
Novel Cell Receptor System of Eukaryotes Formed by Previously Unknown Nucleic Acid-Based Receptors


Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Lines, Culture, and Treatments
2.2. Reagents
2.3. Destruction of Primary TezRs in Planktonic Cultures
2.4. Destruction of Primary TezRs of Mammalian Cells within Monolayers
2.5. Destruction of Secondary TezRs
2.6. Inactivation of TezRs with Propidium Iodine
2.7. Morphology Index
2.8. Growth Curve
2.9. Fungal Viability Test
2.10. Biofilm Morphology
2.11. Light Microscopy-Based Methods
2.12. Fluorescence Microscopy
2.13. Flow Cytometry
2.14. Assays of RNase Internalization
2.15. Virus Release Assay
2.16. Sensitivity of Cells to Opioids
2.17. Sensitivity of Cells to Insulin
2.18. Wound-Healing Assay
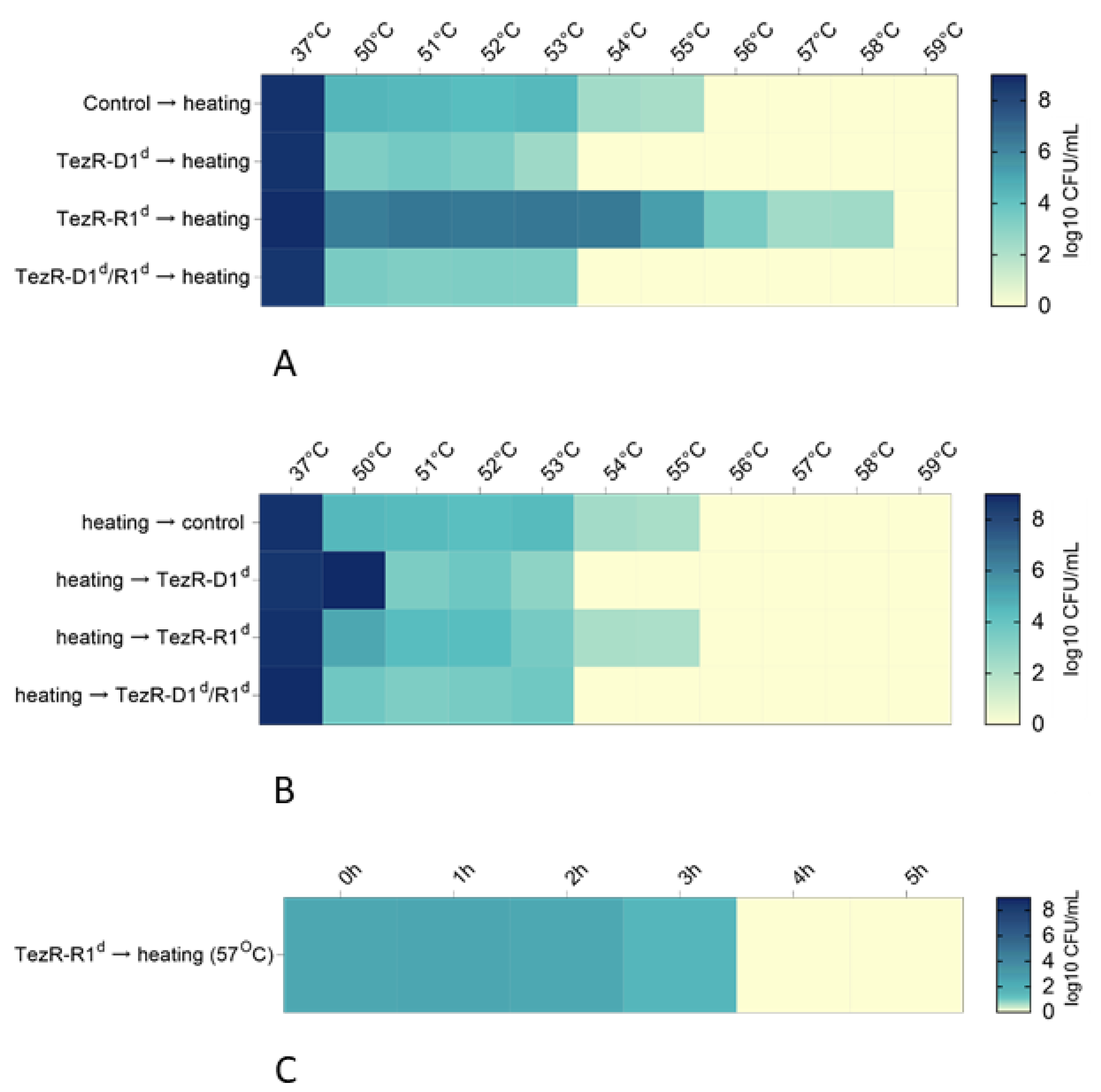
2.19. Modulation of Thermotolerance
2.20. Modulation of Thermotolerance Restoration after TezR Destruction
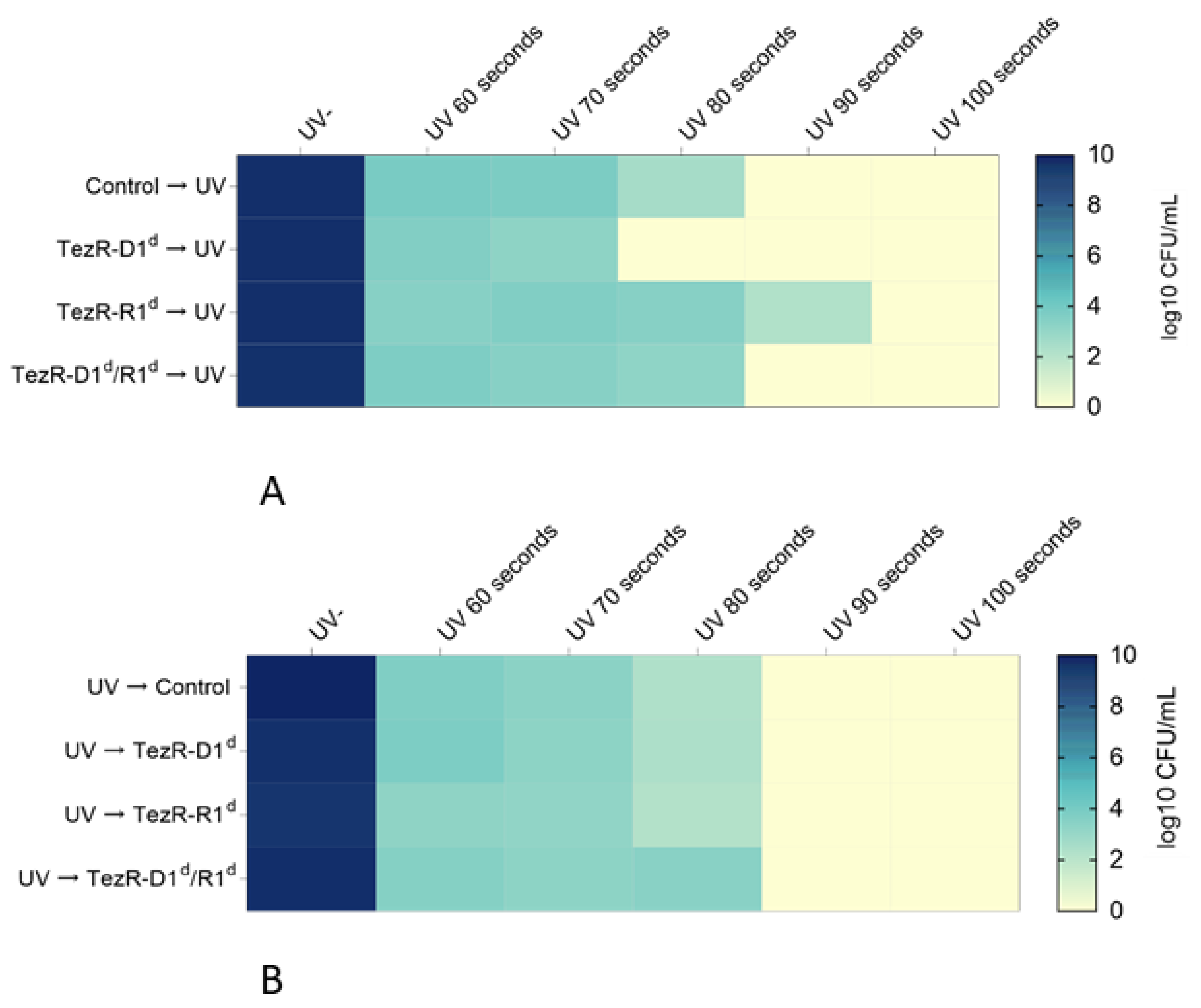
2.21. UV Assay
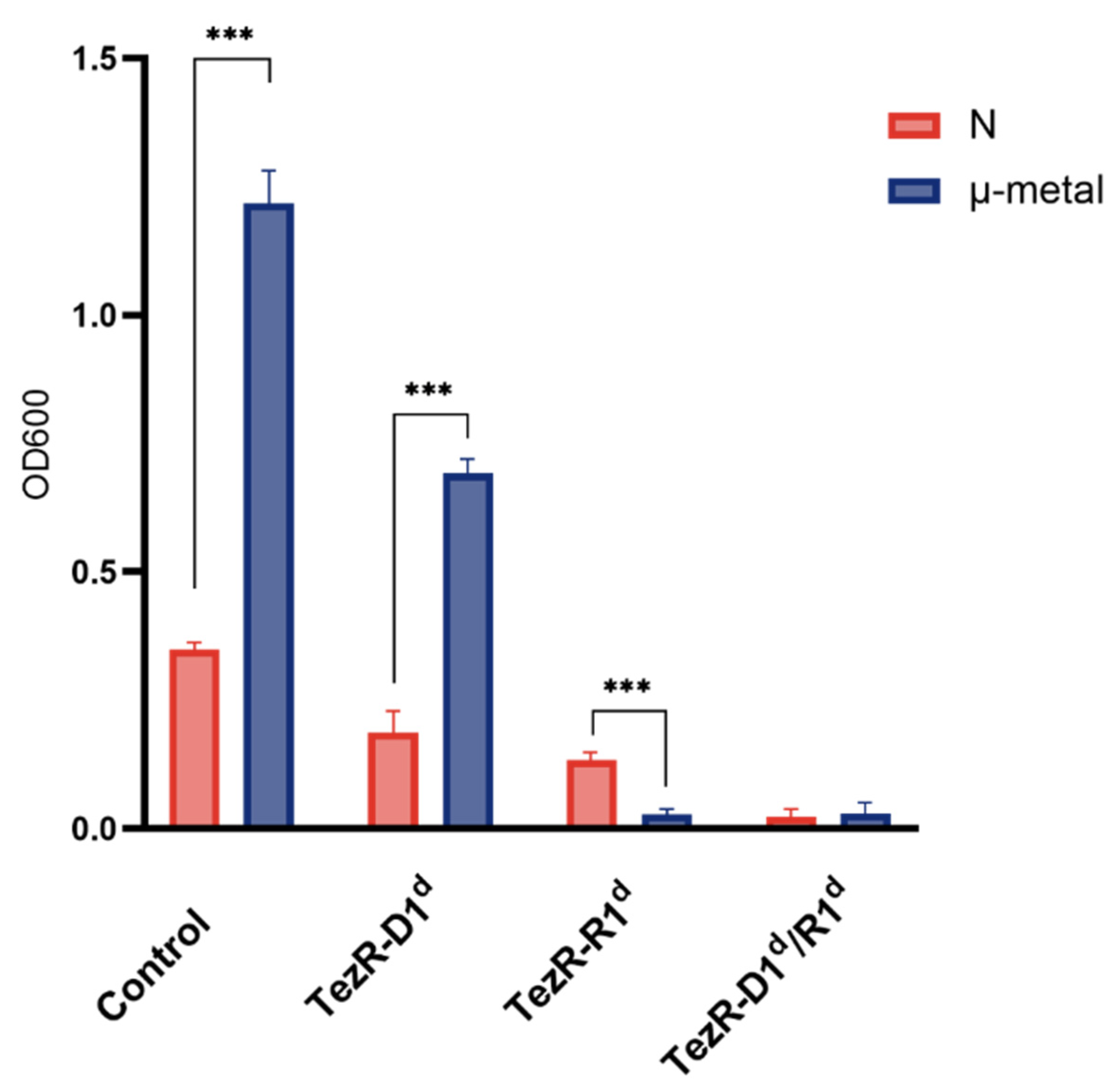
2.22. Magnetic Exposure Conditions
2.23. Light Exposure Experiments
2.24. In Vitro Antifungal Susceptibility Testing
2.25. Effects of RT Inhibitors and Integrase Inhibitors on Cell Growth
2.26. Biochemical Analysis
2.27. Recognition of Maltose
2.28. Cell Memory Formation Experiments
2.29. Evaluation of the Role of TR-System in Memory Formation
2.30. Memory Loss Experiments
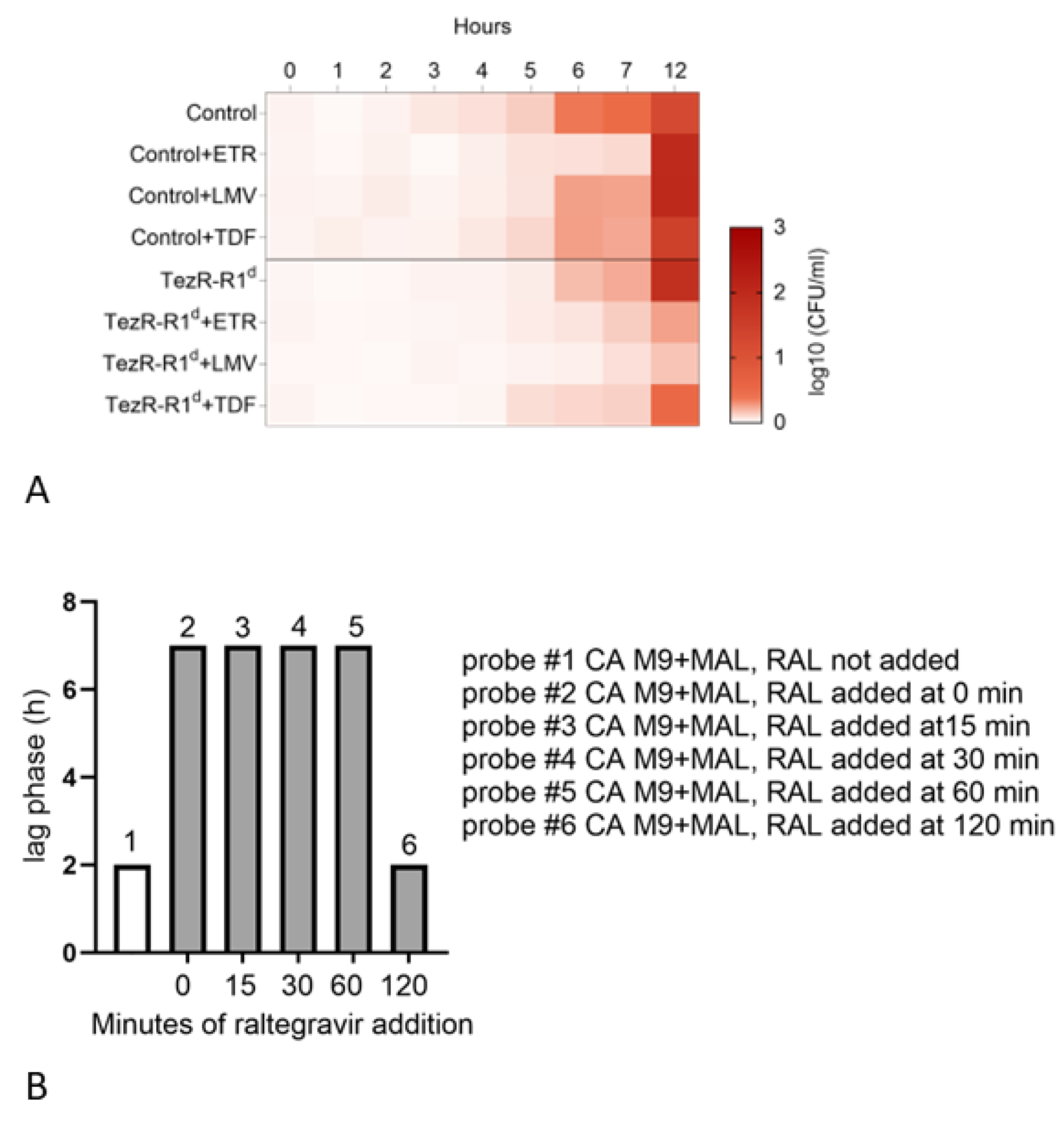
2.31. Raltegravir in Cell Memory Formation Experiments
2.32. Seedling Growth Assay
2.33. Generation of RNA Sequencing Data
2.34. Analysis of RNA Sequencing Data
2.35. Statistics
3. Results
3.1. Classification and Nomenclature of TezRs
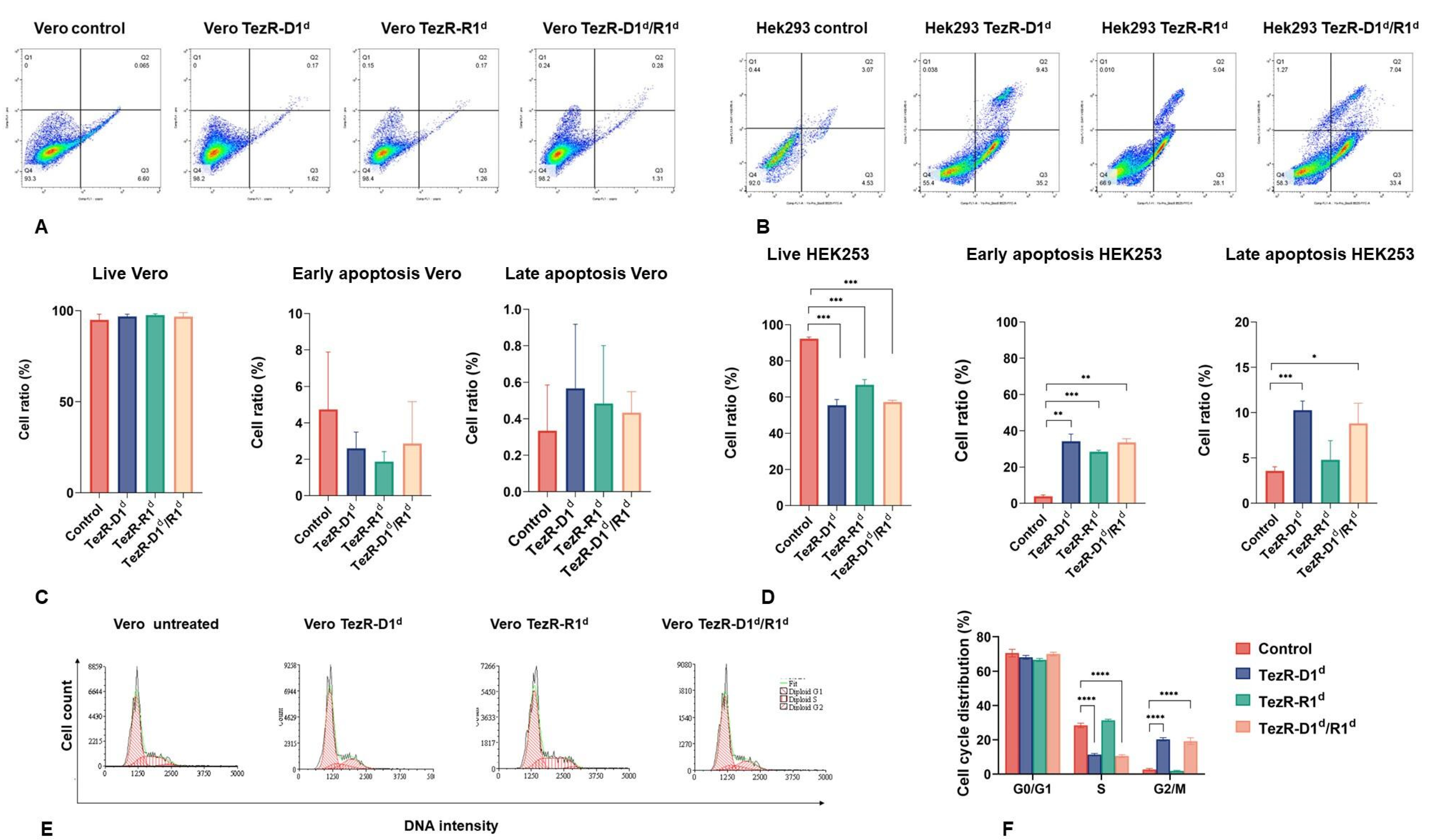
3.2. Cell Cycle and Apoptosis Regulation by TR-System in Mammalian Cells
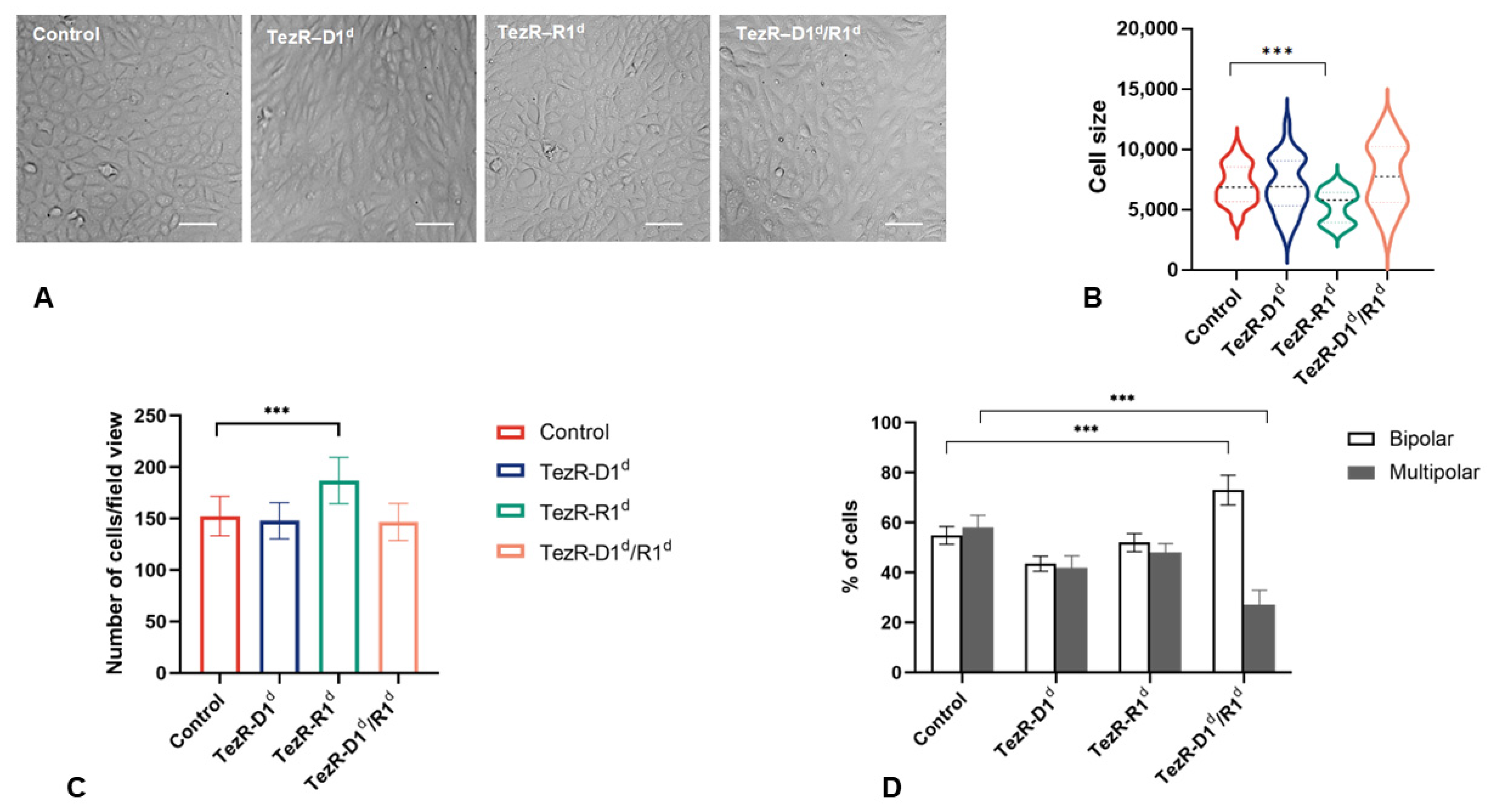
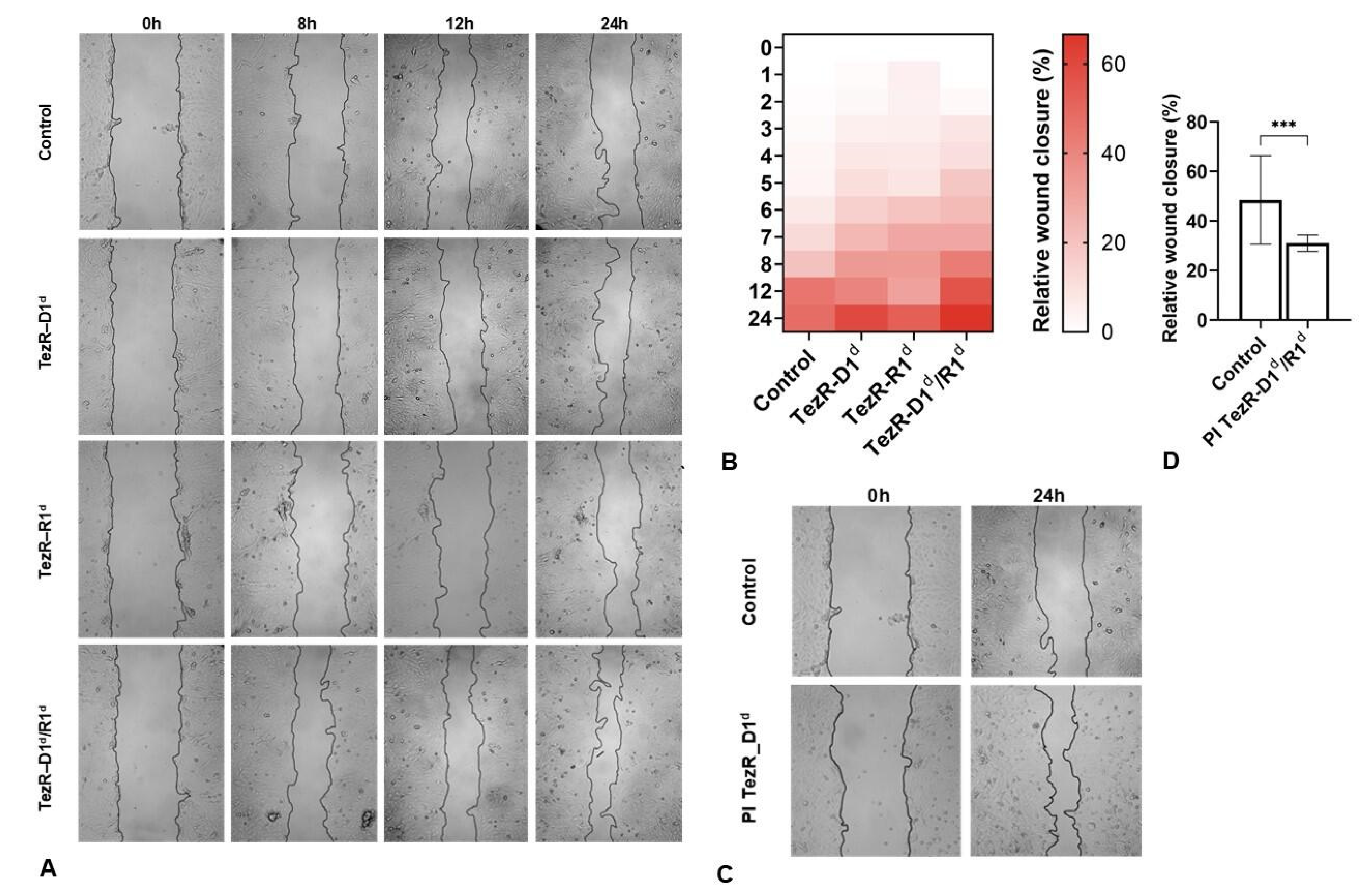
3.3. TezRs Regulate Mammalian Cell Morphology and Migration
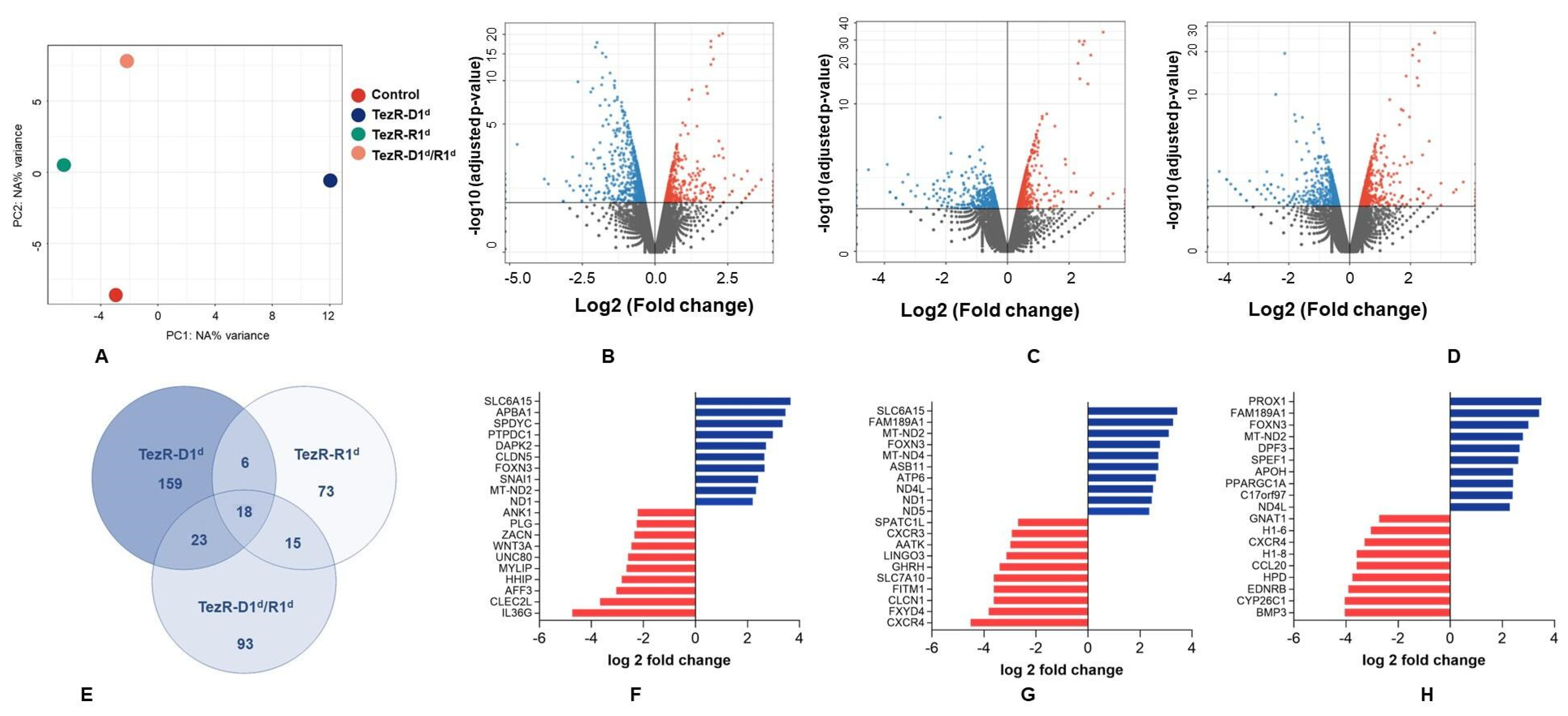
3.4. TezRs Regulate Mammalian Gene Expression
3.5. Role of TezRs in Fungal Growth and Viability
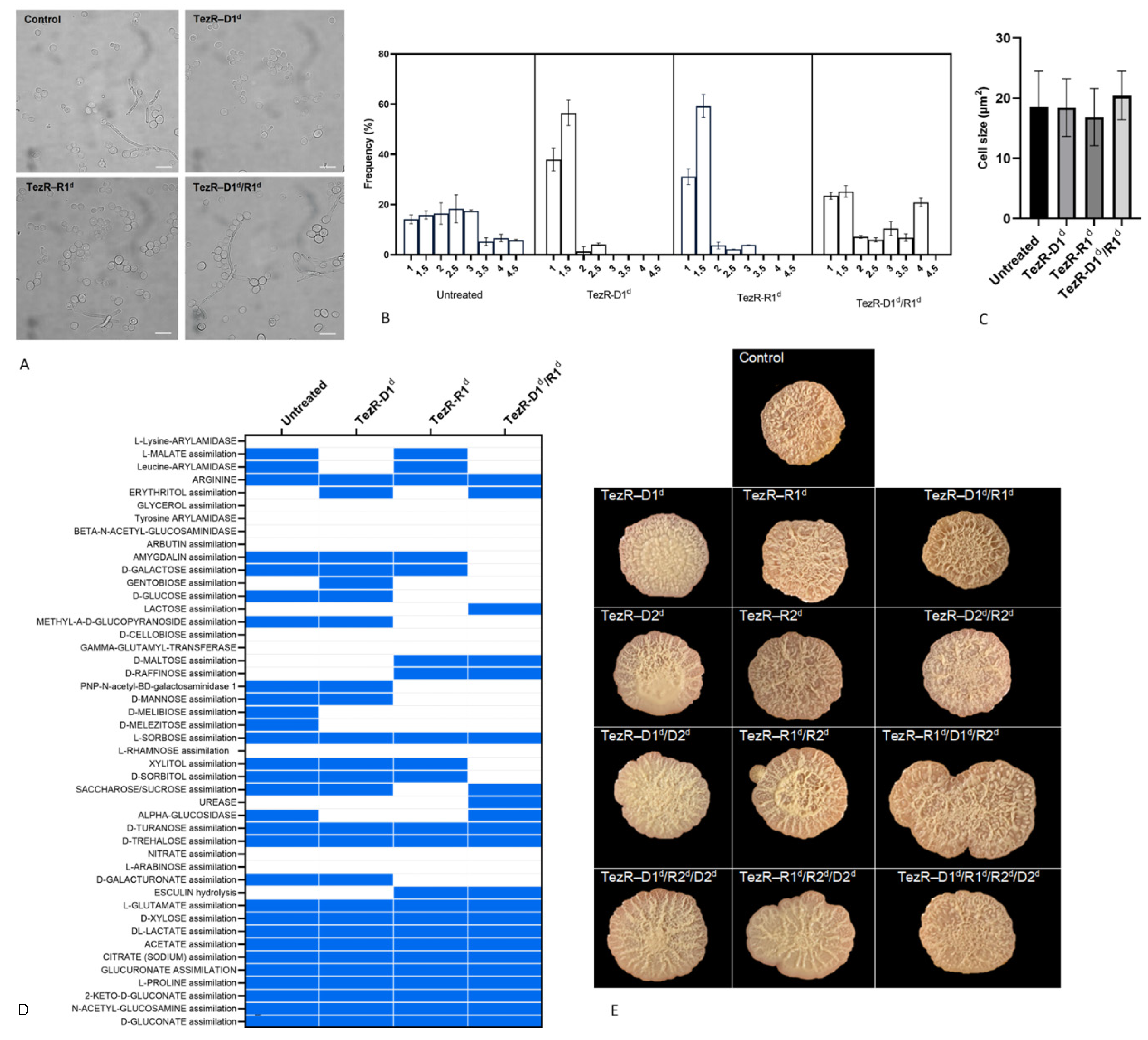
3.6. Dependence of Fungal Cell Morphology and Biochemical Characteristics on TezRs
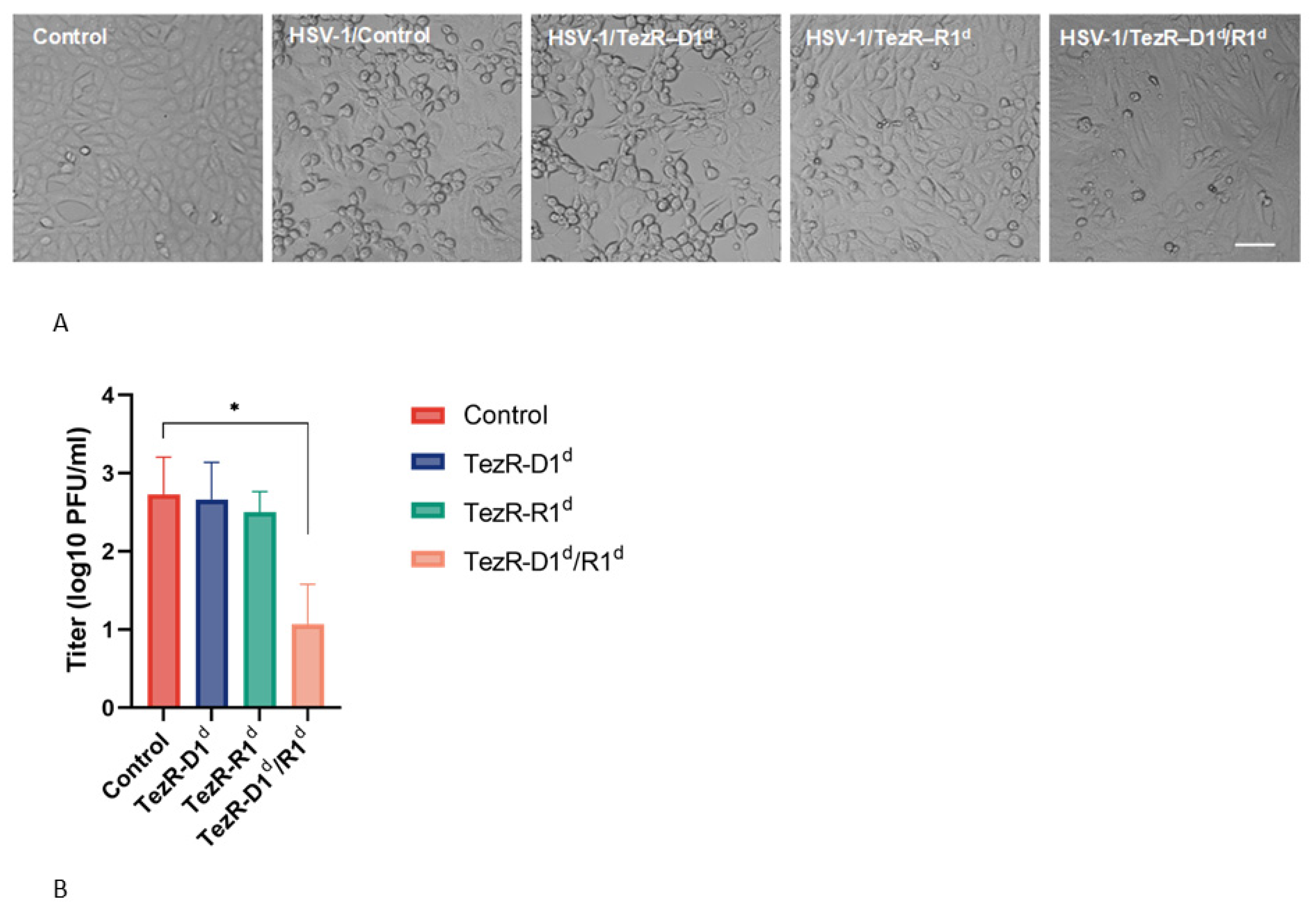
3.7. Control of Viral–Host Interactions by TezRs
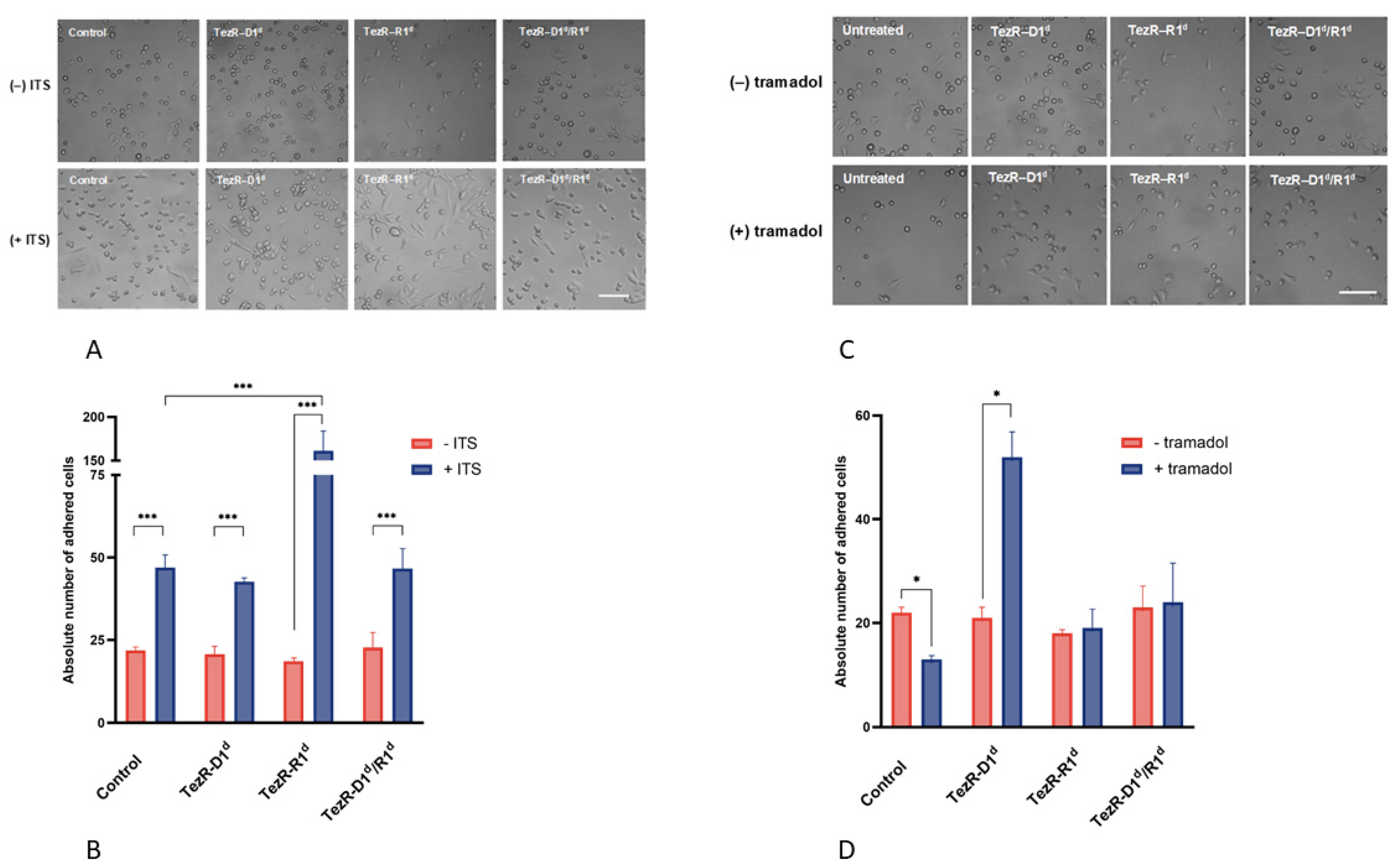
3.8. TezRs Regulate Cell Response to Insulin and Tramadol
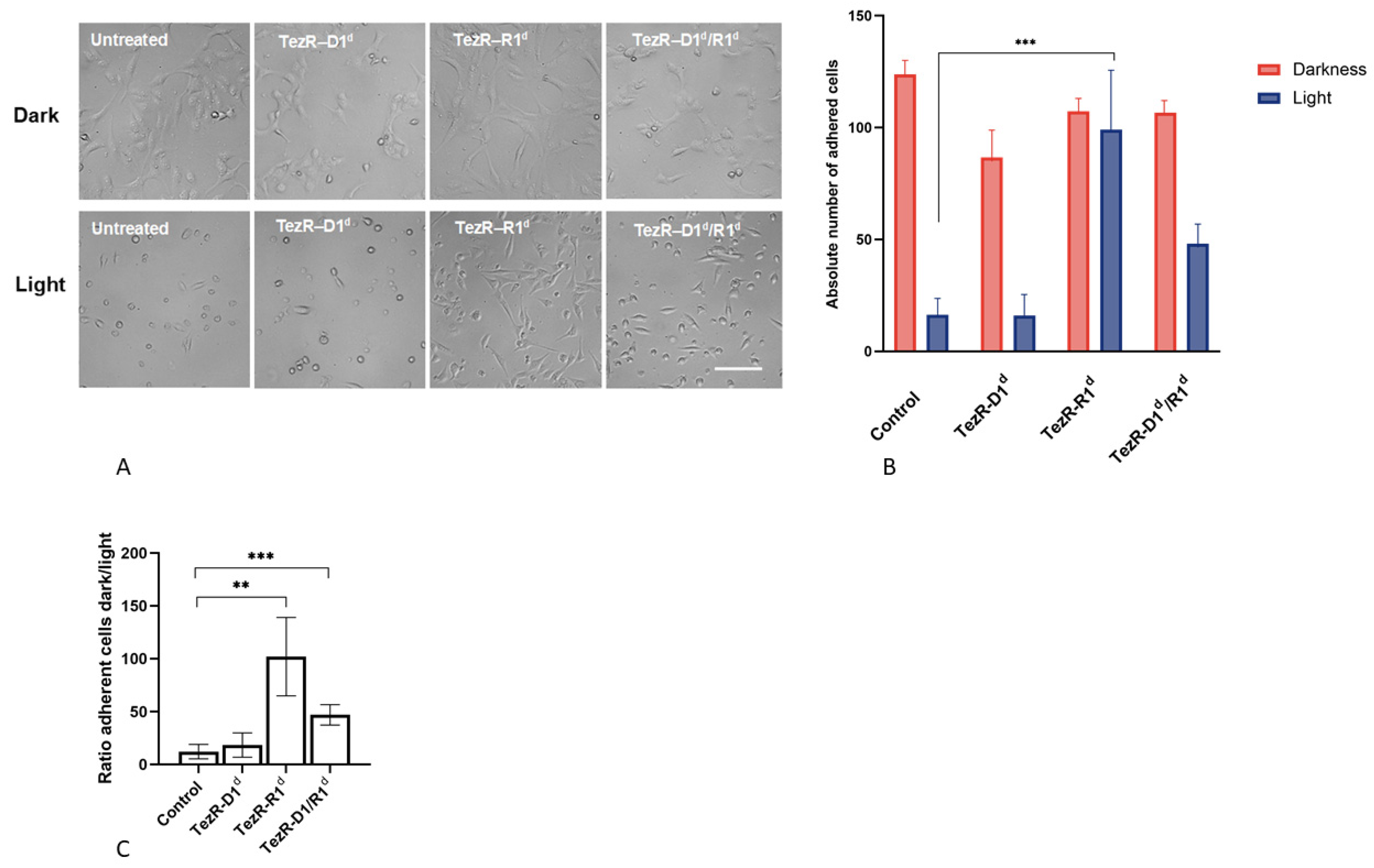
3.9. TezRs Control the Response of Mammalian Cells to Visible Light
3.10. Role of TezRs in Eukaryotic Cell Temperature Tolerance
3.11. TezRs Control Sensitivity to UV
3.12. TezRs Are Implicated in Magnetoreception in Eukaryotic Cells
3.13. TezRs Are Implicated in the Utilization of Nutrients
3.14. Implication of the TR-System in Cell Memory Formation and Forgetting
3.15. Role of Transcriptase Inhibitors in the TR-System of Eukaryotic Cell
3.16. Effects of Integrase Inhibitors on Signal Realization in the TR-System
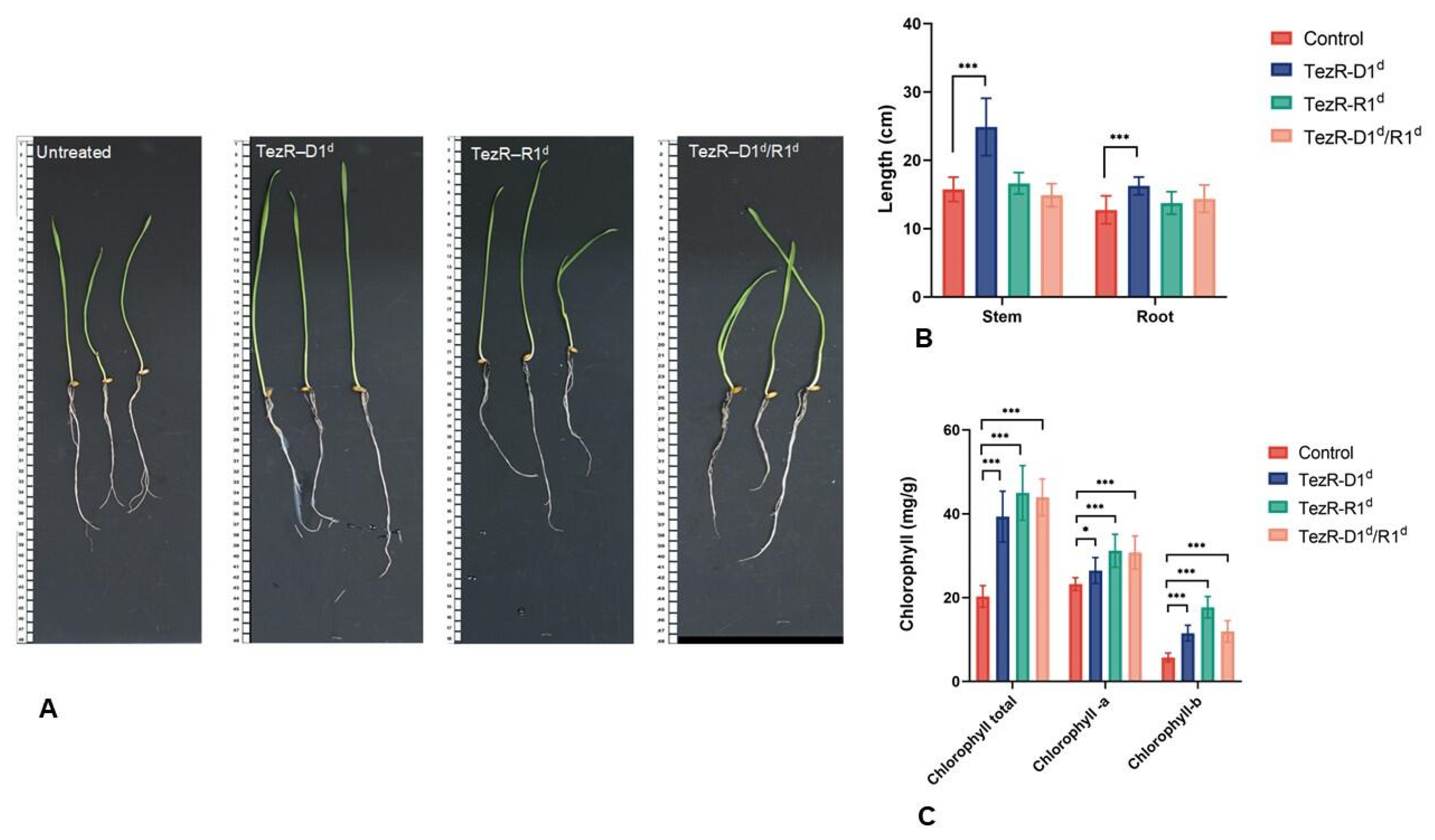
3.17. TezRs Regulate Multicellular Eukaryotic Structures
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Jamora, C.; Dasgupta, R.; Kocieniewski, P.; Fuchs, E. Links between signal transduction, transcription and adhesion in epithelial bud development. Nature 2003, 422, 317–322. [Google Scholar] [CrossRef] [Green Version]
- Swaney, K.F.; Huang, C.-H.; Devreotes, P.N. Eukaryotic Chemotaxis: A Network of Signaling Pathways Controls Motil-ity, Directional Sensing, and Polarity. Annu. Rev. Biophys. 2010, 39, 265–289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saeki, K.; Yokomizo, T. Identification, signaling, and functions of LTB 4 receptors. Semin. Immunol. 2017, 33, 30–36. [Google Scholar] [CrossRef] [PubMed]
- Lomakin, Y.A.; Kaminskaya, A.N.; Stepanov, A.V.; Shmidt, A.A.; Gabibov, A.G.; Belogurov, A.A. Probing Surface Membrane Receptors Using Engineered Bacteriophage Bioconjugates. Bioconjugate Chem. 2019, 30, 1500–1506. [Google Scholar] [CrossRef] [PubMed]
- Lacey, R.F.; Binder, B.M. How plants sense ethylene gas—The ethylene receptors. J. Inorg. Biochem. 2014, 133, 58–62. [Google Scholar] [CrossRef]
- Canton, J.; Neculai, D.; Grinstein, S. Scavenger receptors in homeostasis and immunity. Nat. Rev. Immunol. 2013, 13, 621–634. [Google Scholar] [CrossRef]
- Oswald, D.J.; Lee, A.; Trinidad, M.; Chi, C.; Ren, R.; Rich, C.B.; Trinkaus-Randall, V. Communication between Corneal Epithelial Cells and Trigeminal Neurons Is Facilitated by Purinergic (P2) and Glutamatergic Receptors. PLoS ONE 2012, 7, e44574. [Google Scholar] [CrossRef]
- Valera, S.; Hussy, N.; Evans, R.J.; Adami, N.; North, R.A.; Surprenant, A.; Buell, G. A new class of ligand-gated ion channel defined by P2X receptor for extracellular ATP. Nature 1994, 371, 516–519. [Google Scholar] [CrossRef]
- Rosenbaum, D.M.; Rasmussen, S.G.F.; Kobilka, B.K. The structure and function of G-protein-coupled receptors. Nature 2009, 459, 356–363. [Google Scholar] [CrossRef] [Green Version]
- Roberts, K.; Alberts, B.; Johnson, A.; Walter, P.H.T. Molecular Biology of the Cell; Garland Science: New York, NY, USA, 2002. [Google Scholar]
- Hakak, Y.; Shrestha, D.; Goegel, M.C.; Behan, D.P.; Chalmers, D.T. Global analysis of G-protein-coupled receptor signaling in human tissues. FEBS Lett. 2003, 550, 11–17. [Google Scholar] [CrossRef] [Green Version]
- Salon, J.A.; Lodowski, D.T.; Palczewski, K. The significance of G protein-coupled receptor crystallography for drug dis-covery. Pharm. Rev. 2011, 63, 901–937. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Basith, S.; Cui, M.; Macalino, S.J.Y.; Park, J.; Clavio, N.A.B.; Kang, S.; Choi, S. Exploring G protein-coupled receptors (GPCRs) ligand space via cheminformatics approaches: Impact on ra-tional drug design. Front. Pharm. 2018, 9, 128. [Google Scholar] [CrossRef] [PubMed]
- Kupperman, E.; An, S.; Osborne, N.; Waldron, S.; Stainier, D.Y.R. A sphingosine-1-phosphate receptor regulates cell mi-gration during vertebrate heart development. Nature 2000, 406, 192–195. [Google Scholar] [CrossRef] [PubMed]
- Janku, F.; Yap, T.A.; Meric-Bernstam, F. Targeting the PI3K pathway in cancer: Are we making headway? Nat. Rev. Clin. Oncol. 2018, 15, 273–291. [Google Scholar] [CrossRef]
- Lemmon, M.A.; Schlessinger, J. Cell Signaling by Receptor Tyrosine Kinases. Cell 2010, 141, 1117–1134. [Google Scholar] [CrossRef] [Green Version]
- Beaulieu, J.-M.; Gainetdinov, R.R. The Physiology, Signaling, and Pharmacology of Dopamine Receptors. Pharmacol. Rev. 2011, 63, 182–217. [Google Scholar] [CrossRef] [Green Version]
- Soriano, P. Abnormal kidney development and hematological disorders in PDGF beta-receptor mutant mice. Genes Dev. 1994, 8, 1888–1896. [Google Scholar] [CrossRef] [Green Version]
- Van Duin, D.; Shaw, A.C. Toll-Like Receptors in Older Adults. J. Am. Geriatr. Soc. 2007, 55, 1438–1444. [Google Scholar] [CrossRef]
- Julius, D.; Nathans, J. Signaling by Sensory Receptors. Cold Spring Harb. Perspect. Biol. 2012, 4, a005991. [Google Scholar] [CrossRef] [Green Version]
- Zhou, H.; Yang, S.; Chen, F. The Magnetic Receptor of Monascus ruber M7: Gene Clone and Its Heterologous Expression in Escherichia coli. Front. Microbiol. 2020, 11, 1112. [Google Scholar] [CrossRef]
- Sancar, A. Cryptochrome: The Second Photoactive Pigment in the Eye and Its Role in Circadian Photoreception. Annu. Rev. Biochem. 2000, 69, 31–67. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takeuchi, T.; Mizuno, T.; Higashi, T.; Yamagishi, A.; Date, M. Orientation of red blood cells in high magnetic field. J. Magn. Magn. Mater. 1995, 140–144, 1462–1463. [Google Scholar] [CrossRef]
- Naisbett-Jones, L.C.; Putman, N.F.; Scanlan, M.M.; Noakes, D.L.G.; Lohmann, K.J. Magnetoreception in fishes: The effect of magnetic pulses on orientation of juvenile Pacific salmon. J. Exp. Biol. 2020, 223, jeb222091. [Google Scholar] [CrossRef] [PubMed]
- Ikeya, N.; Woodward, J.R. Cellular autofluorescence is magnetic field sensitive. Proc. Natl. Acad. Sci. USA 2021, 118, e2018043118. [Google Scholar] [CrossRef]
- Blank, M.; Goodman, R. DNA is a fractal antenna in electromagnetic fields. Int. J. Radiat. Biol. 2011, 87, 409–415. [Google Scholar] [CrossRef] [PubMed]
- Berashevich, J.; Chakraborty, T. How the Surrounding Water Changes the Electronic and Magnetic Properties of DNA. J. Phys. Chem. B 2008, 112, 14083–14089. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nikiforov, V.N.; Koksharov, Y.A.; Irkhin, V.Y. Magnetic properties of “doped” DNA. J. Magn. Magn. Mater. 2018, 459, 340–344. [Google Scholar] [CrossRef]
- Cronin, T.W.; Johnsen, S. Extraocular, Non-Visual, and Simple Photoreceptors: An Introduction to the Symposium. Integr. Comp. Biol. 2016, 56, 758–763. [Google Scholar] [CrossRef] [Green Version]
- Yuan, Q.; Metterville, D.; Briscoe, A.; Reppert, S.M. Insect Cryptochromes: Gene Duplication and Loss Define Diverse Ways to Construct Insect Circadian Clocks. Mol. Biol. Evol. 2007, 24, 948–955. [Google Scholar] [CrossRef] [Green Version]
- Stockley, J.H.; Evans, K.; Matthey, M.; Volbracht, K.; Agathou, S.; Mukanowa, J.; Burrone, J.; Káradóttir, R.T. Surpassing light-induced cell damage in vitro with novel cell culture media. Sci. Rep. 2017, 7, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Ramakrishnan, P.; Maclean, M.; MacGregor, S.J.; Anderson, J.G.; Grant, M.H. Cytotoxic responses to 405nm light expo-sure in mammalian and bacterial cells: Involvement of reactive oxygen species. Toxicol. Vitr. 2016, 33, 54–62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ye, L.; Wu, J.; Cohen, P.; Kazak, L.; Khandekar, M.J.; Jedrychowski, M.P.; Zeng, X.; Gygi, S.P.; Spiegelman, B.M. Fat cells directly sense temperature to activate thermogenesis. Proc. Natl. Acad. Sci. USA 2013, 110, 12480–12485. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loh, E.; Righetti, F.; Eichner, H.; Twittenhoff, C.; Narberhaus, F. RNA Thermometers in Bacterial Pathogens. Microbiol. Spectr. 2018, 612. [Google Scholar]
- Meyer, M.; Plass, M.; Pérez-Valle, J.; Eyras, E.; Vilardell, J. Deciphering 3′ss Selection in the Yeast Genome Reveals an RNA Thermosensor that Mediates Alternative Splicing. Mol. Cell 2011, 43, 1033–1039. [Google Scholar] [CrossRef] [Green Version]
- Klinkert, B.; Narberhaus, F. Microbial thermosensors. Experientia 2009, 66, 2661–2676. [Google Scholar] [CrossRef] [PubMed]
- Yoo, H.; Triandafillou, C.; Drummond, D.A. Cellular sensing by phase separation: Using the process, not just the products. J. Biol. Chem. 2019, 294, 7151–7159. [Google Scholar] [CrossRef] [Green Version]
- Turner, J.J.; Ewald, J.C.; Skotheim, J.M. Cell Size Control in Yeast. Curr. Biol. 2012, 22, R350–R359. [Google Scholar] [CrossRef] [Green Version]
- Nordmann, G.C.; Hochstoeger, T.; Keays, D.A. Magnetoreception—A sense without a receptor. PLoS Biol. 2017, 15, e2003234. [Google Scholar] [CrossRef] [Green Version]
- Lyashenko, E.; Niepel, M.; Dixit, P.D.; Lim, S.K.; Sorger, P.K.; Vitkup, D. Receptor-based mechanism of relative sensing and cell memory in mammalian signaling networks. Elife 2020, 9, e50342. [Google Scholar] [CrossRef]
- Pradeu, T.; Du Pasquier, L. Immunological memory: What’s in a name? Immunol. Rev. 2018, 283, 7–20. [Google Scholar] [CrossRef]
- Ahmed, R.; Gray, D. Immunological memory and protective immunity: Understanding their relation. Science 1996, 272, 54–60. [Google Scholar] [CrossRef] [PubMed]
- Wolf, D.M.; Fontaine-Bodin, L.; Bischofs, I.; Price, G.; Keasling, J.; Arkin, A.P. Memory in microbes: Quantifying history-dependent behavior in a bacterium. PLoS ONE 2008, 3, e1700. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lambert, G.; Kussell, E. Memory and Fitness Optimization of bacteria under fluctuating environments. PLoS Genet. 2014, 10, e1004556. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Skoge, M.; Yue, H.; Erickstad, M.; Bae, A.; Levine, H.; Groisman, A.; Loomis, W.F.; Rappel, W.-J. Cellular memory in eukaryotic chemotaxis. Proc. Natl. Acad. Sci. USA 2014, 111, 14448–14453. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akkaya, M.; Kwak, K.; Pierce, S.K. B cell memory: Building two walls of protection against pathogens. Nat. Rev. Immunol. 2020, 20, 229–238. [Google Scholar] [CrossRef]
- Ajo-Franklin, C.M.; Drubin, D.A.; Eskin, J.A.; Gee, E.P.; Landgraf, D.; Phillips, I.; Silver, P.A. Rational design of memory in eukaryotic cells. Genes Dev. 2007, 21, 2271–2276. [Google Scholar] [CrossRef] [Green Version]
- Tetz, V.; Tetz, G. Bacterial DNA induces the formation of heat-resistant disease-associated proteins in human plasma. Sci. Rep. 2019, 9, 17995. [Google Scholar] [CrossRef] [Green Version]
- Tetz, G.; Pinho, M.; Pritzkow, S.; Mendez, N.; Soto, C.; Tetz, V. Bacterial DNA promotes Tau aggregation. Sci. Rep. 2020, 10, 2369. [Google Scholar] [CrossRef] [Green Version]
- Tetz, G.; Tetz, V. Bacterial Extracellular DNA Promotes β-Amyloid Aggregation. Microorganisms 2021, 9, 1301. [Google Scholar] [CrossRef]
- Tetz, V.T.G. Novel prokaryotic sensing and regulatory system employing previously unknown nucleic acids-based recep-tors. bioRxiv 2021. [Google Scholar] [CrossRef]
- Mukaremera, L.; Lee, K.K.; Mora-Montes, H.M.; Gow, N.A.R. Candida albicans Yeast, Pseudohyphal, and Hyphal Mor-phogenesis Differentially Affects Immune Recognition. Front. Immunol. 2017, 8, 629. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B.; et al. Fiji: An open-source platform for biological-image analysis. Nat. Methods 2012, 9, 676–682. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rueden, C.T.; Schindelin, J.; Hiner, M.C.; Dezonia, B.E.; Walter, A.E.; Arena, E.T.; Eliceiri, K.W. ImageJ2: ImageJ for the next generation of scientific image data. BMC Bioinform. 2017, 18, 529. [Google Scholar] [CrossRef] [PubMed]
- Kuang, Z.; Seo, E.J.; Leis, J. Mechanism of Inhibition of Retrovirus Release from Cells by Interferon-Induced Gene ISG15. J. Virol. 2011, 85, 7153–7161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.-J.; Kwon, J.-H.; Park, Y.K.; Kwon, O.; Yoon, T.-W. The effects of various hormones and growth factors on the growth of human insulin-producing cell line in serum-free medium. Exp. Mol. Med. 1997, 29, 209–216. [Google Scholar] [CrossRef] [Green Version]
- Bahar, E.; Yoon, H. Modeling and Predicting the Cell Migration Properties from Scratch Wound Healing Assay on Cispla-tin-Resistant Ovarian Cancer Cell Lines Using Artificial Neural Network. Healthcare 2021, 9, 911. [Google Scholar] [CrossRef]
- Ramenzoni, L.L.; Attin, T.; Schmidlin, P.R. In Vitro Effect of Modified Polyetheretherketone (PEEK) Implant Abutments on Human Gingival Epithelial Keratinocytes Migration and Proliferation. Materials 2019, 12, 1401. [Google Scholar] [CrossRef] [Green Version]
- Rex, J.H.; Alexander, B.D.; Andes, D.; Arthington-Skaggs, B.; Brown, S.D.; Chaturvedi, V.; Ghannoum, M.A.; Espinel-Ingroff, A.; Kiapp, C.C.; Ostrosky-Zeichner, L.; et al. Clinical and Laboratory Standards Institute (CLSI). Reference Method for Broth Dilution Antifungal Susceptibility Testing of Yeasts, Approved Standard-Third Edition, CLSI Document M27-A3; Clinical and Laboratory Standards Institute: Wayne, PA, USA, 2008; ISBN 1-56238-666-2. [Google Scholar]
- Zhu, L.; Yang, Z.; Yang, Q.; Tu, Z.; Ma, L.; Shi, Z.; Li, X. Degradation of dexamethasone by acclimated strain of Pseudomonas Alcaligenes. Int. J. Clin. Exp. Med. 2015, 8, 10971. [Google Scholar]
- Hargarten, A.M.; Nemacheck, J.A.; Subramanyam, S.; Xiao, X.; Schemerhorn, B.J.; Williams, C.E. Physical and metabolic consequences of Hessian fly infestation are more severe on nonhost Brachy-podium distachyon than on host-plant resistant wheat. Arthropod. Plant. Interact. 2017, 11, 767–783. [Google Scholar] [CrossRef]
- Porra, R.J.; Thompson, W.A.; Kriedemann, P.E. Determination of accurate extinction coefficients and simultaneous equa-tions for assaying chlorophylls a and b extracted with four different solvents: Verification of the concentration of chloro-phyll standards by atomic absorption spectroscopy. Biochim. Biophys. Acta Bioenerg. 1989, 975, 384–394. [Google Scholar] [CrossRef]
- Liu, H.; Zhang, Y.-H.; Yin, H.; Wang, W.-X.; Zhao, X.-M.; Du, Y.-G. Alginate oligosaccharides enhanced Triticum aestivum L. tolerance to drought stress. Plant Physiol. Biochem. 2013, 62, 33–40. [Google Scholar] [CrossRef]
- Hulsen, T.; De Vlieg, J.; Alkema, W. BioVenn–a web application for the comparison and visualization of biological lists using area-proportional Venn diagrams. BMC Genom. 2008, 9, 488. [Google Scholar] [CrossRef] [Green Version]
- Jonkman, J.; Cathcart, J.A.; Xu, F.; Bartolini, M.E.; Amon, J.E.; Stevens, K.M.; Colarusso, P. An introduction to the wound healing assay using live-cell microscopy. Cell Adhes. Migr. 2014, 8, 440–451. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Merson-Davies, L.A.; Odds, F.C. A Morphology Index for Characterization of Cell Shape in Candida albicans. Microbiology 1989, 135, 3143–3152. [Google Scholar] [CrossRef] [Green Version]
- Ambaraghassi, G.; Dufresne, P.J.; Dufresne, S.F.; Vallières, É.; Muñoz, J.F.; Cuomo, C.A.; Berkow, E.L.; Lockhart, S.R.; Luong, M.-L. Identification of Candida auris by Use of the Updated Vitek 2 Yeast Identification System, Version 8.01: A Multilaboratory Evaluation Study. J. Clin. Microbiol. 2019, 57, e00884-19. [Google Scholar] [CrossRef] [PubMed]
- Staniszewska, M.; Bondaryk, M.; Ochal, Z. New synthetic sulfone derivatives inhibit growth, adhesion and the leucine arylamidase APE2 gene expression of Candida albicans in vitro. Bioorganic Med. Chem. 2015, 23, 314–321. [Google Scholar] [CrossRef] [PubMed]
- Frade-Pérez, M.D.; Hernández-Cervantes, A.; Flores-Carreón, A.; Mora-Montes, H.M. Biochemical characterization of Candida albicans α-glucosidase I heterologously expressed in Escherichia coli. Antonie van Leeuwenhoek 2010, 98, 291–298. [Google Scholar] [CrossRef]
- Mora-Montes, H.M.; Bates, S.; Netea, M.G.; Díaz-Jiménez, D.F.; Lopez-Romero, E.; Zinker, S.; Ponce-Noyola, P.; Kullberg, B.J.; Brown, A.J.; Odds, F.C.; et al. Endoplasmic Reticulum α-Glycosidases of Candida albicans Are Required for N Glycosylation, Cell Wall Integrity, and Normal Host-Fungus Interaction. Eukaryot. Cell 2007, 6, 2184–2193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rzechonek, D.A.; Szczepańczyk, M.; Wang, G.; Borodina, I.; Mirończuk, A.M. HOG-Independent Osmoprotection by Erythritol in Yeast Yarrowia lipolytica. Genes 2020, 11, 1424. [Google Scholar] [CrossRef]
- Chew, S.Y.; Chee, W.J.Y.; Than, L.T.L. The glyoxylate cycle and alternative carbon metabolism as metabolic adaptation strategies of Candida glabrata: Perspectives from Candida albicans and Saccharomyces cerevisiae. J. Biomed. Sci. 2019, 26, 52. [Google Scholar] [CrossRef]
- Navarathna, D.H.M.L.P.; Harris, S.D.; Roberts, D.D.; Nickerson, K.W. Evolutionary aspects of urea utilization by fungi. FEMS Yeast Res. 2010, 10, 209–213. [Google Scholar] [CrossRef]
- Odds, F.C. Candida and Candidosis: A Review and Bibliography, 2nd ed.; Bailliere-Tindall: London, UK, 1988. [Google Scholar]
- Devadas, S.M. Auxanographic Carbohydrate Assimilation Method for Large Scale Yeast Identification. J. Clin. Diagn. Res. 2017, 11, DC01–DC03. [Google Scholar] [CrossRef] [PubMed]
- Marinho, S.A.; Teixeira, A.B.; Santos, O.S.; Cazanova, R.F.; Ferreira, C.; Cherubini, K.; De Oliveira, S.D. Identification of Candida spp. by phenotypic tests and PCR. Braz. J. Microbiol. 2010, 41, 286–294. [Google Scholar] [CrossRef] [Green Version]
- Jeong, Y.W.; Hossein, M.S.; Bhandari, D.P.; Kim, Y.W.; Kim, J.H.; Park, S.W.; Lee, E.; Park, S.M.; Lee, J.Y.; Kim, S.; et al. Effects of insulin–transferrin–selenium in defined and porcine follicular fluid supplemented IVM media on porcine IVF and SCNT embryo production. Anim. Reprod. Sci. 2008, 106, 13–24. [Google Scholar] [CrossRef]
- Xia, M.; Tong, J.-H.; Ji, N.-N.; Duan, M.; Tan, Y.-H.; Xu, J.-G. Tramadol regulates proliferation, migration and invasion via PTEN/PI3K/AKT signaling in lung adenocarcinoma cells. Eur. Rev. Med Pharmacol. Sci. 2016, 20, 2573–2580. [Google Scholar] [PubMed]
- Lazarczyk, M.; Matyja, E.; Lipkowski, A.W. A comparative study of morphine stimulation and biphalin inhibition of hu-man glioblastoma T98G cell proliferation in vitro. Peptides 2010, 31, 1606–1612. [Google Scholar] [CrossRef]
- Leppert, W. Tramadol as an analgesic for mild to moderate cancer pain. Pharmacol. Rep. 2009, 61, 978–992. [Google Scholar] [CrossRef]
- Liu, S.-D.; Wu, Y.-N.; Wang, T.; Zhang, C.; Xing, X.-H. Maltose Utilization as a Novel Selection Strategy for Continuous Evolution of Microbes with Enhanced Metabolite Production. ACS Synth. Biol. 2017, 6, 2326–2338. [Google Scholar] [CrossRef] [PubMed]
- Van Ende, M.; Wijnants, S.; Van Dijck, P. Sugar Sensing and Signaling in Candida albicans and Candida glabrata. Front. Microbiol. 2019, 10, 99. [Google Scholar] [CrossRef] [PubMed]
- Ostergaard, S.; Olsson, L.; Nielsen, J. Metabolic Engineering of Saccharomyces cerevisiae. Microbiol. Mol. Biol. Rev. 2000, 64, 34–50. [Google Scholar] [CrossRef] [Green Version]
- Zarkan, A.; Cano-Muniz, S.; Zhu, J.; Al Nahas, K.; Cama, J.; Keyser, U.F.; Summers, D.K. Indole Pulse Signalling Regulates the Cytoplasmic pH of E. coli in a Memory-Like Manner. Sci. Rep. 2019, 9, 3868. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lyon, P. The cognitive cell: Bacterial behavior reconsidered. Front. Microbiol. 2015, 6, 264. [Google Scholar] [CrossRef]
- Dai, L.; Huang, Q.; Boeke, J.D. Effect of reverse transcriptase inhibitors on LINE-1 and Ty1 reverse transcriptase activities and on LINE-1 retrotransposition. BMC Biochem. 2011, 12, 18. [Google Scholar] [CrossRef] [Green Version]
- Iyer, L.M.; Koonin, E.V.; Aravind, L. Evolutionary connection between the catalytic subunits of DNA-dependent RNA polymerases and eukaryotic RNA-dependent RNA polymerases and the origin of RNA polymerases. BMC Struct. Biol. 2003, 3, 1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szilvay, A.M.; Stern, B.; Blichenberg, A.; Helland, D.E. Structural and functional similarities between HIV-1 reverse tran-scriptase and the Escherichia coli RNA polymerase β′ subunit. FEBS Lett. 2000, 484, 43–47. [Google Scholar] [CrossRef] [Green Version]
- Sciamanna, I.; De Luca, C.; Spadafora, C. The Reverse Transcriptase Encoded by LINE-1 Retrotransposons in the Genesis, Progression, and Therapy of Cancer. Front. Chem. 2016, 4, 6. [Google Scholar] [CrossRef] [Green Version]
- Spanopoulou, E.; Zaitseva, F.; Wang, F.H.; Santagata, S.; Baltimore, D.; Panayotou, G. The Homeodomain Region of Rag-1 Reveals the Parallel Mechanisms of Bacterial and V(D)J Recom-bination. Cell 1996, 87, 263–276. [Google Scholar] [CrossRef] [Green Version]
- Nishana, M.; Nilavar, N.M.; Kumari, R.; Pandey, M.; Raghavan, S.C. HIV integrase inhibitor, Elvitegravir, impairs RAG functions and inhibits V(D)J recombination. Cell Death Dis. 2017, 8, e2852. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rathjen, J.R.; Strounina, E.V.; Mares, D.J. Water movement into dormant and non-dormant wheat (Triticum aestivum L.) grains. J. Exp. Bot. 2009, 60, 1619–1631. [Google Scholar] [CrossRef] [Green Version]
- Saldivar, J.C.; Hamperl, S.; Bocek, M.J.; Chung, M.; Bass, T.E.; Cisneros-Soberanis, F.; Samejima, K.; Xie, L.; Paulson, J.R.; Earnshaw, W.C.; et al. An intrinsic S/G 2 checkpoint enforced by ATR. Science 2018, 361, 806–810. [Google Scholar] [CrossRef] [Green Version]
- Tao Shen, S.H. The role of Cdc25A in the regulation of cell proliferation and apoptosis. Anticancer. Agents Med. Chem. 2012, 12, 631–639. [Google Scholar] [CrossRef]
- Sansam, C.G.; Goins, D.; Siefert, J.C.; Clowdus, E.A.; Sansam, C.L. Cyclin-dependent kinase regulates the length of S phase through TICRR/TRESLIN phosphorylation. Genes Dev. 2015, 29, 555–566. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.; Hong, D.; Zhou, Q.; Fan, J.-J.; Li, L.; Pan, J.; Hu, S. The Suppression of CCL2 Expression Decrease the Proliferation of HL-60 Cells through Downregulation of Cyclin d1 Via Arresting Cells at the G1 Phase. Blood 2015, 126, 4809. [Google Scholar] [CrossRef]
- Sharma, A.; Jyotsana, N.; Gabdoulline, R.; Heckl, D.; Kuchenbauer, F.; Slany, R.K.; Ganser, A.; Heuser, M. Meningioma 1 is indispensable for mixed lineage leukemia-rearranged acute myeloid leukemia. Haematologica 2020, 105, 1294–1305. [Google Scholar] [CrossRef] [Green Version]
- Etienne-Manneville, S. Polarity proteins in migration and invasion. Oncogene 2008, 27, 6970–6980. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.; Yan, Y.; Cheng, Z.; Hu, Y.; Liu, T. Sotetsuflavone suppresses invasion and metastasis in non-small-cell lung can-cer A549 cells by reversing EMT via the TNF-α/NF-κB and PI3K/AKT signaling pathway. Cell Death Discov. 2018, 4, 26. [Google Scholar] [CrossRef] [PubMed]
- Jiang, L.-P.; Fan, S.-Q.; Xiong, Q.-X.; Zhou, Y.-C.; Yang, Z.-Z.; Li, G.-F.; Huang, Y.-C.; Wu, M.-G.; Shen, Q.-S.; Liu, K.; et al. GRK5 functions as an oncogenic factor in non-small-cell lung cancer. Cell Death Dis. 2018, 9, 295. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.-N.; Lee, H.H.; Chou, C.K.; Yang, W.H.; Wei, Y.; Chen, C.T.; Yao, J.; Hsu, J.L.; Zhu, C.; Ying, H.; et al. Angiogenin/Ribonuclease 5 Is an EGFR Ligand and a Serum Biomarker for Erlotinib Sensitivity in Pan-creatic Cancer. Cancer Cell 2018, 33, 752–769. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, H.-Y.; Chen, C.-Y.; Hung, Y.-F.; Lin, H.-R.; Chao, H.-W.; Shih, P.-Y.; Chuang, C.-N.; Li, W.-P.; Huang, T.-N.; Hsueh, Y.-P. RNase A Promotes Proliferation of Neuronal Progenitor Cells via an ERK-Dependent Pathway. Front. Mol. Neurosci. 2018, 11, 428. [Google Scholar] [CrossRef] [PubMed]
- Shapiro, R.; Robbins, N.; Cowen, L.E. Regulatory Circuitry Governing Fungal Development, Drug Resistance, and Disease. Microbiol. Mol. Biol. Rev. 2011, 75, 213–267. [Google Scholar] [CrossRef] [Green Version]
- Sudbery, P.; Gow, N.; Berman, J. The distinct morphogenic states of Candida albicans. Trends Microbiol. 2004, 12, 317–324. [Google Scholar] [CrossRef]
- Cavalheiro, M.; Teixeira, M.C. Candida Biofilms: Threats, Challenges, and Promising Strategies. Front. Med. 2018, 5, 28. [Google Scholar] [CrossRef] [Green Version]
- Blankenship, J.; Mitchell, A.P. How to build a biofilm: A fungal perspective. Curr. Opin. Microbiol. 2006, 9, 588–594. [Google Scholar] [CrossRef] [PubMed]
- Staniszewska, M.; Gizińska, M.; Kurządkowski, W. The role of leucine arylamidase in the virulence of Candida albicans. Postep. Microbiol. 2013, 52, 373–380. [Google Scholar]
- Ghosh, S.; Navarathna, D.H.; Roberts, D.D.; Cooper, J.T.; Atkin, A.L.; Petro, T.M.; Nickerson, K.W. Arginine-Induced Germ Tube Formation in Candida albicans Is Essential for Escape from Murine Macro-phage Line RAW 264. Infect. Immun. 2009, 77, 1596–1605. [Google Scholar] [CrossRef] [Green Version]
- Veses, V.; Gow, N.A. Pseudohypha budding patterns ofCandida albicans. Med. Mycol. 2009, 47, 268–275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Biswas, S.; Van Dijck, P.; Datta, A. Environmental Sensing and Signal Transduction Pathways Regulating Morphopatho-genic Determinants of Candida albicans. Microbiol. Mol. Biol. Rev. 2007, 71, 348–376. [Google Scholar] [CrossRef] [Green Version]
- Eller, C.H.; Lomax, J.E.; Raines, R.T. Bovine Brain Ribonuclease Is the Functional Homolog of Human Ribonuclease. J. Biol. Chem. 2014, 289, 25996–26006. [Google Scholar] [CrossRef] [Green Version]
- Krajewski, S.S.; Narberhaus, F. Temperature-driven differential gene expression by RNA thermosensors. Biochim. et Biophys. Acta 2014, 1839, 978–988. [Google Scholar] [CrossRef]
- Narberhaus, F. Translational control of bacterial heat shock and virulence genes by temperature-sensing mRNAs. RNA Biol. 2010, 7, 84–89. [Google Scholar] [CrossRef] [Green Version]
- Popitsch, N.; Bilusic, I.; Rescheneder, P.; Schroeder, R.; Lybecker, M. Temperature-dependent sRNA transcriptome of the Lyme disease spirochete. BMC Genom. 2017, 18, 28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gyula, P.; Baksa, I.; Toth, T.; Mohorianu, I.; Dalmay, T.; Szittya, G. Ambient temperature regulates the expression of a small set of sRNAs influencing plant development through NF-YA2 and YUC2. Plant Cell Environ. 2018, 10, 2404–2417. [Google Scholar] [CrossRef]
- Yu, Y.; Teng, Y.; Liu, H.; Reed, S.H.; Waters, R. UV irradiation stimulates histone acetylation and chromatin remodeling at a repressed yeast locus. Proc. Natl. Acad. Sci. USA 2005, 102, 8650–8655. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mao, P.; Wyrick, J.J. Emerging roles for histone modifications in DNA excision repair. FEMS Yeast Res. 2016, 16, fow090. [Google Scholar] [CrossRef] [Green Version]
- Rastogi, R.P.; Kumar, A.R.; Tyagi, M.B.; Sinha, R.P. Molecular Mechanisms of Ultraviolet Radiation-Induced DNA Damage and Repair. J. Nucleic Acids 2010, 2010, 592980. [Google Scholar] [CrossRef] [Green Version]
- Crefcoeur, R.P.; Yin, R.; Ulm, R.; Halazonetis, T.D. Ultraviolet-B-mediated induction of protein–protein interactions in mammalian cells. Nat. Commun. 2013, 4, 1779. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marangoni, R.; Marroni, F.; Ghetti, F.; Gioffré, D.; Colomebtti, G. Effect of UV-B radiation on ciliated protozoa. In Environmental UV Radiation: In Impact on Ecosystems and Human Health and Predictive Models; Springer: Berlin/Heidelberg, Germany, 2001; pp. 231–248. [Google Scholar]
- Monteil, C.L.; Leferve, C.T. Magnetoreception in microorganisms. Trends Microbiol. 2020, 4, 266275. [Google Scholar] [CrossRef]
- Galland, P.; Pazur, A. Magnetoreception in plants. J. Plant Res. 2005, 118, 371–389. [Google Scholar] [CrossRef]
- Zhang, Q.; Throolin, R.; Pitt, S.W.; Serganov, A.; Al-Hashimi, H.M. Probing Motions between Equivalent RNA Domains Using Magnetic Field Induced Residual Dipolar Couplings: Accounting for Correlations between Motions and Alignment. J. Am. Chem. Soc. 2003, 125, 10530–10531. [Google Scholar] [CrossRef]
- Yi, J. Emergent paramagnetism of DNA molecules. Phys. Rev. B 2006, 74, 212406. [Google Scholar] [CrossRef]
- Savelyev, I.V.; Zyryanova, N.V.; Polesskaya, O.O.; Myakishev-Rempel, M. On the Existence of The DNA Resonance Code and Its Possible Mechanistic Connection to The Neural Code. NeuroQuantology 2019, 17, 56. [Google Scholar] [CrossRef]
- Boucher, J.; Kleinridders, A.; Kahn, C.R. Insulin Receptor Signaling in Normal and Insulin-Resistant States. Cold Spring Harb. Perspect. Biol. 2014, 6, a009191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Posung, M.; Promkhatkaew, D.; Borg, J.; Tongta, A. Development of a modified serum-free medium for Vero cell cultures: Effects of protein hydrolysates, l-glutamine and SITE liquid media supplement on cell growth. Cytotechnology 2021, 73, 683–695. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Wei, J.; Gao, X.; Wei, B.; Lin, H.; Huang, R.; Niu, Y.; Lim, K.; Jing, K.; Chu, J. Insulin Promotes the Proliferation of Human Umbilical Cord Matrix-Derived Mesenchymal Stem Cells by Acti-vating the Akt-Cyclin D1 Axis. Stem Cells Int. 2017, 2017, 1–10. [Google Scholar]
- Han, C.; Wei, S.; Song, Q.; He, F.; Xiong, X.; Wan, H.; Liu, D.; Ye, F.; Liu, H.; Li, L.; et al. Insulin Stimulates Goose Liver Cell Growth by Activating PI3K-AKT-mTOR Signal Pathway. Cell Physiol. Bio-Chem. 2016, 38, 558–570. [Google Scholar] [CrossRef]
- Tetz, V.V.; Tetz, G.V. A new biological definition of life. Biomol. Concepts 2020, 11, 1–6. [Google Scholar] [CrossRef]
- Stock, J.B.; Zhang, S. The biochemistry of memory. Curr. Biol. 2013, 23, R741–R745. [Google Scholar] [CrossRef] [Green Version]
- Csermely, P.; Kunsic, N.; Mendik, P.; Kerestély, M.; Faragó, T.; Veres, D.V.; Tompa, P. Learning of Signaling Networks: Molecular Mechanisms. Trends Biochem. Sci. 2020, 45, 284–294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gamble, R.L.; Coonfield, M.L.; Schaller, G.E. Histidine kinase activity of the ETR1 ethylene receptor from Arabidopsis. PNAS 1998, 95, 78257829. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mcheyzerwilliams, M.G.; Okitsu, S.L.; Wang, N.; Mcheyzerwilliams, L.J. Molecular programming of B cell memory. Nat. Rev. Immunol. 2012, 12, 24–34. [Google Scholar] [CrossRef] [Green Version]
- Kurosaki, T.; Kometani, K.; Ise, W. Memory B cells. Nat. Rev. Immunol. 2015, 15, 149–159. [Google Scholar] [CrossRef] [PubMed]
- Raychaudhuri, S. The Problem of Antigen Affinity Discrimination in B-Cell Immunology. ISRN Biomath. 2013, 2013, 845918. [Google Scholar] [CrossRef]
- Gershman, S.J.; Balbi, P.E.; Gallistel, C.R.; Gunawardena, J. Reconsidering the evidence for learning in single cells. eLife 2021, 10, e61907. [Google Scholar] [CrossRef]
- Schott-Verdugo, S.; Müller, L.; Classen, E.; Gohlke, H.; Groth, G. Structural Model of the ETR1 Ethylene Receptor Trans-membrane Sensor Domain. Sci. Rep. 2019, 9, 8869. [Google Scholar] [CrossRef]
- Voegele, A.; Linkies, A.; Müller, K.; Leubner-Metzger, G. Members of the gibberellin receptor gene family GID1 (GIB-BERELLIN INSENSITIVE DWARF1) play distinct roles during Lepidium sativum and Arabidopsis thaliana seed germina-tion. J. Exp. Bot. 2011, 62, 5131–5147. [Google Scholar] [CrossRef] [Green Version]
- Sugiyama, T.; Cam, H.; Verdel, A.; Moazed, D.; Grewal, S.I.S. From the Cover: RNA-dependent RNA polymerase is an essential component of a self-enforcing loop coupling heterochromatin assembly to siRNA production. Proc. Natl. Acad. Sci. USA 2005, 102, 152–157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Indu, P.; Rameshkumar, M.R.; Arunagirinathan, N.; Al-Dhabi, N.A.; Arasu, M.V.; Ignacimuthu, S. Raltegravir, Indinavir, Tipranavir, Dolutegravir, and Etravirine against main protease and RNA-dependent RNA polymerase of SARS-CoV-2: A molecular docking and drug repurposing approach. J. Infect. Public Health 2020, 13, 1856–1861. [Google Scholar] [CrossRef]
- Torres, R.A.; Lewis, W. Aging and HIV/AIDS: Pathogenetic role of therapeutic side effects. Lab. Investig. 2014, 94, 120–128. [Google Scholar] [CrossRef]
- Sciamanna, I.; Landriscina, M.; Pittoggi, C.; Quirino, M.; Mearelli, C.; Beraldi, R.; Mattei, E.; Serafino, A.; Cassano, A.; Sinibaldi-Vallebona, P.; et al. Inhibition of endogenous reverse transcriptase antagonizes human tumor growth. Oncogene 2005, 24, 3923–3931. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Wang, C.; Guan, S.; Liu, Y.; Han, L.; Cheng, Y. Zidovudine, abacavir and lamivudine increase the radiosensitivity of human esophageal squamous cancer cell lines. Oncol. Rep. 2016, 36, 239–246. [Google Scholar] [CrossRef] [Green Version]
- Landriscina, M.; Spadafora, C.; Cignarelli, M.; Barone, C. Anti-Tumor Activity of Non-Nucleosidic Reverse Transcriptase Inhibitors. Curr. Pharm. Des. 2007, 13, 737–747. [Google Scholar] [CrossRef]
- Koczera, P.; Martin, L.; Marx, G.; Schuerholz, T. The Ribonuclease A Superfamily in Humans: Canonical RNases as the Buttress of Innate Immunity. Int. J. Mol. Sci. 2016, 17, 1278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lazarus, R.A.; Wagener, J.S. Recombinant human deoxyribonuclease I. In Pharmaceutical Biotechnology; Springer International Publishing: Berlin/Heidelberg, Germany, 2019; pp. 471–488. [Google Scholar] [CrossRef]
- Lerner, R.A.; Meinke, W.; Goldstein, D.A. Membrane-Associated DNA in the Cytoplasm of Diploid Human Lymphocytes. Proc. Natl. Acad. Sci.USA 1971, 68, 1212–1216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, N.; Fan, X.; Zaleta-Rivera, K.; Nguyen, T.C.; Zhou, J.; Luo, Y.; Gao, J.; Fang, R.H.; Yan, Z.; Chen, Z.B.; et al. Natural display of nuclear-encoded RNA on the cell surface and its impact on cell interaction. Genome Biol. 2020, 21, 225. [Google Scholar] [CrossRef] [PubMed]
- Huleihel, L.; Scarritt, M.E.; Badylak, S.F. The Influence of Extracellular RNA on Cell Behavior in Health, Disease, and Regeneration. Curr. Pathobiol. Rep. 2017, 5, 13–22. [Google Scholar] [CrossRef] [Green Version]
- Cheng, J.; Torkamani, A.; Peng, Y.; Jones, T.M.; Lerner, R.A. Plasma membrane associated transcription of cytoplasmic DNA. Proc. Natl. Acad. Sci.USA 2012, 109, 10827–10831. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fritz, J.V.; Heintz-Buschart, A.; Ghosal, A.; Wampach, L.; Etheridge, A.; Galas, D.; Wilmes, P. Sources and Functions of Extracellular Small RNAs in Human Circulation. Annu. Rev. Nutr. 2016, 36, 301–336. [Google Scholar] [CrossRef] [Green Version]
- Tsatsaronis, J.A.; Franch-Arroyo, S.; Resch, U.; Charpentier, E. Extracellular Vesicle RNA: A Universal Mediator of Micro-bial Communication? Trends Microbiol. 2018, 26, 401–410. [Google Scholar] [CrossRef]
- Fűri, I.; Kalmár, A.; Wichmann, B.; Spisák, S.; Scholler, A.; Bartak, B.; Tulassay, Z.; Molnar, B. Cell Free DNA of Tumor Origin Induces a ‘Metastatic’ Expression Profile in HT-29 Cancer Cell Line. PLoS ONE 2015, 10, e0131699. [Google Scholar] [CrossRef]
- Rayes, R.F.; Vourtzoumis, P.; Rjeily, M.B.; Seth, R.; Bourdeau, F.; Giannias, B.; Berube, J.; Huang, Y.H.; Rousseau, S.; Camilleri-Broet, S.; et al. Neutrophil Extracellular Trap–Associated CEACAM1 as a Putative Therapeutic Target to Prevent Metastat-ic Progression of Colon Carcinoma. J. Immunol. 2020, 204, 2285–2294. [Google Scholar] [CrossRef]
- Zhang, H.; Wang, Y.; Onuma, A.; He, J.; Wang, H.; Xia, Y.; Lal, R.; Cheng, X.; Kasumova, G.; Hu, Z.; et al. Neutrophils Extracellular Traps Inhibition Improves PD-1 Blockade Immunotherapy in Colorectal Cancer. Cancers 2021, 13, 5333. [Google Scholar] [CrossRef] [PubMed]
- Cools-Lartigue, J.; Spicer, J.; McDonald, B.; Gowing, S.; Chow, S.; Giannias, B.; Bourdeau, F.; Kubes, P.; Ferri, L. Neutrophil extracellular traps sequester circulating tumor cells and promote metastasis. J. Clin. Investig. 2013, 123, 3446–3458. [Google Scholar] [CrossRef]
- Joyce, G.F. RNA evolution and the origins of life. Nature 1989, 338, 217–224. [Google Scholar] [CrossRef] [PubMed]
- Buzzo, J.R.; Devaraj, A.; Gloag, E.S.; Jurcisek, J.A.; Robledo-Avila, F.; Kesler, T.; Wilbanks, K.; Mashburn-Warren, L.; Balu, S.; Wickham, J.; et al. Z-form extracellular DNA is a structural component of the bacterial biofilm matrix. Cell 2021, 184, 5740–5758. [Google Scholar] [CrossRef] [PubMed]
- Bacolla, A.; Wang, G.; Vasquez, K.M. New Perspectives on DNA and RNA Triplexes as Effectors of Biological Activity. PLoS Genet. 2015, 11, e1005696. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herbert, A.; Karapetyan, S.; Poptsova, M.; Vasquez, K.; Vicens, Q.; Vögeli, B. Special Issue: A, B and Z: The Structure, Function and Genetics of Z-DNA and Z-RNA. Int. J. Mol. Sci. 2021, 22, 7686. [Google Scholar] [CrossRef] [PubMed]
- Uzzell, T.; Spolsky, C. Mitochondria and plastids as endosymbionts: A revival of special creation? Am. Sci. 1974, 62, 334–343. [Google Scholar] [PubMed]
















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name of the Receptor | Description of the Receptor |
|---|---|
| Primary TezRs | |
| TezR–D1 | DNA-based receptors located outside the membrane; stably associated with the cell surface. |
| TezR–R1 | RNA-based receptors located outside the membrane; stably associated with the cell surface. |
| Secondary TezRs | |
| TezR–D2 | DNA-based receptors located outside the membrane; can be easily washed out along with culture medium or matrix. |
| TezR–R2 | RNA-based receptors located outside the membrane; can be easily washed out along with culture medium or matrix. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tetz, V.; Tetz, G. Novel Cell Receptor System of Eukaryotes Formed by Previously Unknown Nucleic Acid-Based Receptors. Receptors 2022, 1, 13-53. https://doi.org/10.3390/receptors1010003
Tetz V, Tetz G. Novel Cell Receptor System of Eukaryotes Formed by Previously Unknown Nucleic Acid-Based Receptors. Receptors. 2022; 1(1):13-53. https://doi.org/10.3390/receptors1010003
Chicago/Turabian StyleTetz, Victor, and George Tetz. 2022. "Novel Cell Receptor System of Eukaryotes Formed by Previously Unknown Nucleic Acid-Based Receptors" Receptors 1, no. 1: 13-53. https://doi.org/10.3390/receptors1010003