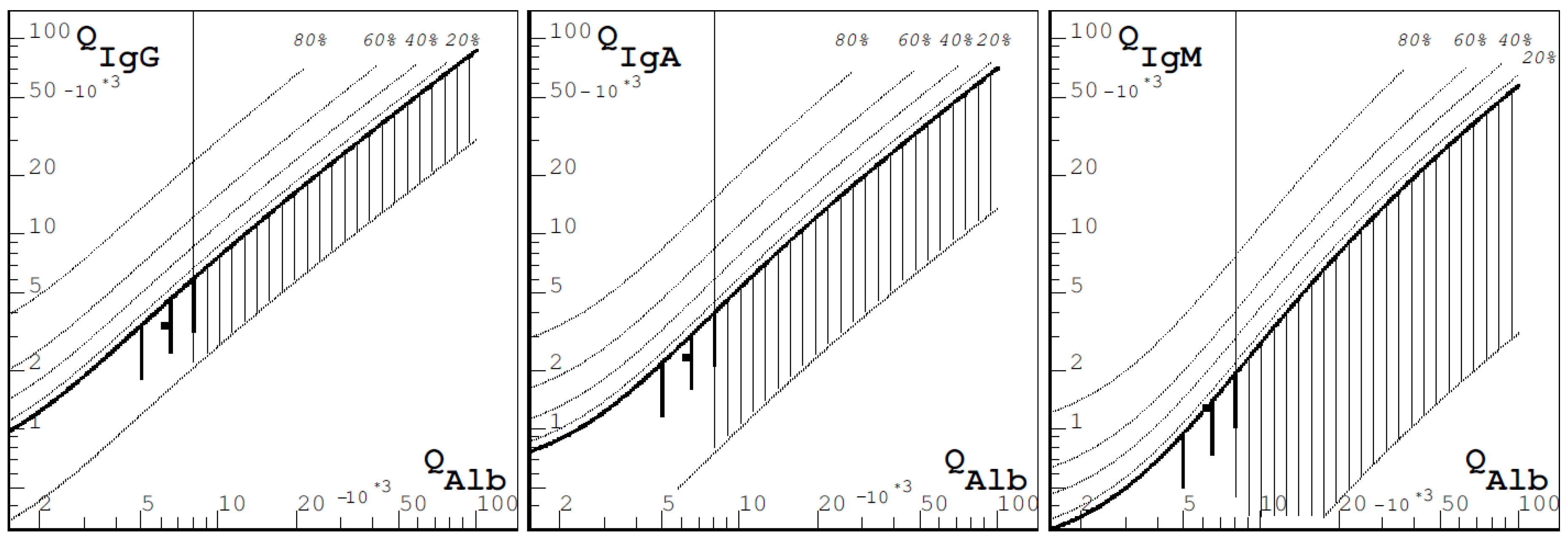
Visual evoked potentials (VEP) revealed no pathologic result. Regarding sensory evoked potentials (SEP), no cortical potentials were derivable from the left and right tibial nerve, whereas L1 was definable, which also affirmed a lesion in the posterior funiculus. The measurement of the motoric evoked potentials (MEP) was considered too painful by the patient, and she terminated the procedure. The examination of CSF showed pleocytosis (20/µL), while there was no alteration in protein, lactate, and glucose. There were discrete signs of autochthonous antibody synthesis regarding immunoglobulin M (IgM), and the oligoclonal bands remained negative (Figure 2).
Diagnostics regarding possible infectious causes showed negative results for neurotropic viruses (including herpes simplex virus, varicella zoster virus, Epstein–Barr virus, cytomegalovirus, and, considering the immunotherapy with hydroxychloroquine, JC virus) as well as bacteria. As the patient had a history of swollen lymph nodes, samples were additionally sent to our Department of Hematology for fluorescence-activated cell sorting (FACS), from which no signs of lymphoma cells were detected. The neuropathological findings also showed no signs of malignant cells, while a lymphocytic dominance with few granulocytes as an expression of possible inflammation was discussed. Additionally antinuclear antibodies (ANA), aPL antibodies (targeted against cardiolipin and beta-2-glycoprotein), and SSA/Ro antibodies were found to be positive, while formerly positive dsDNA antibodies were found to be negative. Serum levels for C4 and C3 complement factors were reduced. Mycobacterium tuberculosis specific T-cells (via FACS-based assay) could not be detected in serum probes, and antibodies against borrelia and treponema pallidum also remained negative. Later, a 1:10,240 serum titer tested positive for aquaporin-4 antibodies, while MOG antibodies were found to be negative. The patient received methylprednisolone for 3 days (2 g per day), and therapy had to be escalated to plasmapheresis (16 sessions in total). As a result, the expanded disability status scale (EDSS) improved from 8.0 to 4.0. Because of the comorbidity, a combined relapse prophylaxis with IL-6 antibody satralizumab and mycophenolate mofetil was established.
Within an observational period of 12 months, the patient has shown no signs of relapse. In the latest clinical examinations, there were still no signs of psychiatric symptoms and no impairment of the cranial nerves. The motor system appeared intact as there were no pareses, no signs of pyramidal track lesions, and physiological muscle tone. Regarding the sensory system, the formerly mentioned paresthesia and hypesthesia had fully regressed, the only remaining symptom being a minor gait ataxia. The EDSS score had improved to 1.0.
4. Discussion
NPSLE was primarily considered the most likely differential diagnosis regarding the patient’s medical history and the observed clinical manifestations. EULAR/ACR criteria [9] for SLE were met via the detection of ANA, dsDNA antibodies, reduced complement factors, positive aPL antibodies, and formerly observed anemia and thrombocytopenia (Table 2).
CSF findings showed signs of inflammation, which also fit the possible NPSLE. The diagnosis was reconsidered after receiving the MRI results with diffuse and extensive longitudinal damage of the spinal cord; even a tumorous genesis was discussed. After receiving negative results for possible infectious causes, an autoinflammatory disorder was still considered most likely. Under empirical treatment with glucocorticoids and, subsequently, plasmapheresis, the symptoms significantly improved, which substantiated the hypothesis. In summary, after receiving positive results for the highly specific AQP4 antibodies, seropositive NMOSD could be diagnosed as well (Table 3).
Former reports have already shown a significant frequency of the simultaneous detection of antiphospholipid antibodies as well as AQP4 antibodies in patients with NMOSD, possibly leading to falsely diagnosed APS-associated myelitis in some cases [10].
Regarding the formerly mentioned myelopathy as a possible manifestation of SLE, older reports have discussed several cases in which SLE patients suffered from myelopathy, even with extensive longitudinal myelitis over four or more spinal segments in some cases, which could also have indicated the presence of NMOSD. Unfortunately, AQP4 antibodies had not been tested or taken in consideration [9,11,12,13,14].
In this case, because of the comorbidity with two competitive autoimmune disorders and severe clinical manifestations, we decided on a combined immunotherapy with 1 g MMF orally twice per day and 120 mg satralizumab subcutaneously every four weeks after an initial loading phase. The decision was made in consultation with the Department of Rheumatology at our hospital.
Satralizumab is a monoclonal antibody that was engineered to reduce the activity of interleukin-6 by binding to its receptors. The drug received approval by the food and drug administration (FDA) in the USA in 2020 for the treatment of adult NMOSD patients who are seropositive for AQP4 antibodies. Compared to IL6-receptor antibody tocilizumab, which has also been studied as an SLE treatment [15], satralizumab shows a fourfold higher affinity to the receptor. The binding of the receptor leads to the inhibition of IL-6 downstream pathways, apparently reducing IL-6-mediated T- and B-cell activation and preventing the differentiation of B cells into AQP4-antibody-secreting plasma cells [16].
Mycophenolate mofetil (MMF) is a prodrug of mycophenolic acid (MPA), which inhibits the enzyme inosine-5’-monophosphate dehydrogenase, leading to the depletion of guanosine nucleotides. This mechanism affects T and B lymphocytes and inhibits their proliferation. It also inhibits the inducible nitric oxide synthase, which is thought to possibly reduce nitric-oxide-associated tissue damage caused by macrophages [17]. MMF is used for the treatment of SLE, especially with the presence of lupus nephritis and extrarenal manifestations [6]. It is also used as relapse prophylaxis in NMOSD patients, although relapse rates seem to be higher in comparison to antibody therapies such as Rituximab (RTX) [18]. In this case, MMF was chosen as an add-on therapy because of the two competing autoimmune diseases and the demonstrated effect in both NMOSD and SLE, as opposed to monotherapy with satralizumab.
As the patient has not shown any signs of relapse within the observation period of twelve months, the combined treatment will be continued as primarily initiated.
5. Conclusions
In this case report, the first detection of aPL antibodies occurred four years prior to the first measurement of AQP4 antibodies. It is unclear whether AQP4 antibody synthesis had already occurred in this regard. However, it should be noted that the previously presented clinical symptoms (hemolysis, thrombopenia, etc.) were not compatible with an NMOSD diagnosis. In our view, the possibility is given that the presence of aPL antibodies may have led to the microvascular damage of the blood–brain barrier and, subsequently, might have triggered an autoinflammatory reaction against AQP4.
Even though the formerly diagnosed SLE could also have sufficiently explained the symptoms of the patient, it is important to reconsider competitive diseases in order to establish an adequate immunotherapy.
In our view, this highlights the importance of thorough differential diagnostics in patients presenting with myelitis and the consideration of other autoimmune diseases.
Author Contributions
Conceptualization, N.A.K. and D.J.; writing—original draft preparation. N.A.K.; writing—review and editing, D.J., M.F. and K.F.; supervision, D.J. and K.F.; visualization, M.K. and N.A.K. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Informed consent has been obtained from the patient to publish this paper.
Data Availability Statement
No new data were created.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Höftberger, R.; Lassmann, H. Inflammatory Demyelinating Diseases of the Central Nervous System. Handb. Clin. Neurol. 2017, 145, 263–283. [Google Scholar] [CrossRef] [PubMed]
- Pandit, L.; Asgari, N.; Apiwattanakul, M.; Palace, J.; Paul, F.; Leite, M.I.; Kleiter, I.; Chitnis, T. GJCF International Clinical Consortium & Biorepository for Neuromyelitis Optica Demographic and Clinical Features of Neuromyelitis Optica: A Review. Mult. Scler. Houndmills Basingstoke Engl. 2015, 21, 845–853. [Google Scholar] [CrossRef]
- Wingerchuk, D.M.; Banwell, B.; Bennett, J.L.; Cabre, P.; Carroll, W.; Chitnis, T.; de Seze, J.; Fujihara, K.; Greenberg, B.; Jacob, A.; et al. International Consensus Diagnostic Criteria for Neuromyelitis Optica Spectrum Disorders. Neurology 2015, 85, 177–189. [Google Scholar] [CrossRef] [PubMed]
- Huda, S.; Whittam, D.; Bhojak, M.; Chamberlain, J.; Noonan, C.; Jacob, A. Neuromyelitis Optica Spectrum Disorders. Clin. Med. Lond. Engl. 2019, 19, 169–176. [Google Scholar] [CrossRef] [PubMed]
- Sechi, E.; Cacciaguerra, L.; Chen, J.J.; Mariotto, S.; Fadda, G.; Dinoto, A.; Lopez-Chiriboga, A.S.; Pittock, S.J.; Flanagan, E.P. Myelin Oligodendrocyte Glycoprotein Antibody-Associated Disease (MOGAD): A Review of Clinical and MRI Features, Diagnosis, and Management. Front. Neurol. 2022, 13, 885218. [Google Scholar] [CrossRef] [PubMed]
- Fava, A.; Petri, M. Systemic Lupus Erythematosus: Diagnosis and Clinical Management. J. Autoimmun. 2019, 96, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, N.; Stock, A.D.; Putterman, C. Neuropsychiatric Lupus: New Mechanistic Insights and Future Treatment Directions. Nat. Rev. Rheumatol. 2019, 15, 137–152. [Google Scholar] [CrossRef] [PubMed]
- Chiganer, E.H.; Hryb, J.P.; Carnero Contentti, E. Myelitis and Lupus: Clinical Manifestations, Diagnosis and Treatment. Review. Reumatol. Clínica Engl. Ed. 2017, 13, 344–348. [Google Scholar] [CrossRef] [PubMed]
- Aringer, M.; Costenbader, K.; Daikh, D.; Brinks, R.; Mosca, M.; Ramsey-Goldman, R.; Smolen, J.S.; Wofsy, D.; Boumpas, D.T.; Kamen, D.L.; et al. 2019 European League Against Rheumatism/American College of Rheumatology Classification Criteria for Systemic Lupus Erythematosus. Ann. Rheum. Dis. 2019, 78, 1151–1159. [Google Scholar] [CrossRef] [PubMed]
- Guerra, H.; Pittock, S.J.; Moder, K.G.; Froehling, D.A.; Flanagan, E.P. Neuromyelitis Optica Spectrum Initially Diagnosed as Antiphospholipid Antibody Myelitis. J. Neurol. Sci. 2016, 361, 204–205. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.-Y.; Yoon, T.-S.; Suh, J.-H. Concomitant Occurrence of Cervical Myelopathy, Cerebral Infarction, and Peripheral Neuropathy in Systemic Lupus Erythematosus: A Case Report. Ann. Rehabil. Med. 2014, 38, 263–268. [Google Scholar] [CrossRef] [PubMed]
- Zotos, P.; Poularas, J.; Karakitsos, D.; Karabinis, A. Lupus Related Longitudinal Myelitis. J. Rheumatol. 2010, 37, 1776. [Google Scholar] [CrossRef] [PubMed]
- Espinosa, G.; Mendizábal, A.; Mínguez, S.; Ramo-Tello, C.; Capellades, J.; Olivé, A.; Cervera, R. Transverse Myelitis Affecting More Than 4 Spinal Segments Associated with Systemic Lupus Erythematosus: Clinical, Immunological, and Radiological Characteristics of 22 Patients. Semin. Arthritis Rheum. 2010, 39, 246–256. [Google Scholar] [CrossRef] [PubMed]
- Ochi, M.G.S.; Shapiro, S.C.; Melamed, E. Lupus and NMOSD: The Blending of Humoral Autoimmunity. Case Rep. Rheumatol. 2020, 2020, 8820071. [Google Scholar] [CrossRef] [PubMed]
- Illei, G.G.; Shirota, Y.; Yarboro, C.H.; Daruwalla, J.; Tackey, E.; Takada, K.; Fleisher, T.; Balow, J.E.; Lipsky, P.E. Tocilizumab in Systemic Lupus Erythematosus: Data on Safety, Preliminary Efficacy, and Impact on Circulating Plasma Cells from an Open-Label Phase I Dosage-Escalation Study. Arthritis Rheum. 2010, 62, 542–552. [Google Scholar] [CrossRef] [PubMed]
- Heo, Y.-A. Satralizumab: First Approval. Drugs 2020, 80, 1477–1482. [Google Scholar] [CrossRef] [PubMed]
- Allison, A.C. Mechanisms of Action of Mycophenolate Mofetil. Lupus 2005, 14 (Suppl. 1), s2–s8. [Google Scholar] [CrossRef] [PubMed]
- Magdalena, C.; Clarissa, A.; Sutandi, N. Comparative Analysis of Treatment Outcomes in Patients with Neuromyelitis Optica Spectrum Disorder Treated with Rituximab, Azathioprine, and Mycophenolate Mofetil: A Systematic Review and Meta-Analysis. Innov. Clin. Neurosci. 2022, 19, 51–64. [Google Scholar] [PubMed]
Figure 1.
MR Imaging of the patient. (a) Exemplary demyelination lesion subcortical right frontal (cranial MRI, 3-tesla, FLAIR transversal, and 4 mm layer thickness). (b) The longitudinal intramedullary signal alteration at thoracic vertebrae 4–11 and a spindle-shaped distension of the spinal cord (spinal MRI, 1.5-tesla, T2TSE sagittal, and 2 mm layer thickness). (c) Intramedullary, partly spotty contrast enhancement with an emphasis on thoracic vertebrae 7–8 (spinal MRI, 1.5-tesla, T1TSE sagittal after contrast injection of Clariscan, and 4 mm layer thickness).
Figure 1.
MR Imaging of the patient. (a) Exemplary demyelination lesion subcortical right frontal (cranial MRI, 3-tesla, FLAIR transversal, and 4 mm layer thickness). (b) The longitudinal intramedullary signal alteration at thoracic vertebrae 4–11 and a spindle-shaped distension of the spinal cord (spinal MRI, 1.5-tesla, T2TSE sagittal, and 2 mm layer thickness). (c) Intramedullary, partly spotty contrast enhancement with an emphasis on thoracic vertebrae 7–8 (spinal MRI, 1.5-tesla, T1TSE sagittal after contrast injection of Clariscan, and 4 mm layer thickness).

Figure 2.
Reibers diagram with slight hints of autochthonic IgM-synthesis. IgG and IgA did not show any pathologic findings.
Figure 2.
Reibers diagram with slight hints of autochthonic IgM-synthesis. IgG and IgA did not show any pathologic findings.


{kind=link}
{kind=link}
Table 1.
Relevant laboratory parameters in the patient (pathological findings highlighted in bold, and values from 2018 marked with asterisk (*)) (CSF = cerebrospinal fluid; IL2 = interleukin-2; dsDNA = double-strand DNA; AQP4 = aquaporin-4).
Table 1.
Relevant laboratory parameters in the patient (pathological findings highlighted in bold, and values from 2018 marked with asterisk (*)) (CSF = cerebrospinal fluid; IL2 = interleukin-2; dsDNA = double-strand DNA; AQP4 = aquaporin-4).
| Parameter | Value | Reference Values |
|---|---|---|
| CSF | ||
| cell count | 20/µL | <5/µL |
| protein | 22 mg/dL | 15–45 mg/dL |
| lactate | 2.3 mmol/L | 1.1–2.4 mmol/L |
| glucose | 65 mg/dL | n.a. |
| hemoglobin | negative | negative |
| blood/serum | ||
| IL2-receptor | 715 U/mL | 158–623 U/mL |
| ANA | 1:640 | <1:80 |
| SSA/Ro Antibodies | 11 U/mL | <7 U/mL |
| dsDNA antibodies | 5.2 U/mL | <10 U/mL |
| 86 U/mL * | ||
| cardiolipin IgG | 11 U/mL | <10 U/mL |
| cardiolipin IgM | 28 U/mL | <10 U/mL |
| β2-Glycoprotein IgG | 14 U/mL | <7 U/mL |
| β2-Glycoprotein IgM | 20 U/mL | <7 U/mL |
| C3c | 80.1 mg/dL | 90–180 mg/dL |
| C4 | 8.0 mg/dL | 10–40 mg/dL |
| hemoglobin | 12.4 g/dL | 12.0–16.0 g/dL |
| 5.2 g/dL * | ||
| thrombocytes | 163,000/µL | 140,000–400,000/µL |
| 63,000/µL | ||
| creatinine | 0.59 mg/dL | 0.5–0.9 mg/dL |
| anti-Sm antibodies | 1.3 U/mL | <7 |
| AQP4 antibodies | 1:10,240 | <1:80 |
| anti-MOG antibodies | negative | negative |
Table 2.
2019 European League Against Rheumatism/American College of Rheumatology classification criteria for systemic lupus erythematosus (simplified) [9]. The patient discussed in our report was seropositive for ANAs, antiphospholipid antibodies, anti-dsDNA antibodies (in 2018), thrombocytopenia, and autoimmune hemolysis (in 2018), and low serum levels of C3 and C4 were detected. Total score = 20.
Table 2.
2019 European League Against Rheumatism/American College of Rheumatology classification criteria for systemic lupus erythematosus (simplified) [9]. The patient discussed in our report was seropositive for ANAs, antiphospholipid antibodies, anti-dsDNA antibodies (in 2018), thrombocytopenia, and autoimmune hemolysis (in 2018), and low serum levels of C3 and C4 were detected. Total score = 20.
| Entry Criterion: Antinuclear Antibodies (ANA) at a Titer of ≥1:80 on HEp-2 Cells or an Equivalent Positive Test (Ever) | ||
|---|---|---|
| Additive Criteria: | ||
| Clinical Domains and Criteria | Weight | Present in the Patient |
| Constitutional • Fever | 2 | |
| Hematologic • Leukopenia • Thrombocytopenia • Autoimmune hemolysis | 3 4 4 | X X |
| Neuropsychiatric • Delirium • Psychosis • Seizure | 2 3 5 | |
| Mucocutaneous • Non-scarring alopecia • Oral ulcers • Subacute cutaneous or discoid lupus • Acute cutaneous lupus | 2 2 4 6 | |
| Serosal • Pleural or pericardial effusion • Acute pericarditis | 5 6 | |
| Musculoskeletal • Joint involvement | 6 | |
| Renal • Proteinuria > 0.5 g/24 h • Renal biopsy class II or V lupus nephritis • Renal biopsy class III or IV lupus nephritis | 4 8 10 | |
| Immunology domains and criteria | ||
| Antiphospholipid antibodies • Anti-cardiolipin antibodies OR • Anti-β2GP1 antibodies OR • Lupus anticoagulant | 2 | X |
| Complement proteins • Low C3 OR low C4 • Low C3 AND low C4 | 3 4 | X |
| SLE-specific antibodies • Anti-dsDNA antibody OR • Anti-Smith antibody | 6 | X |
| Classification as SLE with a score of 10 or more combined with fulfilled entry criterion. Occurrence of a criterion on at least 1 occasion is sufficient. Criteria do not have to occur simultaneously | ||
Table 3.
International consensus criteria for neuromyelitis optica spectrum disorders [3] (NMOSD = neuromyelitis optica spectrum disorders; AQP4-IgG = Aquaporin-4 immunoglobulin G antibodies; MRI = magnetic resonance imaging).
Table 3.
International consensus criteria for neuromyelitis optica spectrum disorders [3] (NMOSD = neuromyelitis optica spectrum disorders; AQP4-IgG = Aquaporin-4 immunoglobulin G antibodies; MRI = magnetic resonance imaging).
| Diagnostic Criteria for NMOSD with AQP4-IgG | |
|---|---|
| 1. At least 1 core clinical characteristic of the following: | 1. Optic neuritis 2. Acute myelitis 3. Area postrema syndrome episode: episode of otherwise unexplained hiccups or nausea and vomiting 4. Acute brainstem syndrome 5. Symptomatic narcolepsy or acute diencephalic syndrome with NMOSD-typical diencephalic MRI lesions 6. Symptomatic cerebral syndrome with NMOSD-typical brain lesions |
| 2. Positive test for AQP4-IgG using best available detection method (cell-based assay strongly recommended) | |
| 3. Exclusion of alternative diagnoses | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Kaempfer, N.A.; Fousse, M.; Kettner, M.; Fassbender, K.; Janitschke, D. Is It Lupus? Is It Neuromyelitis Optica Spectrum Disorder (NMOSD)?—Why Not Both? Sclerosis 2023, 1, 51-59. https://doi.org/10.3390/sclerosis1010006
AMA Style
Kaempfer NA, Fousse M, Kettner M, Fassbender K, Janitschke D. Is It Lupus? Is It Neuromyelitis Optica Spectrum Disorder (NMOSD)?—Why Not Both? Sclerosis. 2023; 1(1):51-59. https://doi.org/10.3390/sclerosis1010006
Chicago/Turabian StyleKaempfer, Niklas Alexander, Mathias Fousse, Michael Kettner, Klaus Fassbender, and Daniel Janitschke. 2023. "Is It Lupus? Is It Neuromyelitis Optica Spectrum Disorder (NMOSD)?—Why Not Both?" Sclerosis 1, no. 1: 51-59. https://doi.org/10.3390/sclerosis1010006