
A Novel Liver X Receptor Inverse Agonist Impairs Cholesterol and Phospholipid Metabolism and Induces Apoptosis and Necroptosis in Pancreatic Ductal Adenocarcinoma Cells




{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Lines and Culture
2.2. Proliferation Assays
2.3. Western Blotting
2.4. RNA-Seq Library Preparation and Sequencing
2.5. RNA-Seq Analysis
2.6. Metabolomics
2.7. Caspase-3/7 Glo Assay
3. Results
3.1. Transcriptomic Analyses Reveal Downregulation of Fatty Acid and Cholesterol Metabolism
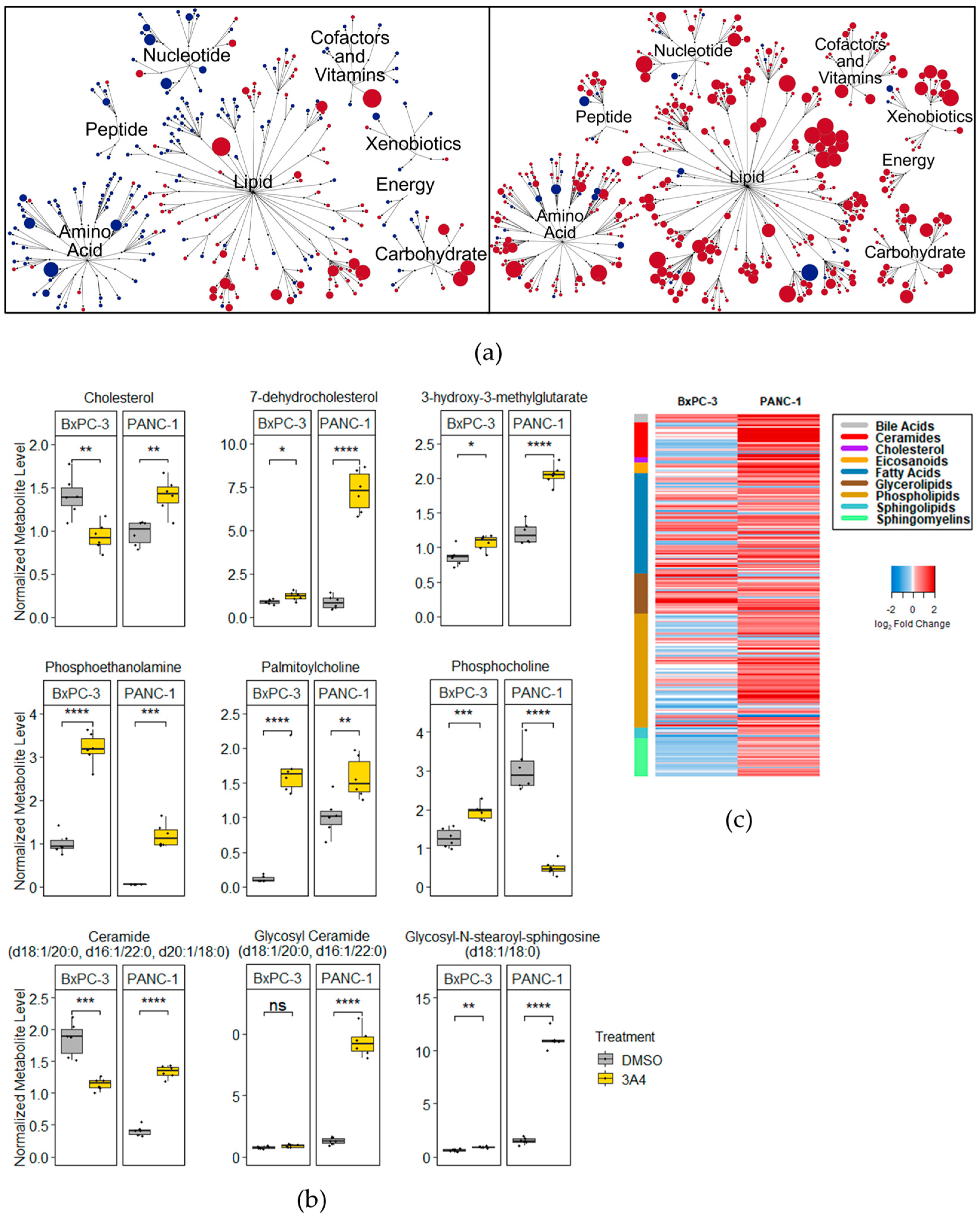
3.2. Metabolomic Analyses Demonstrate Perturbations of Cholesterol, Phospholipid, and Amino Acid Pathways
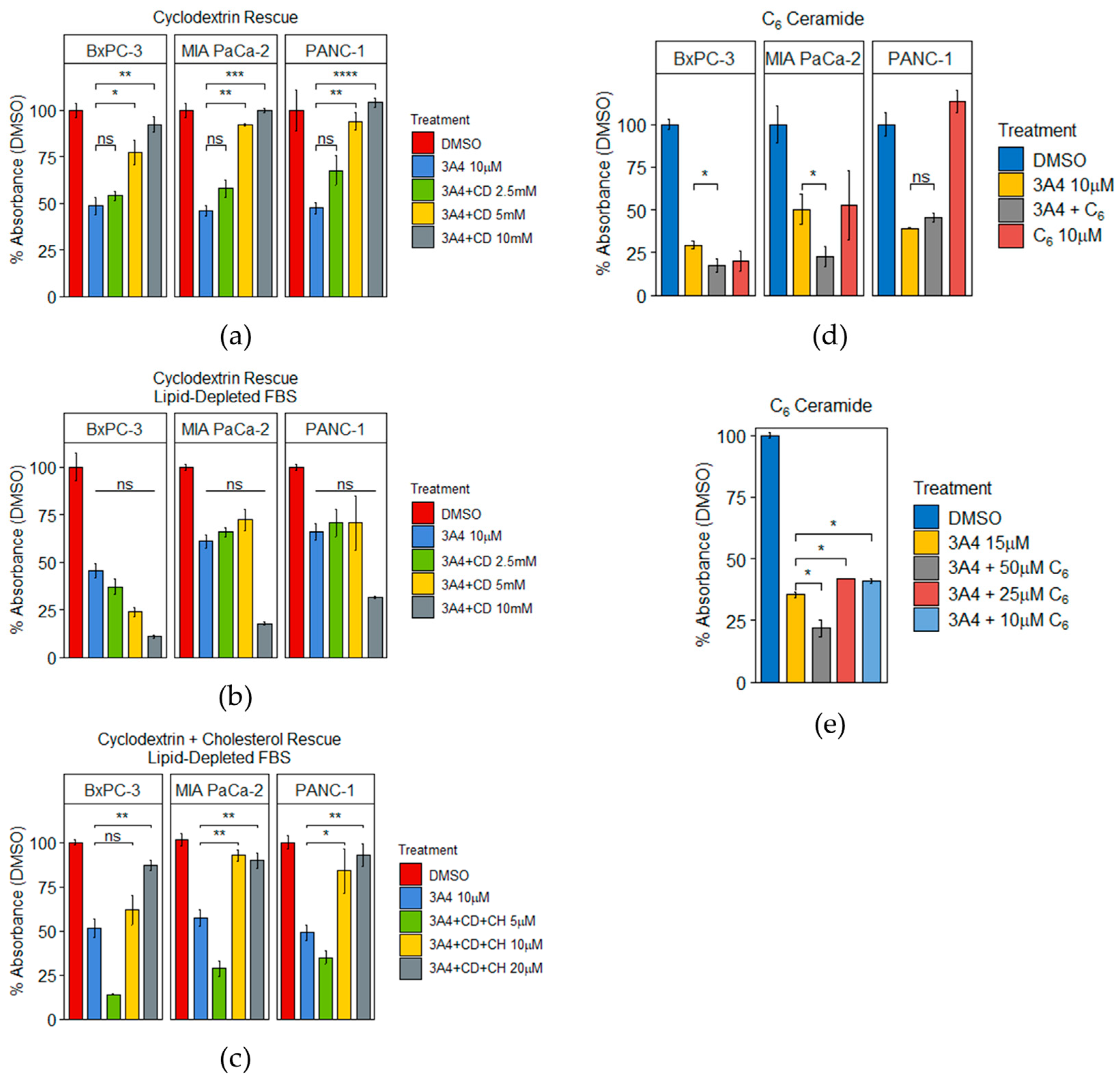
3.3. 3A4 Causes Alterations in Cholesterol and Ceramide Metabolism
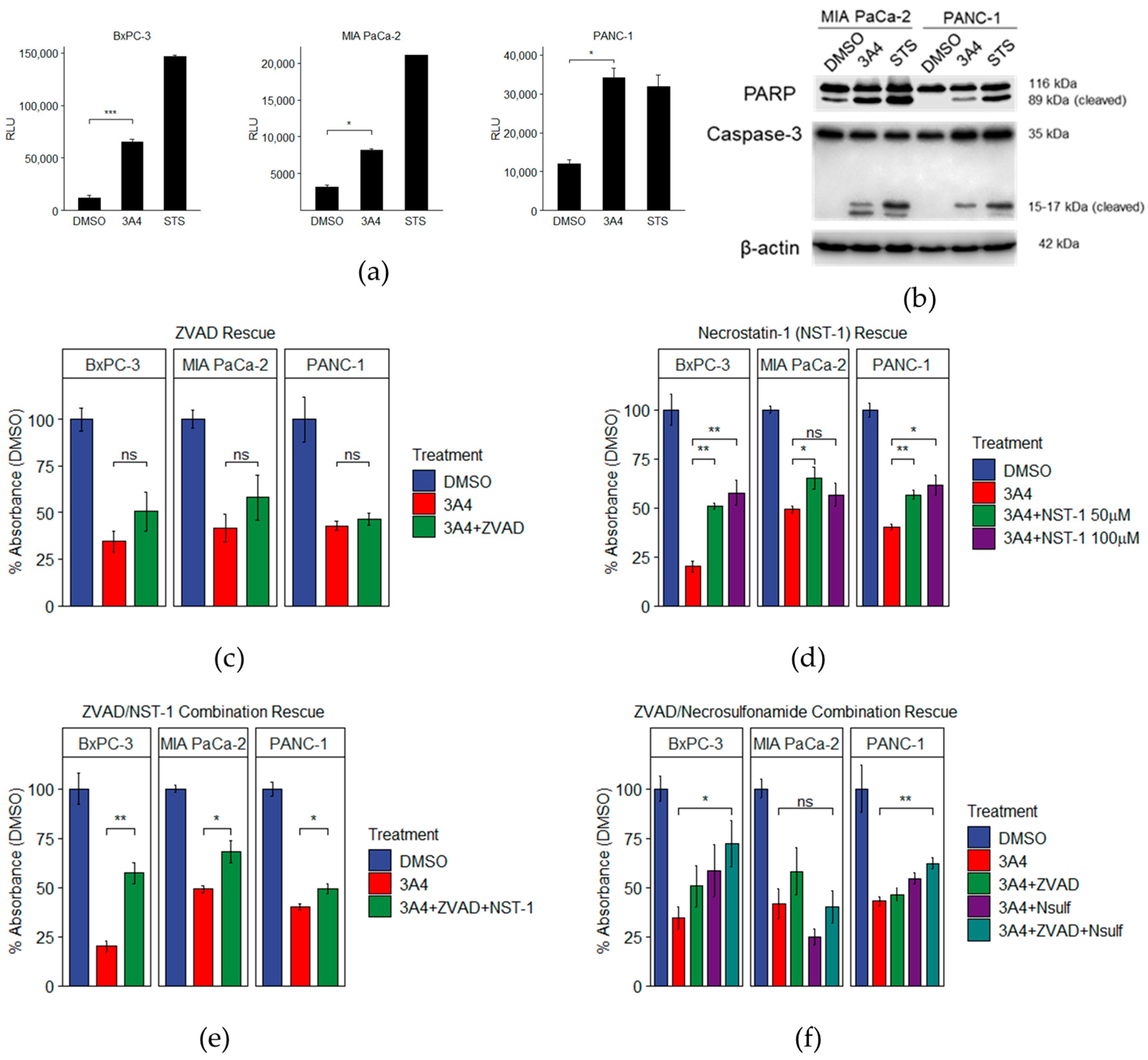
3.4. LXR Inhibition by Novel Ligand Induces Both Apoptotic and Necroptotic Cell Death Pathways
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Rahib, L.; Smith, B.D.; Aizenberg, R.; Rosenzweig, A.B.; Fleshman, J.M.; Matrisian, L.M. Projecting cancer incidence and deaths to 2030: The unexpected burden of thyroid, liver, and pancreas cancers in the United States. Cancer Res. 2014, 74, 2913–2921. [Google Scholar] [CrossRef] [PubMed]
- Adamska, A.; Domenichini, A.; Falasca, M. Pancreatic Ductal Adenocarcinoma: Current and Evolving Therapies. Int. J. Mol. Sci. 2017, 18, 1338. [Google Scholar] [CrossRef]
- Orth, M.; Metzger, P.; Gerum, S.; Mayerle, J.; Schneider, G.; Belka, C.; Schnurr, M.; Lauber, K. Pancreatic ductal adenocarcinoma: Biological hallmarks, current status, and future perspectives of combined modality treatment approaches. Radiat. Oncol. 2019, 14, 141. [Google Scholar] [CrossRef]
- McGuigan, A.; Kelly, P.; Turkington, R.C.; Jones, C.; Coleman, H.G.; McCain, R.S. Pancreatic cancer: A review of clinical diagnosis, epidemiology, treatment and outcomes. World J. Gastroenterol. 2018, 24, 4846–4861. [Google Scholar] [CrossRef] [PubMed]
- Conroy, T.; Hammel, P.; Hebbar, M.; Ben Abdelghani, M.; Wei, A.C.; Raoul, J.-L.; Choné, L.; Francois, E.; Artru, P.; Biagi, J.J.; et al. FOLFIRINOX or Gemcitabine as Adjuvant Therapy for Pancreatic Cancer. N. Engl. J. Med. 2018, 379, 2395–2406. [Google Scholar] [CrossRef] [PubMed]
- Long, J.; Zhang, Y.; Yu, X.; Yang, J.; LeBrun, D.G.; Chen, C.; Yao, Q.; Li, M. Overcoming drug resistance in pancreatic cancer. Expert Opin. Ther. Targets 2011, 15, 817–828. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2020. CA Cancer J. Clin. 2020, 70, 7–30. [Google Scholar] [CrossRef]
- Barberis, I.; Martini, M.; Iavarone, F.; Orsi, A. Hallmarks of Cancer: The Next Generation. J. Prev. Med. Hyg. 2016, 57, E41–E46. [Google Scholar]
- Swierczynski, J.; Hebanowska, A.; Sledzinski, T. Role of abnormal lipid metabolism in development, progression, diagnosis and therapy of pancreatic cancer. World J. Gastroenterol. 2014, 20, 2279–2303. [Google Scholar] [CrossRef]
- Medes, G.; Thomas, A.; Weinhouse, S. Metabolism of neoplastic tissue. IV. A study of lipid synthesis in neoplastic tissue slices in vitro. Cancer Res. 1953, 13, 27–29. [Google Scholar]
- Milgraum, L.Z.; Witters, L.A.; Pasternack, G.R.; Kuhajda, F.P. Enzymes of the fatty acid synthesis pathway are highly expressed in in situ breast carcinoma. Clin. Cancer Res. 1997, 3, 2115–2120. [Google Scholar]
- Joseph, S.B.; Laffitte, B.A.; Patel, P.H.; Watson, M.A.; Matsukuma, K.E.; Walczak, R.; Collins, J.L.; Osborne, T.F.; Tontonoz, P. Direct and Indirect Mechanisms for Regulation of Fatty Acid Synthase Gene Expression by Liver X Receptors. J. Biol. Chem. 2002, 277, 11019–11025. [Google Scholar] [CrossRef]
- Repa, J.J.; Liang, G.; Ou, J.; Bashmakov, Y.; Lobaccaro, J.-M.A.; Shimomura, I.; Shan, B.; Brown, M.S.; Goldstein, J.L.; Mangelsdorf, D.J. Regulation of mouse sterol regulatory by oxysterol receptors, LXR and LXR. Genes Dev. 2000, 36, 2819–2830. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chu, K.; Miyazaki, M.; Man, W.C.; Ntambi, J.M. Stearoyl-Coenzyme A Desaturase 1 Deficiency Protects against Hypertriglyceridemia and Increases Plasma High-Density Lipoprotein Cholesterol Induced by Liver X Receptor Activation. Mol. Cell. Biol. 2006, 26, 6786–6798. [Google Scholar] [CrossRef] [PubMed]
- Song, C.; Kokontis, J.M.; Hiipakka, R.A.; Liao, S. Ubiquitous receptor: A receptor that modulates gene activation by retinoic acid and thyroid hormone receptors. Proc. Natl. Acad. Sci. USA 1994, 91, 10809–10813. [Google Scholar] [CrossRef] [PubMed]
- Jakobsson, T.; Treuter, E.; Gustafsson, J.-Å.; Steffensen, K.R. Liver X receptor biology and pharmacology: New pathways, challenges and opportunities. Trends Pharmacol. Sci. 2012, 33, 394–404. [Google Scholar] [CrossRef]
- Lin, C.-Y.; Vedin, L.-L.; Steffensen, K.R. The emerging roles of liver X receptors and their ligands in cancer. Expert Opin. Ther. Targets 2015, 20, 61–71. [Google Scholar] [CrossRef] [PubMed]
- Nguyen-Vu, T.; Vedin, L.-L.; Liu, K.; Jonsson, P.; Lin, J.Z.; Candelaria, N.R.; Candelaria, L.P.; Addanki, S.; Williams, C.; Gustafsson, J.; et al. Liver × receptor ligands disrupt breast cancer cell proliferation through an E2F-mediated mechanism. Breast Cancer Res. 2013, 15, R1–R12. [Google Scholar] [CrossRef] [PubMed]
- Vedin, L.L.; Gustafsson, J.A.; Steffensen, K.R. The oxysterol receptors lxrα and lxrβ suppress proliferation in the colon. Mol. Carcinog. 2013, 52, 835–844. [Google Scholar] [CrossRef]
- Pencheva, N.; Buss, C.; Posada, J.; Merghoub, T.; Tavazoie, S.F. Broad-Spectrum Therapeutic Suppression of Metastatic Melanoma through Nuclear Hormone Receptor Activation. Cell 2014, 156, 986–1001. [Google Scholar] [CrossRef]
- Guo, D.; Reinitz, F.; Youssef, M.; Hong, C.; Nathanson, D.; Akhavan, D.; Kuga, D.; Amzajerdi, A.N.; Soto, H.; Zhu, S.; et al. An LXR agonist promotes GBM cell death through inhibition of an EGFR/AKT/SREBP-1/LDLR-dependent pathway. Cancer Discov. 2011, 1, 442–456. [Google Scholar] [CrossRef] [PubMed]
- Candelaria, N.R.; Addanki, S.; Zheng, J.; Nguyen-Vu, T.; Karaboga, H.; Dey, P.; Gabbi, C.; Vedin, L.-L.; Liu, K.; Wu, W.; et al. Antiproliferative Effects and Mechanisms of Liver X Receptor Ligands in Pancreatic Ductal Adenocarcinoma Cells. PLoS ONE 2014, 9, e106289. [Google Scholar] [CrossRef] [PubMed]
- Karaboga, H.; Huang, W.; Srivastava, S.; Widmann, S.; Addanki, S.; Gamage, K.T.; Mazhar, Z.; Ebalunode, J.O.; Briggs, J.M.; Gustafsson, J.; et al. Screening of Focused Compound Library Targeting Liver X Receptors in Pancreatic Cancer Identified Ligands with Inverse Agonist and Degrader Activity. ACS Chem. Biol. 2020, 15, 2916–2928. [Google Scholar] [CrossRef]
- Pertea, M.; Kim, D.; Pertea, G.M.; Leek, J.T.; Salzberg, S.L. Transcript-level expression analysis of RNA-seq experiments with HISAT, StringTie and Ballgown. Nat. Protoc. 2016, 11, 1650–1667. [Google Scholar] [CrossRef] [PubMed]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef]
- Evans, A.; Bridgewater, B.R.; Liu, Q.; Mitchell, M.W.; Robinson, R.J.; Dai, H.; Stewart, S.J.; DeHaven, C.D.; Miller, L.A.D. High Resolution Mass Spectrometry Improves Data Quantity and Quality as Compared to Unit Mass Resolution Mass Spectrometry in High-Throughput Profiling Metabolomics. Metabolomics 2014, 4, 1000132. [Google Scholar]
- Evans, A.M.; DeHaven, C.D.; Barrett, T.; Mitchell, M.; Milgram, E. Integrated, Nontargeted Ultrahigh Performance Liquid Chromatography/Electrospray Ionization Tandem Mass Spectrometry Platform for the Identification and Relative Quantification of the Small-Molecule Complement of Biological Systems. Anal. Chem. 2009, 81, 6656–6667. [Google Scholar] [CrossRef]
- DeHaven, C.D.; Evans, A.M.; Dai, H.; Lawton, K.A. Organization of GC/MS and LC/MS metabolomics data into chemical libraries. J. Chemin. 2010, 2, 9. [Google Scholar] [CrossRef]
- Van Beek, M.; Oravecz-Wilson, K.I.; Delekta, P.C.; Gu, S.; Li, X.; Jin, X.; Apel, I.J.; Konkle, K.S.; Feng, Y.; Teitelbaum, D.H.; et al. Bcl10 Links Saturated Fat Overnutrition with Hepatocellular NF-κB Activation and Insulin Resistance. Cell Rep. 2012, 1, 444–452. [Google Scholar] [CrossRef] [PubMed]
- Bilotta, M.T.; Petillo, S.; Santoni, A.; Cippitelli, M. Liver X Receptors: Regulators of Cholesterol Metabolism, Inflammation, Autoimmunity, and Cancer. Front. Immunol. 2020, 11, 584303C. [Google Scholar] [CrossRef] [PubMed]
- Hall, Z.; Wilson, C.H.; Burkhart, D.L.; Ashmore, T.; Evan, G.I.; Griffin, J.L. Myc linked to dysregulation of cholesterol transport and storage in nonsmall cell lung cancer. J. Lipid Res. 2020, 61, 1390–1399. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Liu, H.; Qing, G. Targeting oncogenic Myc as a strategy for cancer treatment. Signal Transduct. Target. Ther. 2018, 3, 5. [Google Scholar] [CrossRef]
- Pelengaris, S.; Khan, M.; Evan, G. c-MYC: More than just a matter of life and death. Nat. Rev. Cancer 2002, 2, 764–776. [Google Scholar] [CrossRef] [PubMed]
- Christian, A.E.; Haynes, M.P.; Phillips, M.C.; Rothblat, G.H. Use of cyclodextrins for manipulating cellular cholesterol content. J. Lipid Res. 1997, 38, 2264–2272. [Google Scholar] [CrossRef] [PubMed]
- Flowers, M.; Fabrias, G.; Delgado, A.; Casas, J.; Abad, J.L.; Cabot, M.C. C6-Ceramide and targeted inhibition of acid ceramidase induce synergistic decreases in breast cancer cell growth. Breast Cancer Res. Treat. 2011, 133, 447–458. [Google Scholar] [CrossRef] [PubMed]
- Griffiths, B.; Lewis, C.A.; Bensaad, K.; Ros, S.; Zhang, Q.; Ferber, E.C.; Konisti, S.; Peck, B.; Miess, H.; East, P.; et al. Sterol regulatory element binding protein-dependent regulation of lipid synthesis supports cell survival and tumor growth. Cancer Metab. 2013, 1, 3. [Google Scholar] [CrossRef]
- Fritz, V.; Benfodda, Z.; Henriquet, C.; Hure, S.; Cristol, J.-P.; Michel, F.; Carbonneau, M.-A.; Casas, F.; Fajas, L. Metabolic intervention on lipid synthesis converging pathways abrogates prostate cancer growth. Oncogene 2012, 32, 5101–5110. [Google Scholar] [CrossRef]
- Ariyama, H.; Kono, N.; Matsuda, S.; Inoue, T.; Arai, H. Decrease in Membrane Phospholipid Unsaturation Induces Unfolded Protein Response. J. Biol. Chem. 2010, 285, 22027–22035. [Google Scholar] [CrossRef]
- Greer, Y.E.; Porat-Shliom, N.; Nagashima, K.; Stuelten, C.; Crooks, D.; Koparde, V.N.; Gilbert, S.F.; Islam, C.; Ubaldini, A.; Ji, Y.; et al. ONC201 kills breast cancer cells in vitro by targeting mitochondria. Oncotarget 2018, 9, 18454–18479. [Google Scholar] [CrossRef]
- McComb, S.; Aguadé-Gorgorió, J.; Harder, L.; Marovca, B.; Cario, G.; Eckert, C.; Schrappe, M.; Stanulla, M.; von Stackelberg, A.; Bourquin, J.-P.; et al. Activation of concurrent apoptosis and necroptosis by SMAC mimetics for the treatment of refractory and relapsed ALL. Sci. Transl. Med. 2016, 8, 339ra70. [Google Scholar] [CrossRef] [PubMed]
- Collins, J.L.; Fivush, A.M.; Watson, M.A.; Galardi, C.M.; Lewis, M.C.; Moore, L.B.; Parks, D.J.; Wilson, J.G.; Tippin, T.K.; Binz, J.G.; et al. Identification of a Nonsteroidal Liver X Receptor Agonist through Parallel Array Synthesis of Tertiary Amines. J. Med. Chem. 2002, 45, 1963–1966. [Google Scholar] [CrossRef] [PubMed]
- Schultz, J.R.; Tu, H.; Luk, A.; Repa, J.J.; Medina, J.C.; Li, L.; Schwendner, S.; Wang, S.; Thoolen, M.; Mangelsdorf, D.J.; et al. Role of LXRs in control of lipogenesis. Genes Dev. 2000, 14, 2831–2838. [Google Scholar] [CrossRef] [PubMed]
- Repa, J.J.; Liang, G.; Ou, J.; Bashmakov, Y.; Lobaccaro, J.M.A.; Shimomura, I.; Shan, B.; Brown, M.S.; Goldstein, J.L.; Mangelsdorf, D.J. Regulation of mouse sterol regulatory element-binding protein-1c gene (SREBP-1c) by oxysterol receptors, LXRα and LXRβ. Genes Dev. 2000, 14, 2819–2830. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katz, A.; Udata, C.; Ott, E.; Hickey, L.; Burczynski, M.E.; Burghart, P.; Vesterqvist, O.; Meng, X. Safety, pharmacokinetics, and pharmacodynamics of single doses of lxr-623, a novel liver X-receptor agonist, in healthy participants. J. Clin. Pharmacol. 2009, 49, 643–649. [Google Scholar] [CrossRef]
- Flaveny, C.A.; Griffett, K.; El-Gendy, B.E.-D.M.; Kazantzis, M.; Sengupta, M.; Amelio, A.L.; Chatterjee, A.; Walker, J.; Solt, L.A.; Kamenecka, T.M.; et al. Broad Anti-tumor Activity of a Small Molecule that Selectively Targets the Warburg Effect and Lipogenesis. Cancer Cell 2015, 28, 42–56. [Google Scholar] [CrossRef]
- Deer, E.L.; González-Hernández, J.; Coursen, J.D.; Shea, J.E.; Ngatia, J.; Scaife, C.L.; Firpo, M.A.; Mulvihill, S.J. Phenotype and Genotype of Pancreatic Cancer Cell Lines. Pancreas 2010, 39, 425–435, Erratum in Pancreas 2018, 47, e37. [Google Scholar] [CrossRef]
- Gu, L.; Saha, S.T.; Thomas, J.; Kaur, M. Targeting cellular cholesterol for anticancer therapy. FEBS J. 2019, 286, 4192–4208. [Google Scholar] [CrossRef]
- Li, Y.C.; Park, M.J.; Ye, S.-K.; Kim, C.-W.; Kim, Y.-N. Elevated Levels of Cholesterol-Rich Lipid Rafts in Cancer Cells Are Correlated with Apoptosis Sensitivity Induced by Cholesterol-Depleting Agents. Am. J. Pathol. 2006, 168, 1107–1118. [Google Scholar] [CrossRef] [PubMed]
- Miller, D.M.; Thomas, S.D.; Islam, A.; Muench, D.; Sedoris, K. c-Myc and Cancer Metabolism. Clin. Cancer Res. 2012, 18, 5546–5553. [Google Scholar] [CrossRef] [PubMed]
- Klefstrom, J.; Västrik, I.; Saksela, E.; Valle, J.; Eilers, M.; Alitalo, K. c-Myc induces cellular susceptibility to the cytotoxic action of TNF-alpha. EMBO J. 1994, 13, 5442–5450. [Google Scholar] [CrossRef] [PubMed]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Widmann, S.; Srivastava, S.; Lin, C.-Y. A Novel Liver X Receptor Inverse Agonist Impairs Cholesterol and Phospholipid Metabolism and Induces Apoptosis and Necroptosis in Pancreatic Ductal Adenocarcinoma Cells. Receptors 2023, 2, 34-46. https://doi.org/10.3390/receptors2010003
Widmann S, Srivastava S, Lin C-Y. A Novel Liver X Receptor Inverse Agonist Impairs Cholesterol and Phospholipid Metabolism and Induces Apoptosis and Necroptosis in Pancreatic Ductal Adenocarcinoma Cells. Receptors. 2023; 2(1):34-46. https://doi.org/10.3390/receptors2010003
Chicago/Turabian StyleWidmann, Scott, Shivangi Srivastava, and Chin-Yo Lin. 2023. "A Novel Liver X Receptor Inverse Agonist Impairs Cholesterol and Phospholipid Metabolism and Induces Apoptosis and Necroptosis in Pancreatic Ductal Adenocarcinoma Cells" Receptors 2, no. 1: 34-46. https://doi.org/10.3390/receptors2010003