
COQ7-Related Juvenile-Onset Motor Neuronopathy: A New Pathogenetic Dysfunction Associated with Motor Neuron Disease
 , , and
, , and 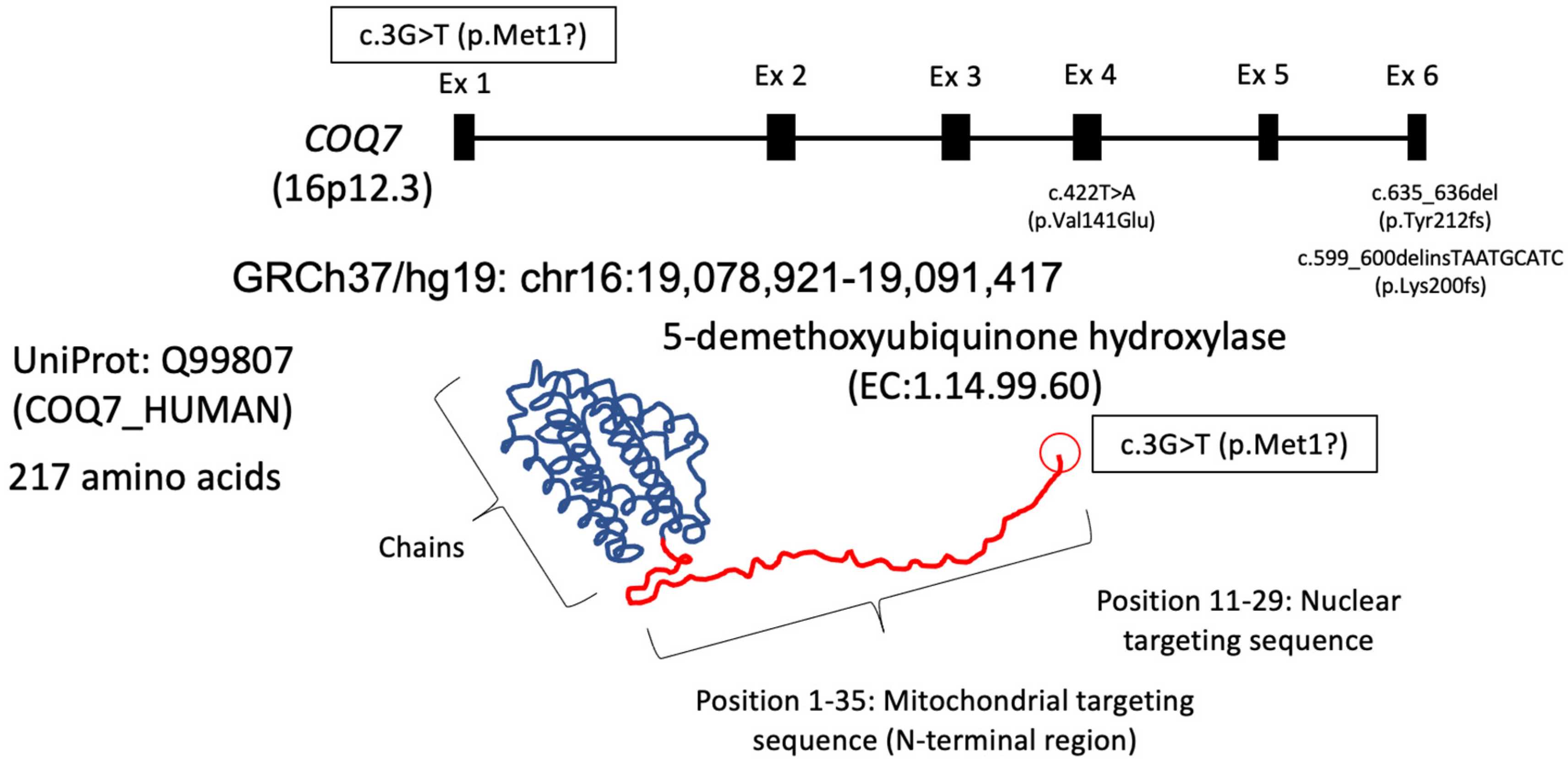{kind=link}
Abstract
:1. Introduction
2. Case Presentation
3. Discussion
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| ACMG | American College of Medical Genetics and Genomics. |
| ALS | Amyotrophic Lateral Sclerosis |
| ALSFRS-R | ALS Functional Rating Scale-Revised. |
| CMAP | Compound Motor Action Potential |
| dHMN | Distal Hereditary Motor Neuronopathy |
| MND | Motor Neuron Disease |
| ROS | Reactive Oxygen Species |
| SMA | Spinal Muscular Atrophy |
References
- Souza, P.V.S.; Bortholin, T.; Naylor, F.G.M.; Chieia, M.A.T.; Pinto, W.B.V.R.; Oliveira, A.S.B. Motor neuron disease in inherited neurometabolic disorders. Rev. Neurol. 2018, 174, 115–124. [Google Scholar] [CrossRef] [PubMed]
- Jacquier, A.; Theuriet, J.; Fontaine, F.; Mosbach, V.; Lacoste, N.; Ribault, S.; Risson, V.; Carras, J.; Coudert, L.; Simonet, T.; et al. Homozygous COQ7 mutation: A new cause of potentially treatable distal hereditary motor neuropathy. Brain 2022, awac453, Epub ahead of print. [Google Scholar] [CrossRef] [PubMed]
- Smith, I.C.; Pileggi, C.A.; Wang, Y.; Kernohan, K.; Hartley, T.; McMillan, H.J.; Sampaio, M.L.; Melkus, G.; Woulfe, J.; Parmar, G.; et al. Novel homozygous variant in COQ7 in siblings with hereditary motor neuropathy. Neurol. Genet. 2023, 9, e200048. [Google Scholar] [CrossRef]
- Wang, Y.; Gumus, E.; Hekimi, S. A novel COQ7 mutation causing primarily neuromuscular pathology and its treatment options. Mol. Genet. Metab. Rep. 2022, 31, 100877. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Smith, C.; Parboosingh, J.S.; Khan, A.; Innes, M.; Hekimi, S. Pathogenicity of two COQ7 mutations and responses to 2,4-dihydroxybenzoate bypass treatment. J. Cell. Mol. Med. 2017, 21, 2329–2343. [Google Scholar] [CrossRef] [PubMed]
- Awad, A.M.; Bradley, M.C.; Fernández-del-Río, L.; Nag, A.; Tsui, H.S.; Clarke, C.F. Coenzyme Q10 deficiencies: Pathways in yeast and humans. Essays. Biochem. 2018, 62, 361–376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakai, D.; Shimizu, T.; Nojiri, H.; Uchiyama, S.; Koike, H.; Takahashi, M.; Hirokawa, K.; Shirasawa, T. coq7/clk-1 regulates mitochondrial respiration and the generation of reactive oxygen species via coenzyme Q. Aging Cell 2004, 3, 273–281. [Google Scholar] [CrossRef] [PubMed]
- Kaufmann, P.; Thompson, J.L.; Levy, G.; Buchsbaum, R.; Shefner, J.; Krivickas, L.S.; Katz, J.; Rollins, Y.; Barohn, R.J.; Jackson, C.E.; et al. Phase II trial of CoQ10 for ALS finds insufficient evidence to justify phase III. Ann. Neurol. 2009, 66, 235–244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cunha-Oliveira, T.; Montezinho, L.; Mendes, C.; Firuzi, O.; Saso, L.; Oliveira, P.J.; Silva, F.S. Oxidative stress in Amyotrophic Lateral Sclerosis: Pathophysiology and opportunities for pharmacological intervention. Oxid. Med. Cell. Longev. 2020, 2020, 5021694. [Google Scholar] [CrossRef] [PubMed]
- Lehky, T.; Grunseich, C. Juvenile Amyotrophic Lateral Sclerosis: A Review. Genes 2021, 12, 1935. [Google Scholar] [CrossRef] [PubMed]
- Orlacchio, A.; Babalini, C.; Borreca, A.; Patrono, C.; Massa, R.; Basaran, S.; Munhoz, R.P.; Rogaeva, E.A.; St George-Hyslop, P.H.; Bernardi, G.; et al. SPATACSIN mutations cause autosomal recessive juvenile amyotrophic lateral sclerosis. Brain 2010, 133, 591–598. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ben Hamida, M.; Hentati, F.; Ben Hamida, C. Hereditary motor system diseases (chronic juvenile amyotrophic lateral sclerosis). Conditions combining a bilateral pyramidal syndrome with limb and bulbar amyotrophy. Brain 1990, 113, 347–363. [Google Scholar] [CrossRef] [PubMed]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Souza, P.V.S.d.; Farias, I.B.; Serrano, P.d.L.; Badia, B.d.M.L.; Jorge, A.C.d.S.; Barros, G.B.; Oliveira, H.B.d.; Calil, S.R.; Fernandes, I.D.; Ribeiro, R.C.; et al. COQ7-Related Juvenile-Onset Motor Neuronopathy: A New Pathogenetic Dysfunction Associated with Motor Neuron Disease. Sclerosis 2023, 1, 22-26. https://doi.org/10.3390/sclerosis1010004
Souza PVSd, Farias IB, Serrano PdL, Badia BdML, Jorge ACdS, Barros GB, Oliveira HBd, Calil SR, Fernandes ID, Ribeiro RC, et al. COQ7-Related Juvenile-Onset Motor Neuronopathy: A New Pathogenetic Dysfunction Associated with Motor Neuron Disease. Sclerosis. 2023; 1(1):22-26. https://doi.org/10.3390/sclerosis1010004
Chicago/Turabian StyleSouza, Paulo Victor Sgobbi de, Igor Braga Farias, Paulo de Lima Serrano, Bruno de Mattos Lombardi Badia, Ana Carolina dos Santos Jorge, Glenda Barbosa Barros, Hélvia Bertoldo de Oliveira, Samia Rogatis Calil, Isabela Danziato Fernandes, Roberta Correa Ribeiro, and et al. 2023. "COQ7-Related Juvenile-Onset Motor Neuronopathy: A New Pathogenetic Dysfunction Associated with Motor Neuron Disease" Sclerosis 1, no. 1: 22-26. https://doi.org/10.3390/sclerosis1010004