

Lobe X of the Cerebellum: A Natural Neuro-Resistant Region

1
Laboratory of Neuronal Plasticity and Neurorepair, Institute for Neuroscience of Castilla y León (INCyL), Universidad de Salamanca, 37007 Salamanca, Spain
2
Institute of Biomedical Research of Salamanca, IBSAL, 37007 Salamanca, Spain
*
Author to whom correspondence should be addressed.
Anatomia 2023, 2(1), 43-62; https://doi.org/10.3390/anatomia2010005
Submission received: 28 November 2022
/
Revised: 23 December 2022
/
Accepted: 19 January 2023
/
Published: 23 January 2023
(This article belongs to the Special Issue State-of-the-Art Anatomical Research in the Mediterranean Region 2022)
Abstract
:The cerebellum is an encephalic region classically known for its central role in the control of movement, although recent research has revealed its involvement in other cognitive and affective tasks. Several different pathologies are known to affect this structure, causing a wide range of behavioral and gait impairments. Intriguingly, although the neurodegenerative factors affect all Purkinje cells of the cerebellum uniformly, certain neurodegeneration patterns can be distinguished, in which some Purkinje cells persist longer than other cell types. Specifically, there is a cerebellar region, lobe X, which is more resistant to different types of neurodegeneration, regardless of the injury. Degeneration patterns of the cerebellum have been described in several models, but this review goes further, as it aims at describing a phenomenon not so described: the resistance of the lobe X to neurodegeneration. For this purpose, the main models of cerebellar degeneration will be reviewed and a common origin for the lobe X resistance will be sought.
1. Introduction. The Cerebellum
Cerebellum is a Latin word that means “small brain”. This structure was initially considered to be a less significant addition to the main brain because this organ only represents 10% of the total weight of the encephalon. Its side location to different nervous centers and pathways led to the traditional belief that its role was solely focused on the coordination and refinement of motor control [1]; however, this classical concept has changed in recent decades. The cerebellum is currently considered one of the most important encephalic structures and its involvement in several cognitive and affective tasks is widely accepted.
The cerebella in mammals and birds, the most developed among vertebrates, have several structures that can be distinguished macroscopically (Figure 1). At a first glance, a narrow and long structure called the vermis can be observed in the most central area, and throughout the entire cerebellar sagittal plane. The cerebellar hemispheres are located on the sides of the vermis. Two small structures called the flocculus and the paraflocculus protrude from the hemispheres, and lastly, the entire cerebellar structure is attached to the brain via the cerebellar peduncles [2].
The outermost part of the cerebellum is called the cerebellar cortex, which is extensive but small, comprising numerous convolutions or folia, which, unlike other structures of the central nervous system, cross the midline completely and are perpendicular to the sagittal plane [4]. These folds of the cerebellar cortex are grouped into lobes separated by fissures. In birds and mammals, up to 10 lobes can be distinguished in the most central part (i.e., vermis; Figure 1), which are numbered rostrocaudally with Roman numerals from I to X [5].
This arrangement in the lobes allows the different cerebellar regions to be distinguished. The most primitive of all is the archicerebellum or vestibulocerebellum, associated with the flocculonodular area (lobe X and cerebellar floccules and parafloccules; Figure 1), which is equivalent to the folia that only appears in the most primitive vertebrates. This structure receives vestibular and visual inputs and sends projections to the vestibular nuclei, which are related to balance, vestibular reflexes, and eye movements. The second region is the paleocerebellum or spinocerebellum, comprised of the vermis (lobes I–IX) and part of the hemispheres (Figure 1). The term spinocerebellum was chosen because it receives somatosensory and proprioceptive inputs of spinal origin, and projects these inputs towards descending pathways controlling the axial muscles. Finally, the phylogenetically youngest part of the cerebellum is the neocerebellum or cerebrocerebellum, which is the most evolved in primates, and corresponds to the large lateral hemispheres of the cerebellum (Figure 1). Its main inputs come from the cerebral cortex, and its outputs also return to the motor, premotor, and prefrontal areas of the cortex. The best-known of the neocerebellar functions is the planning and execution of fine and precise movements [6].
Although the role of the cerebellum is linked to motor control, it has recently been accepted that this organ could be associated with cognitive functions and emotional control [7]. The clues suggesting the involvement of these new functions began with electrophysiology experiments, as the activation of the fastigial nucleus activated neurons of the hippocampus and vice versa [8]. Later on, using new imaging techniques, it was possible to verify the activation of different cerebellar regions during different affective/cognitive tasks [9]. Moreover, similar to how damage in the cerebellum may cause motor impairments, the existence of non-motor cerebellar disorders and pathologies have been confirmed, as in the case of autism and schizophrenia.
1.1. Cell Types and Cerebellar Pathways
The cerebellar cortex comprises an exquisite cellular organization, as its neurons present an arrangement and connectivity that is extremely well conserved from the most primitive vertebrates, with little variation (Figure 2). The cortex is divided into 3 layers, from the innermost to the outermost part, called the granular layer, the Purkinje cell layer, and the molecular layer (Figure 2).
The granular layer is so named because it is made up of a large number of cells. The small granule cells contain a nucleus that stains intensely and constitute the most abundant neuronal cell type in the brain. In this layer, other cell types can also be found, such as Golgi cells (interneurons larger than granule cells), Lugaro cells, and brush cells [10]. Curiously, the latter cell type is only found in lobes I, IX, and X in mammals [11].
The Purkinje cell layer, as its name suggests, is made up of Purkinje cell somata. Purkinje cell axons contact deep cerebellar nuclei passing through the granular layer and their dendritic trees extend throughout the molecular layer. Purkinje cells are one of the largest cell types in the brain and are the only efferent neurons of the cerebellar cortex. In this layer, chandelier cells [12] and the somata of the Bergmann glia [2] can also be found. The latter cell type is related to radial glia and is responsible for guiding Purkinje and granule cells to their final location during embryonic development [13].
The outermost stratum of the cerebellar cortex is the 500-µm thick molecular layer that contains the dendritic arborizations of the Purkinje cells oriented in a sagittal plane, although the cells are narrow considering their transverse axis. In addition to these huge dendrites, other interneurons such as the stellate cells are located here, towards the surface, as well as basket cells located toward the inside of the cerebellum. The repetitive structure of the cerebellar cortex is reinforced because the dendrites of both basket and stellate cells share the orientation of the Purkinje cell dendritic arbor [2]. Lastly, parallel fibers, the axons of granule cells that branch out and make contact with the dendrites of Purkinje cells, are also located in the molecular layer [3].
The connectivity of the cerebellum has also been characterized in detail (Figure 3) [14]. As previously shown, afferent information can originate from the spinal cord, the brainstem, and even from the cerebral cortex [15]. This information arrives mainly via the middle peduncle as mossy or climbing fibers. Additionally, the mossy fibers, originating from different regions, arrive at the granular layer, forming structures called cerebellar glomeruli. In these formations, the mossy fibers make excitatory synapses on dendrites of granule cells in a ratio of 1 fiber to 400–600 granule cells. In these same structures, Golgi cell axons make inhibitory synapses on the dendrites of granule cells [16]. The climbing fibers project directly to the molecular layer and contact dendrites of the Purkinje cell in a 1:1 ratio, and both mossy and climbing fibers send collaterals to neurons of the deep cerebellar nuclei [2].
The granule cells, on the other hand, emit their axons into the molecular layer where they branch and give rise to parallel fibers, which extend several millimeters and contact hundreds of Purkinje cells [3]. In turn, stellate and basket cells contact the dendrites and the soma of Purkinje cells, respectively [17].
Finally, the only efferent fibers of the cerebellar cortex are Purkinje cell axons. Thus, all the information processed here leaves these fibers and reaches the deep cerebellar nuclei, from where it travels to the different motor and cognitive centers in the brainstem and cerebral cortex [10].
1.2. Regions of the Cerebellar Cortex
The cerebellum has a structure that has been highly conserved throughout evolution and a histology that appears uniform throughout its different lobes [18]. However, beyond this uniformity, there is some heterogeneity in the form of parasagittal bands [2,19,20]. These bands can be distinguished by using certain immunocytochemical markers such as Zebrin II, named after its striped pattern in terms of its distribution across the cerebellar cortex [21]. As a result of these banding patterns, the four aforementioned transversal regions can be distinguished (Figure 1): anterior (lobes I–V), central (VI–VII), posterior (VIII–IX), and nodular (X). These banding patterns consist of clusters of Purkinje cells that are highly immunoreactive to Zebrin II and areas with little or no immunoreactivity. The anterior zone has hardly any cells that are strongly positive for Zebrin II. However, half of the Purkinje cells in the posterior zone are usually strongly labeled with Zebrin II, and the other half are not. Lastly, all Purkinje cells in the central and nodular areas are highly positive for this protein (Figure 4) [22]. This cerebellar topography appears in prenatal development and remains stable during postnatal growth [23,24]. Finally, similar compartmentalization exists when using other markers for granule cells [25,26], interneurons [27,28], mossy fibers [29], and climbing fibers [30].
In addition to Zebrin II in mice, other parasagittal banding patterns have been generated using other markers, and in several different species. For this reason, in 2011, a study was carried out on the colocalization of some of the most used markers in more than 20 different species of mammals and birds [31]. In particular, this analysis was carried out on the postero-nodular area of the cerebellum, because the largest number of Zebrin II-positive cells is known to exist in this area. Through this work, it was concluded that this banding pattern is highly conserved in mammals and in some birds. Concerning the other antigens studied, it was confirmed that the parasagittal bands revealed by the other markers were sometimes related with the Zebrin II-positive bands. However, on other occasions, their labeling appeared in those bands that did not express Zebrin II intensely. Finally, other markers formed mixed patterns in which they sometimes coincided, or did not, with Zebrin II expression (i.e., HSP25, as will be discussed below; Figure 4).
We have highlighted the heterogeneity of the cerebellar cortex, classically considered to be extremely uniform, due to the different regions in which it can be divided, which will help us to ultimately distinguish other biological functions and characteristics. It can be anticipated that some of these additional expression patterns are also found by analyzing heat shock proteins (HSPs). More importantly, related to the objectives of this work, these regions also show a selective vulnerability to neurodegeneration.
2. Objective
The cerebellar cortex is a fascinating region whose degeneration has generated even more interest in the scientific community. Neurodegeneration patterns in the cerebellum are widely described and, moreover, several reviews about such patterns have also been published. However, under these patterns of neuronal death, there is few information about a striking phenomenon: the resistance to neurodegeneration of the cerebellar lobe X.
Several models of cerebellar neurodegeneration will be described here, as well as their own patterns of neuronal loss. This review aims at demonstrating how the lobe X always presents a higher resistance to neuronal death. Apart from the description of this resistance, this review also aims to reveal a common cause for it, and enlighten the reader on why lobe X is more protected against different types of damage.
3. Models of Cerebellar Degeneration
The damaging factor causing different neurodegenerative diseases is usually uniform throughout the central nervous system or in one of its regions and can be either a mutation, a toxin, or the effects of aging, among others. However, some neuronal populations rapidly degenerate in the face of these factors, while others are less vulnerable and retain their functions [34]. In relation to this selective vulnerability, a given tissue may be highly susceptible to certain neurodegenerative factors while not being susceptible to others. However, as we will see below, there are regions of the central nervous system that show a lower constant vulnerability to all the neurodegenerative factors mentioned; that is why we have called this phenomenon neuroresistance.
Specifically, a very striking example of neuroresistance can be found in the cerebellum [35]. Despite the similarity of all Purkinje cells and the repetitive structures in their connectivity, neurodegeneration does not occur uniformly throughout the cerebellar cortex, giving rise to patterns of neuronal death that are not specific to a particular pathology, but common among a wide range of pathologies [28]. Previously, we discussed the existence of different regions exhibiting different neurochemical phenotypes, such as those positive or negative for Zebrin II staining. Furthermore, we mentioned that these regions were related to selective vulnerability to neurodegeneration. Hence, a common pattern of neurodegeneration exists depending on the cerebellar region: the anterior area is the most sensitive and generally the first to degenerate; an intermediate susceptibility can be found in the central and posterior areas; and, lastly, the greatest resistance appears in the nodular region, mainly comprising lobe X. The diseases that show this pattern of anteroposterior vulnerability are extremely varied. Some are derived from mutations, such as spinocerebellar ataxia [36], saposin C deficiency (one of the causes of Gaucher disease) [37], ataxia telangiectasia [38], Niemann-Pick A/B and C disease [22,39], and multiple system atrophy [40]. Others may be due to toxins, such as alcohol [41], hypoxia-ischemia [42], or even normal aging [43]. For some of these diseases, there are animal models that reproduce them, such as Niemann-Pick C1 (NPC1) disease, which shows the same pattern of neurodegeneration as in humans. In addition to these pathologies, this selective vulnerability has also been found in specific rodent models such as the Leaner mouse [44], the Toppler mouse [45], the Robotic mouse [46], the shaker rat [47], the Lurcher mouse [48], and the PCD (Purkinje cell degeneration) mouse [49,50].
As we are unable to cover all animal models with cerebellar damage, we will only refer to the genetic models for which the neurodegeneration patterns have been clearly described.
3.1. Tottering, Leaner, and Nagoya Models
The Tottering mutant mouse presents a pathology caused by a spontaneous mutation in the α1A subunit of a Ca2+ channel. The mutation tg is recessive and there are two variants of it, which define two additional models homologous to the previous one: the tgla variant corresponding to the Leaner mutant and the tgrol that has given rise to the Nagoya mouse model [51]. These three models are slightly different and overlap in some features, and although the original mutant, the Tottering mouse, is a model of absence epilepsy, they all suffer from ataxia. They differ in the severity of the symptoms of ataxia, with the mildest occurring in the Tottering mouse, followed by the Nagoya mouse, and, finally, the most severe symptoms appearing in the Leaner mouse [52]. Although the integrity of Purkinje cells is altered, the Nagoya mouse does not present quantitative changes in Purkinje cell numbers [53]. For this reason and because it presents the most aggressive symptomatology of the tg mutation, the Leaner mouse will be described in more detail.
Like its homolog, the Leaner mutant mouse presents a pathology due to a spontaneous mutation in the α1A subunit of a Ca2+ channel. The tgla mutation is recessive and is caused by a base substitution that disrupts the transcription of the open reading frame, resulting in a truncated and nonfunctional protein [54]. The affected subunit is mainly expressed in Purkinje cells, and this mutation is the genetic basis of the Leaner mice, which suffer from ataxia. In addition, this same defect also exists in humans and is known to cause hemiplegic migraines, episodic ataxia type 2, and spinocerebellar ataxia type 6 [55].
Leaner mice present cerebellar ataxia as early as postnatal day 10 (P10). Morphological analyses of the cerebellum of Leaner mice showed that the degeneration affects the Purkinje cells as well as granule and Golgi cells, especially in the anterior region. The onset of ataxia, at P10, coincides temporally with the degeneration of granule cells, although Purkinje cell loss begins at P40 [52]. Also, a banded expression pattern of a vitamin D-dependent calcium-binding protein has been verified in this model: the anterior zones present a lower expression of this protein, while the nodular zone has the highest expression of the entire cerebellar cortex [44]. This is a pattern comparable with that of Zebrin II, which coincides with a lower vulnerability to neurodegeneration in the posterior and nodular zones, compared with the more anterior regions.
Regarding the Tottering mouse, it is worth highlighting that its Purkinje cell degeneration is observed in Zebrin II negative cells in the anterior regions. By contrast, in the posterior region, the degenerating Purkinje cells are Zebrin II positive [56]. This fact demonstrates that Zebrin II does not present full neuroprotective properties, as will be discussed later.
Finally, although the three aforementioned models suffer a mutation in the α1A subunit of a Ca2+ channel, they are not unique. Another model that suffers a mutation of this gene is the Pogo mouse. Like the previous models, this mutant suffers Purkinje cell death and, once more, cell loss is more severe in the anterior lobes than in the posterior lobes [57].
3.2. Toppler Model
The discovery of the Toppler mouse is relatively recent compared to other cerebellar degeneration mutants. Hence, this model was described for the first time in 2004 as a result of a spontaneous mutation in the FVB (Friend leukemia virus B) strain of mice [45]. FVB mice had been bred for many generations without abnormalities, but suddenly four pups of the same litter began to show ataxia and abnormal posture at 4–5 weeks of age. These offspring were used as parents for other crosses, revealing that their pathology was hereditary. As their impairments were motor and postural, a cerebellar or demyelinating origin was suspected. Later, histological studies revealed an evident loss of Purkinje cells [45]. Surprisingly, at P30, all the Purkinje cells of lobe X survived (Figure 5A), although their morphology was abnormal and similar to that of neurons in the regions most affected by the neurodegeneration [45].
3.3. Robotic Model
The origin of the Robotic mouse is particular, as it is an autosomal dominant mutant model that is spontaneous in origin, but induced. Like the other models described here, the Robotic mouse has an ataxic gait and shows a loss of Purkinje cells in early adulthood, and also develops cataracts [46].
Mutant mice are a useful tool for understanding the functions of different genes in mammals, and one way to develop these mice is by using a mutagen. In the case of the Robotic model, male C3Heb/Fej mice were injected with the mutagen N-ethyl-N-nitrosourea and were crossed with females of the same strain. The resulting mice were tested for dominant or codominant pathologies and crossed with each other to discover additional recessive traits [60]. This experiment gave rise to the Robotic mouse and, after carrying out genetic mapping, it was found that its pathology was due to a nonsense mutation, a base substitution that results in a stop codon, in the Af4 gene [46]. The involvement of Af4 is an additional unique feature of this model, as it is a gene involved in leukemia, and, although the relationship between the gene and the disease is not fully understood, Af4 knock-out mice present an abnormal development of B and T lymphocytes [61]. In terms of gene function, the resulting protein belongs to a family of transcription cofactors that are usually translocated in childhood leukemia [62].
To study the relationship between Af4 and neurodegeneration, several studies were carried out to show that Af4 was expressed mainly in Purkinje cells. Its truncated form in Robotic mice accumulates and gives rise to neurodegeneration. In addition, to study how neuronal loss progressed in Robotic mice using immunohistochemical techniques, calbindin expression was analyzed in coronal and sagittal slices of the cerebellum. Once again, a pattern of bands was observed, as in the case of Zebrin II and/or other calcium-binding proteins in other models. Moreover, an anteroposterior progression of neurodegeneration could be observed in the sagittal planes, with the anterior regions degenerating much earlier than the nodular and posterior areas (Figure 5B).
3.4. Shaker Model
This is a rat model rather than a mouse model. The Shaker rat was first described in 1992 as a hereditary model that is not sex-linked and develops severe ataxia with age. After studying the cause of this disorder, an absence of Purkinje cells was found, especially in the anterior lobes, which were 52% smaller in Shaker rats than in the controls [63].
A few years after the first publication, using the 5th and 6th generation of the original rats, two parallel research papers were published: in one of them, behavioral studies were performed [64] and in the other, sections of the cerebellum were marked with calbindin by immunoprecipitation [47]. In the first paper, two variants in the pathology of the shaker rat were found, termed mild or strong. The mild variant corresponded to 77% of the total number of rats, being those that only presented ataxia. The strong variant constituted the remaining 23%, suffering from both ataxia and whole-body tremors at 3 months of age. In addition, rats with mild tremors never developed the tremor characteristic of rats with severe tremors [64]. The second article analyzed the distribution of Purkinje cells at different ages and confirmed at 11 months of age what we have already described in previous models: an anteroposterior loss of Purkinje cells in lobes I to IX, however, lobe X remained undamaged, and the authors literally said, “In lobule X, the distribution of Purkinje Cells appeared very similar to that seen in normal rats” (Figure 5C) [47].
3.5. Lurcher Model
The Lurcher model presents a cerebellar degeneration caused by a mutation that was first described in 1960 [65]. Heterozygous Lurcher (+/Lc) mutants suffer the death of all Purkinje cells, among other cell types, from P10 to P65, resulting in the loss of cerebellar function. The homozygous Lurcher mutation is lethal; the embryos appear to develop normally, however, the pups die within a few days of birth as they are unable to suckle [66].
This murine model has a base-shift mutation in the δ2 glutamate receptor gene, which causes the receptor to behave like a small Ca2+ channel, a function hypothetically lost during evolution [67]. The possible mechanisms of Purkinje cell death that have been described are varied: necrotic, autophagic, apoptotic, and excitotoxicity due to high levels of glutamate derived from high levels of Ca2+ [67]. However, the exact cause of this neuronal loss is not known [68].
What is known, however, is the degenerative process it undergoes. Neurodegeneration in the Lurcher mouse begins around the second postnatal week in the entire cerebellum. However, at P25-P36 this cell death is accelerated in the anterior zone (lobes I to V), becoming evident later in the central and posterior zones (lobes VI to VII and VIII to IX, respectively). Finally, the neurodegeneration of the nodular area (lobe X) is somewhat delayed with respect to the rest of the regions (Figure 5D). In addition, and parallel to this selective neurodegeneration, the existence of parasagittal expression bands of the heat shock protein HSP25 in surviving Purkinje cells has been described [58].
3.6. NPC1 Model
NPC1 disease is an inherited recessive lipid storage disorder caused by a defect in intracellular cholesterol transport and homeostasis [69]. In humans, the disease causes hepatosplenomegaly at birth, and children with the disease develop ataxia, psychomotor impairments, and/or dementia, dying at 5–15 years of age [70]. Fortunately, for the study of this pathology, there are two murine models generated by a spontaneous mutation in the same gene as in humans, the Npc1 gene. Furthermore, the murine mutated gene corresponds to the same complementary group as the human NPC1 gene [71].
NPC1 mouse pups are indistinguishable from their wild-type littermates. However, as they reach adulthood, they begin to develop ataxia, with Purkinje cell degeneration evident from P40. However, like in the other models, neurodegeneration is not uniform throughout the cerebellum: the more rostral areas of the vermis are more sensitive to neurodegeneration than the rostral areas, with the X lobe being the most resistant region of all (Figure 5E) [22].
As mentioned above, the nodular zone has a substantial expression of Zebrin II, and different patterns of degeneration can be defined accordingly. In the case of the NPC1 model, when analyzing the degeneration of Purkinje cells in coronal sections, it was observed that the first neurons to die are those that do not express Zebrin II [33]. In addition, a recent bioinformatic study cross-checked available data from other in situ hybridization experiments to find genes that were more highly expressed in the nodular zone, and therefore could influence the increased neuroresistance of this region [28]. The result of this research showed that the expression of the heat shock protein HSP25 (referred to as HSPB1 in this review) was higher in lobe X than in the rest of the lobes of NPC1 mice. Moreover, the phosphorylation of HSP25 significantly increased its neuroprotective properties [28].
3.7. Nervous Model
The Nervous mouse is a model that suffers from Purkinje cell degeneration due to an autosomal recessive mutation [72]. Its degeneration starts rapidly and then slows down after two months [72]. The Nervous mutation is known to be located on chromosome 8 [73], but the specific gene altered is still unknown. When this model was first studied, the membrane-bound protein P400 was found to be absent in nerve cells [74], and it was then suspected that the Nervous mutation was related to this protein. However, as P400 is found in dendrites and the cell bodies of Purkinje cells [75], most of the cerebellar degeneration models show low levels of this protein [75], as well as in the Nervous model.
Although this mouse model undergoes severe neurodegeneration, about 10% of its Purkinje cells remain alive after the acute neurodegenerative phase, except for some sporadic death [76]. Again, the distribution of resistant Purkinje cells is not random: in the hemispheres, 90% of Purkinje cells die, but in the vermis, only 50% are lost [72]. Furthermore, apart from this distribution, the most ventral part of the vermis is an additional area of surviving Purkinje cells, this region being composed of the lobe I, lobe X, and the ventral part of the lobe IX [77]. These results are consistent with a later study on the compartmentalization of Purkinje cell death and Zebrin II expression [78]. Zebrin II was found to be expressed in the most vulnerable Purkinje cells in Nervous mice [78]. This explained why lobe I was less vulnerable to neurodegeneration, due to its null expression of Zebrin II. Furthermore, in this case, the posterior lobes would theoretically be more susceptible to neurodegeneration due to their higher expression of Zebrin II. Despite this presumed vulnerability, Purkinje cells of lobe X do not degenerate as expected, thus supporting an additional source of resistance of this region apart from the distribution of Zebrin II (see below).
3.8. PCD Model
This is a model of cerebellar degeneration caused by an autosomal mutation in the Ccp1 gene. Recent research shows that this genetic defect is also present in humans, causing childhood-onset neurodegeneration with cerebellar atrophy (CONDCA), a disease with the same pathogenic symptoms as the animal model [79,80,81].
The pcd/pcd pups are somewhat smaller than their wild-type littermates, although they show no other anomalous signs until P20, when they begin to develop ataxia due to progressive Purkinje cell death [82]. The PCD model also suffers from the death of other neuronal types such as some thalamic populations [83], retinal photoreceptors [73], and the mitral cells of the olfactory bulb [84]. In addition, the spermatozoa of PCD males have an abnormal morphology that causes sterility. Females, on the other hand, are fertile, but unable to adequately care for their offspring [49]. It is also worth noting that, although cerebellar neurodegeneration is evident in PCD mice from P20 onwards, two stages of neuronal damage in the cerebellum have been shown to exist: the pre-neurodegenerative and the degenerative stage [85]. The first stage starts at P15 and ends at P18 and is characterized by nuclear, cytological, and morphological alterations in the still-living Purkinje cells [86]. Then, with the onset of death of these neurons, the degenerative stage is considered to begin [85].
The pcd mutation arose spontaneously in the C57BR/cdJ strain and affects a regulatory region of the Ccp1 gene, located on chromosome 13 [82,87], leading to the almost complete disappearance of its transcription [87,88]. The protein affected by this mutation is called “ATP/GTP-binding protein 1” or “axotomy-induced nervous system nuclear protein 1” (AGTPB1 or NNA1, respectively). The second name is due to the origin of its discovery, as it was found to be expressed after an axotomy of the sciatic nerve, and its expression was associated with axonal differentiation and regeneration processes [89]. Considering its function, this enzyme is a peptidase capable of hydrolyzing the carboxy-terminal ends of the glutamate chains of α and β tubulins, and because of this, it belongs to the family of cytosolic carboxypeptidases. Thus, its most frequent name is “cytosolic carboxypeptidase type 1” or CCP1. This enzyme regulates the glutamylation of tubulins and if it fails, microtubules become hyper-glutamylated, thus causing an excessive instability of the cytoskeleton, and ultimately Purkinje cell death [85,90].
Once again, in the PCD mouse, a pattern of selective neuronal degeneration is observed, such that lobe X of the cerebellum emerges as a neuroprotected region. In this sense, at P30, when the rest of the Purkinje cells have almost disappeared, lobe X remains virtually intact [91]. Furthermore, at 9 months of age, long after the end of degeneration, some Purkinje cells can still be detected in this lobe [82].
3.9. Tambaleante Model
The Tambaleante model is another mouse that suffers a recessive mutation (see below) [77]. This mouse differs from the previous models in that it does not show a clear pattern of neurodegeneration. In this case, the death of Purkinje cells occurs randomly, with some groups of these neurons remaining alive in bands at 2.5 months of age [77]. Although the resistant properties of lobe X of the Tambaleante model are not as marked as in the previous models, the surviving Purkinje cells of this lobe remain alive for a few days longer than in the rest of the cerebellar cortex.
As for the mutation, it is located on chromosome 9 and consists of an adenosine-to-guanine transition, resulting in a glycine-to-glutamate substitution that alters the function of the E3 ubiquitin ligase protein HERC1 [92]. The mutation causes overexpression of the altered protein, the accumulation of which leads to Purkinje cell death [92]. The E3 ubiquitin ligase HERC1 belongs to the ubiquitin-proteasome system, which plays a role in protein degradation, a key component in neuronal homeostasis [93].
Table 1 collects most relevant data from the different models previously described.
3.10. Other Non-Genetic Models
In addition to the aforementioned mutants, other non-genetic models also show antero-posterior neurodegeneration, with lobe X being the most resistant region of the cerebellum. The list of these examples would be excessively long, but it is worth mentioning that this resistance has been reported with toxins, some of which have the same effects in humans. For example, alcoholics have reduced Purkinje cell populations in the superior and middle regions of the cerebellum [41]. Cytosine arabinoside is another example: high doses of this drug were given to patients with leukemia or lymphoma and four of them developed cerebellar degeneration. Postmortem analyses revealed that Purkinje cells were relatively preserved in the posterior lower portions of the cerebellum [94]. Methotrexate is another chemotherapeutic drug that was administered to patients with acute lymphoblastic leukemia. Survivors of the disease showed hypoplasia, caused by the drug, of the cerebellar vermis of lobes I-VII, with lobes VI and VII being the most affected. Although no specific data on lobe X degeneration were shown, the most caudal part of the cerebellum remained undamaged [95]. Another well-known chemical to induce ataxia is 3-acetyl pyridine (3-AP). An intraperitoneal injection of this substance can cause signs of cerebellar ataxia in just 24 h [96], related to damage within the inferior olive [97]. However, there is no information about its effects over the nodular zone of the cerebellum.
In addition to toxicants, another example of resistance to neurodegeneration in the posterior lobes is observed in hypoxia-ischemia models: rat pups subjected to hypoxia-ischemia developed cerebellar injuries. This degeneration was studied in two groups: lobes III–IV and lobes VIII–IX. Again, the posterior lobes were found to suffer less than the anterior lobes [42], although lobe X was not explicitly studied. Finally, the cerebella of 19 Caucasian males (19–84 years old) were studied, and the anterior lobes were found to be the most affected by aging [43].
4. Possible Causes of the Neuroresistance of Lobe X
It has been shown in these and other models of cerebellar degeneration that Purkinje cell death does not occur uniformly. Indeed, there are well-established patterns of degeneration in the cerebellum, and there are almost no examples in which Purkinje cells die randomly. Specifically, the banded patterns of neurodegeneration were first clearly described in the PCD model, together with Nervous and Tambaleante mice [77]. Moreover, several patterns of Purkinje cell vulnerability have been shown [35], with the relationship between Zebrin II expression patterns and neurodegeneration being one of the most striking examples. However, this relationship is not rigorously consistent with a putative neuroprotective function for Zebrin II. Thus, in some cases, it is true that Purkinje cells that do not express Zebrin II are more sensitive to neuronal degeneration, as in the Tambaleante mouse [51] or the anterior region of the Tottering mouse [56]. By contrast, there are other examples where Purkinje cells expressing Zebrin II are more vulnerable to cell death, as in the Nervous mouse, described above [77] or the posterior region of the Tottering mouse [56]. In this sense, it seems that the presence of Zebrin II can only inform us about the specific pathways of degeneration that converge (or not) in Purkinje cell death, and not inform us on the presence of this protein as a strict marker of resistance [35], as traditionally thought. This variable pattern of degeneration related to Zebrin II expression may be caused by a specific stress that acts uniformly on all Purkinje cells of the cerebellum, and depending on the nature of this weakening, some neurons are more sensitive than others to neuronal degeneration. In this case, the presence or absence of Zebrin II could be beneficial or detrimental. A clear example of uniform stress throughout the cerebellum, but with heterogeneous neurodegeneration, is found in the Tambaleante mouse. This model suffers from a mutation in a gene encoding a calcium channel [51]. Several studies have shown that all Purkinje cells express similar levels of mRNA of this mutated gene, but not all of them suffer the same neuronal degeneration, and in this case, only those that do not express Zebrin II die [51]. This also agrees with the case of the NPC1 mouse, in which the neurodegenerative factor, the accumulation of cholesterol vesicles, is uniform in all Purkinje cells, and the first to degenerate are negative to Zebrin II. Furthermore, Purkinje cells positive for Zebrin II that degenerate are accompanied by ectopic expression of tyrosine hydroxylase [22], a fact that could be relevant later in this discussion. In these two cases, Zebrin II-positive Purkinje cells are less vulnerable to neurodegeneration, which may lead us to consider Zebrin II as a neuroprotective factor, especially considering that the Purkinje cells of lobe X express it abundantly [35]. Nevertheless, in the Nervous mouse Zebrin, II-positive Purkinje cells are more susceptible to neuronal degeneration [77], which rules out possible neuroprotective effects of this protein, at least in general. In this sense, it would be reasonable to think that all the Purkinje cells in lobe X of the Nervous mouse would die, since they all express Zebrin II. However, there are some exceptions in areas with Zebrin II-positive cells that do not degenerate, specifically in lobes IX and X [78], and to a lesser extent in the floccules and parafloccules and in lobe VI. Therefore, as previously mentioned, Zebrin II expression bands define different Purkinje cell populations that are different from others, showing different susceptibilities to different neurodegenerative processes and types of cellular stress, but not a strict predisposition toward survival or cell death.
By contrast, the nodular zone always shows more resistant Purkinje cells regardless of Zebrin II-related patterns and sensitivity (see above for the case of Nervous mice). Therefore, it cannot be said that the neurons of this region are more susceptible to some impairments but not to others: lobe X is consistently more resistant, regardless of the neurodegenerative factor. Hence, factors in addition to Zebrin II must be present to confer such resistance, and although not fully understood, HSP25 and tyrosine hydroxylase expression is proposed as a key mechanism for neuroprotection.
Indeed, HSP25 expression has been confirmed in the resistant regions of several models of cerebellar degeneration, such as Weaver [98], Lurcher [58], and NPC1 mice [35]. Moreover, Chung et al. [28] confirmed the potent neuroprotective effect of HSP25 when phosphorylated at some of its serine residues. This enzymatic reaction occurs naturally in some neurodegenerative processes, especially in lobe X [28]. The final evidence for the neuroprotective properties of HSP25 is its slight expression in the central zone of the cerebellum, in particular in lobe VI [59]. In this lobe, a certain resistance to Purkinje cell death has been detected in models such as the NPC1 [59]. This low vulnerability is not always detected and is not as evident as that of lobe X. Thus, there is a cause-effect relationship for HSP25 expression levels and neuroprotection. Interestingly, HSP25 has several functions, but as a small heat shock protein, one of them should be acting as a chaperone [99]. We hypothesize that its constitutive expression shown in lobe X [28] might indicate that its functions in this particular lobe are more important than those of the other lobes. This idea will be developed further below. Besides, tyrosine hydroxylase is expressed with HSP25 in degenerating Purkinje cells positive for Zebrin II, the last ones that degenerate. For this reason, the former proteins are associated with surviving cells [22]. Furthermore, HSP25 expression patterns are complementary to ectopic tyrosine hydroxylase expression in the cerebellum of Rolling mice [100].In any case, the underlying cause of the resistance of lobe X may be complex and multifactorial. Therefore, in addition to the differential expression of either Zebrin II, HSP25 or tyrosine hydroxylase, many other peculiarities differentiate lobe X from the rest. In terms of functionality, Purkinje cells in lobe X have been found to exhibit more regular impulse firing and less adaptation to repeated stimuli than lobes III–V [101]. In addition, a transcriptomic study compared mRNA expression between lobes III, VI, and X in both wild-type animals and NPC1 mutants [102]. It was found that between lobes III and VI there were around 180–350 differentially expressed genes. Surprisingly, when comparing lobe III or VI versus lobe X, the difference in expression increased to 1300–1500 genes [102]. This gives us an idea of the myriad of possible factors why lobe X Purkinje cells survive longer. Thus, some of the additional potential reasons responsible for such neuroprotection are increased calcium signaling, increased Sonic Hedgehog signaling, and increased glutamate buffering [102]. Last but not least, when comparing gene expression in NPC1 and wild-type mice, a generalized increase in immune and inflammatory response-related genes was observed, but independently of the lobes [102]. These data, together with other studies, show that it is not the inflammatory response that underlies Purkinje cell resistance patterns, but some of the factors discussed above, which emphasizes the question of why lobe X is so different from the rest, while being more protected.
5. Why Lobe X Is Different from Other Lobes?
We have found that lobe X of the cerebellum is more resistant than the other lobes in numerous animal models suffering Purkinje cell death. As described above, some examples are the PCD [82], Leaner [44], Shaker [47], Robotic [46], Toppler [45], Lurcher [58] and NPC1 [59] models. In addition, in some of these animals, such as NPC1 [22], Weaver [98], and Lurcher [58] mice, an increased expression of HSP25 has been detected in lobe X, which suggests that this protein is a clear candidate responsible for such resistance. However, the million-dollar question remains: Why is lobe X more protected against neurodegeneration than the rest of the lobes?
To try to answer this question, we can start by recalling the abovementioned expression of HSP25, specifically in the surviving Purkinje cells of the NPC1 mouse [35]. The functions of HSP25 in the cerebellum are unclear, but it has been shown in non-neuronal cell lines that this protein acts as a chaperone during heat stress [103] and also regulates the organization of actin filaments during oxidative stress [104,105]. In this sense, to date, the only functions that have been described for HSP25 point to a protective nature, similar to that of the rest of the small HSPs, which are involved in stabilizing other proteins under stress conditions [106,107,108]. It stands to reason, that its expression should be specifically induced by a damage/stress factor. However, expression of HSP25 in lobe X of wild-type mice is evident, unlike in other lobes [31,58,109]. Thus, if the only function of HSP25 were to act as a chaperone against cellular stress, it would not make sense for it to be constitutively expressed in the nodular, or, to a lesser extent, in the central region (mainly in lobes X and VI, respectively), where no neurodegeneration exists. A dichotomy arises here: on the one hand, HSP25 may have other functions not yet described (besides being a chaperone) or, on the other hand, these regions need to be further protected because they may be more important than the rest of the cerebellar cortex.
In the introduction, we mentioned that lobe X, together with the cerebellar flocculi, constitute the flocculonodular zone [6]. This zone is the most primitive region of the cerebellum and corresponds to the only cerebellar folia possessed by the most primitive vertebrates [6]. In addition, the flocculonodular zone receives vestibular and visual inputs, and its outputs are projected to the vestibular nuclei, which defines its functions: it participates in balance, vestibular reflexes, and eye movements [6]. In this sense, it is logical to think that, evolutionarily, the protection of the flocculonodular zone has been more of a priority than that of other cerebellar areas, as it is one of the most primitive regions and is responsible for the most basic functions of the cerebellum. In other words, it is possible that during evolution, the primary functions of maintaining body posture or balance (paleocerebellum) have tended to be more protected than “additional” fine motor skills (cerebrocerebellum).
Moreover, we could also attribute the resistance of lobe X to its simplicity or its different functioning. Phylogenetically, more advanced cerebellar regions perform more complex processing, enabling fine and precise movements to be planned and executed [6]. This processing involves greater neuronal complexity and richness in gene expression, and this requirement may make the more specialized Purkinje cell phylogenetically in more advanced regions more susceptible to neuronal degeneration factors. Neuronal activity requires chromatin to be active and arranged as euchromatin to be read [110]. Euchromatin is more exposed than heterochromatin, the former being more susceptible to DNA damage [111] and the latter more protected [112] because the compaction of DNA together with non-histone proteins acts as a shield against damage [113]. Thus, more complex Purkinje cells carrying out more elaborate processing are likely to have higher gene expression. In these neurons, the euchromatin/heterochromatin ratio would be higher, and would therefore be more susceptible to different neurodegenerative factors. In fact, it has been proven that the accumulation of DNA damage in Purkinje cells of PCD mice is one of the causes of their death, and that chromatin compaction is a defense mechanism, although it eventually prevents its repair [86]. Another example of this relationship between cell complexity and vulnerability is found in the mitral cells of the olfactory bulb. These neurons are highly susceptible to DNA damage because of their high metabolic and bioelectric activity [114,115,116]. Therefore, a future line of research could be to study the relationship between euchromatin and heterochromatin in Purkinje cells of lobe X compared with the other lobes.
The selective resistance to neuronal damage of the lobe X is especially remarkable, as it is present in many animal models of different natures. The study of its natural protection or reduced vulnerability may constitute an interesting piece of research to find putative therapies against neurodegenerative diseases, which are becoming one of the main health problems in our society.
Author Contributions
Conceptualization, C.H.-P. and D.D.; investigation, C.H.-P.; resources, E.W.; writing—original draft preparation, C.H.-P. and D.D.; writing—review and editing, C.H.-P. and D.D.; project administration, E.W.; funding acquisition, E.W. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by the Ministry of Economy, Industry and Competitiveness (MINECO; SAF2016-79668-R), the Ministry of Science and Innovation (MICINN; PID2019-106943RB-I00), the Regional Government of Castile and Leon (SA030P17, SA129P20 and EDU/529/2017), the Centre for Regenerative Medicine and Cell Therapy of Castile and Leon, and the University of Salamanca.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Acknowledgments
We thank Emma Keck for revising the English.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Freeland, K.R.; Anderson, G.H.; Thomas, T.W. Chapter 42. The Cerebellum. In Principles of Neural Science, 5th ed.; Springer: Berlin/Heidelberg, Germany, 2000. [Google Scholar]
- Voogd, J.; Glickstein, M. The anatomy of the cerebellum. Trends Cogn. Sci. 1998, 2, 307–313. [Google Scholar] [CrossRef] [PubMed]
- White, J.J.; Sillitoe, R.V. Development of the cerebellum: From gene expression patterns to circuit maps. Wiley Interdiscip. Rev. Dev. Biol. 2012, 2, 149–164. [Google Scholar] [CrossRef] [PubMed]
- Delgado-García, J.M. Structure and function of the cerebellum. Rev. Neurol. 2002, 33, 635–642. [Google Scholar]
- Kandel, E.R. Principles of Neural Science; McGraw-Hill: New York, NY, USA, 2013. [Google Scholar]
- Brooks, V.B.; Thach, W.T. Cerebellar Control of Posture and Movement. Compr. Physiol. 1981, 10, 877–946. [Google Scholar] [CrossRef]
- Galliano, E.; Potters, J.-W.; Elgersma, Y.; Wisden, W.; Kushner, S.A.; De Zeeuw, C.I.; Hoebeek, F.E. Synaptic Transmission and Plasticity at Inputs to Murine Cerebellar Purkinje Cells Are Largely Dispensable for Standard Nonmotor Tasks. J. Neurosci. 2013, 33, 12599–12618. [Google Scholar] [CrossRef] [Green Version]
- Newman, P.P.; Reza, H. Functional relationships between the hippocampus and the cerebellum: An electrophysiological study of the cat. J. Physiol. 1979, 287, 405–426. [Google Scholar] [CrossRef] [PubMed]
- Petersen, S.E.; Fox, P.T.; Posner, M.I.; Mintun, M.; Raichle, M.E. Positron emission tomographic studies of the cortical anatomy of single-word processing. Nature 1988, 331, 585–589. [Google Scholar] [CrossRef]
- Sillitoe, R.V.; Joyner, A.L. Morphology, Molecular Codes, and Circuitry Produce the Three-Dimensional Complexity of the Cerebellum. Annu. Rev. Cell Dev. Biol. 2007, 23, 549–577. [Google Scholar] [CrossRef]
- Mugnaini, E.; Dio, M.R.; Jaarsma, D. Chapter 8 The unipolar brush cells of the mammalian cerebellum and cochlear nucleus: Cytology and microcircuitry. In Progress in Brain Research; De Zeeuw, C.I., Strata, P., Voogd, J., Eds.; Elsevier: Amsterdam, The Netherlands, 1997. [Google Scholar]
- Allin, M.; Matsumoto, H.; Santhouse, A.M.; Nosarti, C.; AlAsady, M.H.S.; Stewart, A.L.; Rifkin, L.; Murray, R.M. Cognitive and motor function and the size of the cerebellum in adolescents born very pre-term. Brain 2001, 124, 60–66. [Google Scholar] [CrossRef] [Green Version]
- Levitt, P.; Rakic, P. Immunoperoxidase localization of glial fibrillary acidic protein in radial glial cells and astrocytes of the developing rhesus monkey brain. J. Comp. Neurol. 1980, 193, 815–840. [Google Scholar] [CrossRef]
- Beckinghausen, J.; Sillitoe, R.V. Insights into cerebellar development and connectivity. Neurosci. Lett. 2018, 688, 2–13. [Google Scholar] [CrossRef]
- Ito, M. Cerebellar circuitry as a neuronal machine. Prog. Neurobiol. 2006, 78, 272–303. [Google Scholar] [CrossRef] [PubMed]
- Barmack, N.H.; Baughman, R.W.; Eckenstein, F.P. Cholinergic innervation of the cerebellum of rat, rabbit, cat, and monkey as revealed by choline acetyltransferase activity and immunohistochemistry. J. Comp. Neurol. 1992, 317, 233–249. [Google Scholar] [CrossRef]
- Miall, R.C. Cerebellum: Anatomy and Function. Neuroscience in the 21st Century: From Basic to Clinical; Springer: Berlin/Haidelberg, Germany, 2013; pp. 1149–1167. [Google Scholar]
- Larsell, O. The morphogenesis and adult pattern of the lobules and fissures of the cerebellum of the white rat. J. Comp. Neurol. 1952, 97, 281–356. [Google Scholar] [CrossRef] [PubMed]
- Hawkes, R. Chapter 3 An anatomical model of cerebellar modules. Prog. Brain Res. 1997, 114, 39–52. [Google Scholar] [CrossRef] [PubMed]
- Apps, R.; Hawkes, R. Cerebellar cortical organization: A one-map hypothesis. Nat. Rev. Neurosci. 2009, 10, 670–681. [Google Scholar] [CrossRef]
- Brochu, G.; Maler, L.; Hawkes, R. Zebrin II: A polypeptide antigen expressed selectively by purkinje cells reveals compartments in rat and fish cerebellum. J. Comp. Neurol. 1990, 291, 538–552. [Google Scholar] [CrossRef] [PubMed]
- Sarna, J.R.; Larouche, M.; Marzban, H.; Sillitoe, R.V.; Rancourt, D.E.; Hawkes, R. Patterned Purkinje cell degeneration in mouse models of Niemann-Pick type C disease. J. Comp. Neurol. 2003, 456, 279–291. [Google Scholar] [CrossRef] [PubMed]
- Larouche, M.; Che, P.M.; Hawkes, R. Neurogranin expression identifies a novel array of Purkinje cell parasagittal stripes during mouse cerebellar development. J. Comp. Neurol. 2005, 494, 215–227. [Google Scholar] [CrossRef]
- Marzban, H.; Chung, S.; Watanabe, M.; Hawkes, R. Phospholipase Cbeta4 expression reveals the continuity of cerebellar topography through development. J. Comp. Neurol. 2007, 502, 857–871. [Google Scholar] [CrossRef]
- Hawkes, R.; Turner, R.W. Compartmentation of NADPH-diaphorase activity in the mouse cerebellar cortex. J. Comp. Neurol. 1994, 346, 499–516. [Google Scholar] [CrossRef] [PubMed]
- Ozol, K.O.; Hawkes, R. Compartmentation of the granular layer of the cerebellum. Histol. Histopathol. 1997, 12, 171–184. [Google Scholar] [PubMed]
- Sillitoe, R.V.; Chung, S.H.; Fritschy, J.M.; Hoy, M.; Hawkes, R. Golgi cell dendrites are restricted by Purkinje cell stripe boundaries in the adult mouse cerebellar cortex. J. Neurosci. 2008, 28, 2820–2826. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chung, C.; Elrick, M.J.; Dell’Orco, J.M.; Qin, Z.S.; Kalyana-Sundaram, S.; Chinnaiyan, A.M.; Shakkottai, V.G.; Lieberman, A.P. Heat Shock Protein Beta-1 Modifies Anterior to Posterior Purkinje Cell Vulnerability in a Mouse Model of Niemann-Pick Type C Disease. PLoS Genet. 2016, 12, e1006042. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Armstrong, C.L.; Chung, S.H.; Armstrong, J.N.; Hochgeschwender, U.; Jeong, Y.G.; Hawkes, R. A novel somatostatin-immunoreactive mossy fiber pathway associated with HSP25-immunoreactive purkinje cell stripes in the mouse cerebellum. J. Comp. Neurol. 2009, 517, 524–538. [Google Scholar] [CrossRef]
- Voogd, J.; Pardoe, J.; Ruigrok, T.J.H.; Apps, R. The Distribution of Climbing and Mossy Fiber Collateral Branches from the Copula Pyramidis and the Paramedian Lobule: Congruence of Climbing Fiber Cortical Zones and the Pattern of Zebrin Banding within the Rat Cerebellum. J. Neurosci. 2003, 23, 4645–4656. [Google Scholar] [CrossRef]
- Marzban, H.; Hawkes, R. On the Architecture of the Posterior Zone of the Cerebellum. Cerebellum 2010, 10, 422–434. [Google Scholar] [CrossRef]
- Armstrong, C.L.; Krueger-Naug, A.M.; Currie, R.W.; Hawkes, R. Expression of heat-shock proteina Hsp25 in mouse Purkinje cells during development reveals novel features of cerebellar compartmentation. J. Comp. Neurol. 2001, 429, 7–21. [Google Scholar] [CrossRef]
- Sillitoe, R.V.; Hawkes, R. Whole-mount Immunohistochemistry: A High-throughput Screen for Patterning Defects in the Mouse Cerebellum. J. Histochem. Cytochem. 2002, 50, 235–244. [Google Scholar] [CrossRef] [Green Version]
- Double, K.L.; Reyes, S.; Werry, E.L.; Halliday, G.M. Selective cell death in neurodegeneration: Why are some neurons spared in vulnerable regions? Prog. Neurobiol. 2010, 92, 316–329. [Google Scholar] [CrossRef]
- Sarna, J.R.; Hawkes, R. Patterned Purkinje cell death in the cerebellum. Prog. Neurobiol. 2003, 70, 473–507. [Google Scholar] [CrossRef] [PubMed]
- Clark, H.B.; Burright, E.N.; Yunis, W.S.; Larson, S.; Wilcox, C.; Hartman, B.; Matilla, A.; Zoghbi, H.Y.; Orr, H.T. Purkinje cell expression of a mutant allele of SCA1 in transgenic mice leads to disparate effects on motor behaviors, followed by a progressive cerebellar dysfunction and histological alterations. J. Neurosci. 1997, 17, 7385–7395. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoneshige, A.; Suzuki, K.; Suzuki, K.; Matsuda, J. A mutation in the saposin C domain of the sphingolipid activator protein (Prosaposin) gene causes neurodegenerative disease in mice. J. Neurosci. Res. 2010, 88, 2118–2134. [Google Scholar] [CrossRef] [PubMed]
- Tavani, F.; Zimmerman, R.A.; Berry, G.; Sullivan, K.; Gatti, R.; Bingham, P. Ataxia-telangiectasia: The pattern of cerebellar atrophy on MRI. Neuroradiology 2003, 45, 315–319. [Google Scholar] [CrossRef] [PubMed]
- Sarna, J.; Miranda, S.R.P.; Schuchman, E.H.; Hawkes, R. Patterned cerebellar Purkinje cell death in a transgenic mouse model of Niemann Pick type A/B disease. Eur. J. Neurosci. 2001, 13, 1873–1880. [Google Scholar] [CrossRef]
- Kume, A.; Takahashi, A.; Hashizume, Y.; Asai, J. A histometrical and comparative study on Purkinje cell loss and olivary nucleus cell loss in multiple system atrophy. J. Neurol. Sci. 1991, 101, 178–186. [Google Scholar] [CrossRef] [PubMed]
- Torvik, A.; Torp, S. The prevalence of alcoholic cerebellar atrophy: A morphometric and histological study of an autopsy material. J. Neurol. Sci. 1986, 75, 43–51. [Google Scholar] [CrossRef]
- Biran, V.; Heine, V.M.; Verney, C.; Sheldon, R.A.; Spadafora, R.; Vexler, Z.S.; Rowitch, D.H.; Ferriero, D.M. Cerebellar abnormalities following hypoxia alone compared to hypoxic–ischemic forebrain injury in the developing rat brain. Neurobiol. Dis. 2011, 41, 138–146. [Google Scholar] [CrossRef] [Green Version]
- Andersen, B.B.; Pakkenberg, B. Aging of the human cerebellum: A stereological study. J. Comp. Neurol. 2003, 466, 356–365. [Google Scholar] [CrossRef]
- Heckroth, J.A.; Abbott, L.C. Purkinje cell loss from alternating sagittal zones in the cerebellum of leaner mutant mice. Brain Res. 1994, 658, 93–104. [Google Scholar] [CrossRef]
- Duchala, C.S.; Shick, H.E.; Garcia, J.; Deweese, D.M.; Sun, X.; Stewart, V.J.; Macklin, W.B. The toppler mouse: A novel mutant exhibiting loss of Purkinje cells. J. Comp. Neurol. 2004, 476, 113–129. [Google Scholar] [CrossRef] [PubMed]
- Isaacs, A.M.; Oliver, P.L.; Jones, E.L.; Jeans, A.; Potter, A.; Hovik, B.H.; Nolan, P.M.; Vizor, L.; Glenister, P.; Simon, A.K.; et al. A Mutation inAf4Is Predicted to Cause Cerebellar Ataxia and Cataracts in the Robotic Mouse. J. Neurosci. 2003, 23, 1631–1637. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tolbert, D.L.; Ewald, M.; Gutting, J.; La Regina, M.C. Spatial and temporal pattern of Purkinje cell degeneration in shaker mutant rats with hereditary cerebellar ataxia. J. Comp. Neurol. 1995, 355, 490–507. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, C.L.; Duffin, C.A.; McFarland, R.; Vogel, M.W. Mechanisms of Compartmental Purkinje Cell Death and Survival in the Lurcher Mutant Mouse. Cerebellum 2010, 10, 504–514. [Google Scholar] [CrossRef]
- Wang, T.; Morgan, J.I. The Purkinje cell degeneration (pcd) mouse: An unexpected molecular link between neuronal degeneration and regeneration. Brain Res. 2007, 1140, 26–40. [Google Scholar] [CrossRef]
- Baltanás, F.C.; Berciano, M.T.; Santos, E.; Lafarga, M. The Childhood-Onset Neurodegeneration with Cerebellar Atrophy (CONDCA) Disease Caused by AGTPBP1 Gene Mutations: The Purkinje Cell Degeneration Mouse as an Animal Model for the Study of this Human Disease. Biomedicines 2021, 9, 1157. [Google Scholar] [CrossRef]
- Fletcher, C.F.; Lutz, C.M.; O’Sullivan, T.; Shaughnessy, J.D.; Hawkes, R.; Frankel, W.N.; Copeland, N.G.; Jenkins, N.A. Absence Epilepsy in Tottering Mutant Mice Is Associated with Calcium Channel Defects. Cell 1996, 87, 607–617. [Google Scholar] [CrossRef] [Green Version]
- Herrup, K.; Wilczynski, S. Cerebellar cell degeneration in the leaner mutant mouse. Neuroscience 1982, 7, 2185–2196. [Google Scholar] [CrossRef]
- Sawada, K.; Haga, H.; Fukui, Y. Ataxic mutant mice with defects in Ca2+ channel α1A subunit gene: Morphological and functional abnormalities in cerebellar cortical neurons. Congenit. Anom. 2000, 40, 99–107. [Google Scholar] [CrossRef]
- Doyle, J.; Ren, X.; Lennon, G.; Stubbs, L. Mutations in the Cacnl1a4 calcium channel gene are associated with seizures, cerebellar degeneration, and ataxia in tottering and leaner mutant mice. Mamm. Genome 1997, 8, 113–120. [Google Scholar] [CrossRef]
- Lorenzon, N.M.; Lutz, C.M.; Frankel, W.N.; Beam, K.G. Altered calcium channel currents in Purkinje cells of the neurological mutant mouse leaner. J. Neurosci. 1998, 18, 4482–4489. [Google Scholar] [CrossRef] [PubMed]
- Sawada, K.; Azad, A.K.; Sakata-Haga, H.; Lee, N.-S.; Jeong, Y.-G.; Fukui, Y. Striking pattern of Purkinje cell loss in cerebellum of an ataxic mutant mouse, tottering. Acta Neurobiol. Exp. 2009, 69, 138–145. [Google Scholar]
- Jeong, Y.-G.; Hyun, B.-H.; Hawkes, R. Abnormalities in cerebellar Purkinje cells in the novel ataxic mutant mouse, pogo. Dev. Brain Res. 2000, 125, 61–67. [Google Scholar] [CrossRef]
- Duffin, C.A.; Mcfarland, R.; Sarna, J.R.; Vogel, M.W.; Armstrong, C.L. Heat shock protein 25 expression and preferential Purkinje cell survival in the lurcher mutant mouse cerebellum. J. Comp. Neurol. 2010, 518, 1892–1907. [Google Scholar] [CrossRef] [PubMed]
- Praggastis, M.; Tortelli, B.; Zhang, J.; Fujiwara, H.; Sidhu, R.; Chacko, A.; Chen, Z.; Chung, C.; Lieberman, A.P.; Sikora, J.; et al. A murine Niemann-Pick C1 I1061T knock-in model recapitulates the pathological features of the most prevalent human disease allele. J. Neurosci. 2015, 35, 8091–8106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Angelis, M.H.; Flaswinkel, H.; Fuchs, H.; Rathkolb, B.; Soewarto, D.; Marschall, S.; Heffner, S.; Pargent, W.; Wuensch, K.; Jung, M.; et al. Genome-wide, large-scale production of mutant mice by ENU mutagenesis. Nat. Genet. 2000, 25, 444–447. [Google Scholar] [CrossRef] [PubMed]
- Isnard, P.; Coré, N.; Naquet, P.; Djabali, M. Altered lymphoid development in mice deficient for the mAF4 proto-oncogene. Blood 2000, 96, 705–710. [Google Scholar] [CrossRef] [PubMed]
- Bitoun, E.; Davies, K.E. The Robotic Mouse: Understanding the Role of AF4, a Cofactor of Transcriptional Elongation and Chromatin Remodelling, in Purkinje Cell Function. Cerebellum 2009, 8, 175–183. [Google Scholar] [CrossRef]
- La Regina, M.C.; Yates-Siilata, K.; Woods, L.; Tolbert, D. Preliminary characterization of hereditary cerebellar ataxia in rats. Lab. Anim. Sci. 1992, 42, 19–26. [Google Scholar]
- Wolf, L.W.; LaRegina, M.C.; Tolbert, D.L. A behavioral study of the development of hereditary cerebellar ataxia in the shaker rat mutant. Behav. Brain Res. 1996, 75, 67–81. [Google Scholar] [CrossRef]
- Phillips, R.J.S. ‘Lurcher’, a new gene in linkage group XI of the house mouse. J. Genet. 1960, 57, 35–42. [Google Scholar] [CrossRef]
- Cheng, S.S.-W.; Heintz, N. Massive Loss of Mid- and Hindbrain Neurons during Embryonic Development of Homozygous Lurcher Mice. J. Neurosci. 1997, 17, 2400–2407. [Google Scholar] [CrossRef] [PubMed]
- Vogel, M.W.; Caston, J.; Yuzaki, M.; Mariani, J. The Lurcher mouse: Fresh insights from an old mutant. Brain Res. 2007, 1140, 4–18. [Google Scholar] [CrossRef] [PubMed]
- MCfarland, R.; Blokhin, A.; Sydnor, J.; Mariani, J.; Vogel, M.W. Oxidative stress, nitric oxide, and the mechanisms of cell death in Lurcher Purkinje cells. Dev. Neurobiol. 2007, 67, 1032–1046. [Google Scholar] [CrossRef] [PubMed]
- Pentchev, P.G.; Comly, M.E.; Kruth, H.S.; Patel, S.; Proestel, M.; Weintroub, H. The cholesterol storage disorder of the mutant BALB/c mouse. A primary genetic lesion closely linked to defective esterification of exogenously derived cholesterol and its relationship to human type C Niemann-Pick disease. J. Biol. Chem. 1986, 261, 2772–2777. [Google Scholar] [CrossRef] [PubMed]
- Vanier, M.T.; Rodriguez-Lafrasse, C.; Rousson, R.; Duthel, S.; Harzer, K.; Pentchev, P.G.; Revol, A.; Louisot, P. Type C Niemann-Pick Disease: Biochemical Aspects and Phenotypic Heterogeneity. Dev. Neurosci. 1991, 13, 307–314. [Google Scholar] [CrossRef]
- Akaboshi, S.; Yano, T.; Miyawaki, S.; Ohno, K.; Takeshita, K. A C57BL/KsJ mouse model of Niemann-Pick disease (spm) belongs to the same complementation group as the major childhood type of Niemann-Pick disease type C. Hum. Genet. 1997, 99, 350–353. [Google Scholar] [CrossRef]
- Sidman, R.L.; Green, M.C. “Nervous,” a new mutant mouse with cerebellar disease. In Les Mutants Pathologiques chez l’Animal; Sabourdy, M., Ed.; Centre National de la Reserche Scientifique: Paris, France, 1970; pp. 69–79. [Google Scholar]
- Mullen, R.J.; Lavail, M.M. Two new types of retinal degeneration in cerebellar mutant mice. Nature 1975, 258, 528–530. [Google Scholar] [CrossRef]
- Mallet, J.; Huchet, M.; Pougeois, R.; Changeux, J.-P. Anatomical, physiological and biochemical studies on the cerebellum from mutant mice. III. Protein differences associated with the weaver, staggerer and nervous mutations. Brain Res. 1976, 103, 291–312. [Google Scholar] [CrossRef]
- Mikoshiba, K.; Okano, H.; Tsukada, Y. P400 Protein Characteristic to Purkinje Cells and Related Proteins in Cerebella from Neuropathological Mutant Mice: Autoradiographic Study by 14C-Leucine and Phosphorylation. Dev. Neurosci. 1985, 7, 179–187. [Google Scholar] [CrossRef]
- Sotelo, C.; Triller, A. Fate of presynaptic afferents to Purkinje cells in the adult nervous mutant mouse: A model to study presynaptic stabilization. Brain Res. 1979, 175, 11–36. [Google Scholar] [CrossRef] [PubMed]
- Wassef, M.; Sotelo, C.; Cholley, B.; Brehier, A.; Thomasset, M. Cerebellar mutations affecting the postnatal survival of Purkinje cells in the mouse disclose a longitudinal pattern of differentially sensitive cells. Dev. Biol. 1987, 124, 379–389. [Google Scholar] [CrossRef] [PubMed]
- Edwards, M.A.; Crandall, J.E.; Leclerc, N.; Yamamoto, M. Effects of nervous mutation on purkinje cell compartments defined by Zebrin II and 9-O-acetylated gangliosides expression. Neurosci. Res. 1994, 19, 167–174. [Google Scholar] [CrossRef]
- Shashi, V.; Magiera, M.M.; Klein, D.; Zaki, M.; Schoch, K.; Rudnik-Schöneborn, S.; Norman, A.; Neto, O.L.A.; Dusl, M.; Yuan, X.; et al. Loss of tubulin deglutamylaseCCP1 causes infantile-onset neurodegeneration. EMBO J. 2018, 37, e100540. [Google Scholar] [CrossRef] [PubMed]
- Karakaya, M.; Paketci, C.; Altmueller, J.; Thiele, H.; Hoelker, I.; Yis, U.; Wirth, B. Biallelic variant in AGTPBP1 causes infantile lower motor neuron degeneration and cerebellar atrophy. Am. J. Med. Genet. Part A 2019, 179, 1580–1584. [Google Scholar] [CrossRef] [PubMed]
- Sheffer, R.; Gur, M.; Brooks, R.; Salah, S.; Daana, M.; Fraenkel, N.; Eisenstein, E.; Rabie, M.; Nevo, Y.; Jalas, C.; et al. Biallelic variants in AGTPBP1, involved in tubulin deglutamylation, are associated with cerebellar degeneration and motor neuropathy. Eur. J. Hum. Genet. 2019, 27, 1419–1426. [Google Scholar] [CrossRef]
- Mullen, R.J.; Eicher, E.M.; Sidman, R.L. Purkinje cell degeneration, a new neurological mutation in the mouse. Proc. Natl. Acad. Sci. USA 1976, 73, 208–212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Gorman, S.; Sidman, R.L. Degeneration of thalamic neurons in “Purkinje cell degeneration” mutant mice. I. Distribution of neuron loss. J. Comp. Neurol. 1985, 234, 277–297. [Google Scholar] [CrossRef] [PubMed]
- Greer, C.A.; Shepherd, G.M. Mitral cell degeneration and sensory function in the neurological mutant mouse Purkinje cell degeneration (PCD). Brain Res. 1982, 235, 156–161. [Google Scholar] [CrossRef]
- Muñoz-Castañeda, R.; Díaz, D.; Peris, L.; Andrieux, A.; Bosc, C.; Muñoz-Castañeda, J.M.; Janke, C.; Alonso, J.R.; Moutin, M.-J.; Weruaga, E. Cytoskeleton stability is essential for the integrity of the cerebellum and its motor- and affective-related behaviors. Sci. Rep. 2018, 8, 3072. [Google Scholar] [CrossRef] [Green Version]
- Baltanás, F.C.; Casafont, I.; Weruaga, E.; Alonso, J.R.; Berciano, M.T.; Lafarga, M. Nucleolar Disruption and Cajal Body Disassembly are Nuclear Hallmarks of DNA Damage-Induced Neurodegeneration in Purkinje Cells. Brain Pathol. 2010, 21, 374–388. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Gonzalez, A.; La Spada, A.R.; Treadaway, J.; Higdon, J.C.; Harris, B.S.; Sidman, R.L.; Morgan, J.I.; Zuo, J. Purkinje cell degeneration (pcd) phenotypes caused by mutations in the axotomy-induced gene, Nna1. Science 2002, 295, 1904–1906. [Google Scholar] [CrossRef]
- Chakrabarti, L.; Neal, J.T.; Miles, M.; Martinez, R.A.; Smith, A.C.; Sopher, B.L.; La Spada, A.R. The Purkinje cell degeneration 5J mutation is a single amino acid insertion that destabilizes Nna1 protein. Mamm. Genome 2006, 17, 103–110. [Google Scholar] [CrossRef] [PubMed]
- Harris, A.; Morgan, J.I.; Pecot, M.; Soumare, A.; Osborne, A.; Soares, H.D. Regenerating motor neurons express Nna1, a novel ATP/GTP-binding protein related to zinc carboxypeptidases. Mol. Cell Neurosci. 2000, 16, 578–596. [Google Scholar] [CrossRef] [PubMed]
- Rogowski, K.; Van Dijk, J.; Magiera, M.M.; Bosc, C.; Deloulme, J.C.; Bosson, A.; Peris, L.; Gold, N.D.; Lacroix, B.; Bosch Grau, M.; et al. A family of protein-deglutamylating enzymes associated with neurodegeneration. Cell 2010, 143, 564–578. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baltanás, F.C.; Berciano, M.T.; Valero, J.; Gómez, C.; Díaz, D.; Alonso, J.R.; Lafarga, M.; Weruaga, E. Differential glial activation during the degeneration of Purkinje cells and mitral cells in the PCD mutant mice. Glia 2012, 61, 254–272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mashimo, T.; Hadjebi, O.; Amair-Pinedo, F.; Tsurumi, T.; Langa, F.; Serikawa, T.; Sotelo, C.; Guénet, J.-L.; Rosa, J.L. Progressive Purkinje Cell Degeneration in tambaleante Mutant Mice Is a Consequence of a Missense Mutation in HERC1 E3 Ubiquitin Ligase. PLoS Genet. 2009, 5, e1000784. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruiz, R.; Pérez-Villegas, E.M.; Bachiller, S.; Rosa, J.L.; Armengol, J.A. HERC 1 Ubiquitin Ligase Mutation Affects Neocortical, CA3 Hippocampal and Spinal Cord Projection Neurons: An Ultrastructural Study. Front. Neuroanat. 2016, 10, 42. [Google Scholar] [CrossRef] [Green Version]
- Winkelman, M.D.; Hines, J.D. Cerebellar degeneration caused by high-dose cytosine arabinoside: A clinicopathological study. Ann. Neurol. 1983, 14, 520–527. [Google Scholar] [CrossRef]
- Ciesielski, K.T.; Yanofsky, R.; Ludwig, R.N.; Hill, D.E.; Hart, B.L.; Astur, R.S.; Snyder, T. Hypoplasia of the Cerebellar Vermis and Cognitive Deficits in Survivors of Childhood Leukemia. Arch. Neurol. 1994, 51, 985–993. [Google Scholar] [CrossRef]
- Butterworth, R.F.; Hamel, E.; Landreville, F.; Barbeau, A. Cerebellar ataxia produced by 3-acetyl pyridine in rat. Can. J. Neurol. Sci. 1978, 5, 131–133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hess, B.H.; Krewet, J.A.; Tolbert, D.L. Olivocerebellar projections are necessary for exogenous trophic factors to delay heredo-Purkinje cell degeneration. Brain Res. 2003, 986, 54–62. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, C.; Hawkes, R. Selective Purkinje cell ectopia in the cerebellum of the Weaver mouse. J. Comp. Neurol. 2001, 439, 151–161. [Google Scholar] [CrossRef] [PubMed]
- Kostenko, S.; Moens, U. Heat shock protein 27 phosphorylation: Kinases, phosphatases, functions and pathology. Cells Mol. Life Sci. 2009, 66, 3289–3307. [Google Scholar] [CrossRef]
- Sawada, K.; Haga, H.; Fukui, Y. Alternating array of tyrosine hydroxylase and heat shock protein 25 immunopositive Purkinje cell stripes in zebrin II-defined transverse zoneof the cerebellum of rolling mouse Nagoya. Brain Res. 2010, 9, 46–53. [Google Scholar] [CrossRef]
- Kim, C.-H.; Shin, J.J.; Kim, J.; Kim, S.J. Reduced spike frequency adaptation in Purkinje cells of the vestibulocerebellum. Neurosci. Lett. 2013, 535, 45–50. [Google Scholar] [CrossRef]
- Martin, K.B.; Williams, I.M.; Cluzeau, C.V.; Cougnoux, A.; Dale, R.K.; Iben, J.R.; Cawley, N.X.; Wassif, C.A.; Porter, F.D. Identification of Novel Pathways Associated with Patterned Cerebellar Purkinje Neuron Degeneration in Niemann-Pick Disease, Type C1. Int. J. Mol. Sci. 2019, 21, 292. [Google Scholar] [CrossRef] [Green Version]
- Jakob, U.; Gaestel, M.; Engel, K.; Buchner, J. Small heat shock proteins are molecular chaperones. J. Biol. Chem. 1993, 268, 1517–1520. [Google Scholar] [CrossRef]
- Lavoie, J.N.; Hickey, E.; Weber, L.A.; Landry, J. Modulation of actin microfilament dynamics and fluid phase pinocytosis by phosphorylation of heat shock protein 27. J. Biol. Chem. 1993, 268, 24210–24214. [Google Scholar] [CrossRef]
- Huot, J.; Houle, F.; Spitz, D.; Landry, J. HSP27 phosphorylation-mediated resistance against actin fragmentation and cell death induced by oxidative stress. Cancer Res 1996, 56, 273–279. [Google Scholar]
- Carver, J.A.; Aquilina, J.A.; Cooper, P.G.; Williams, G.A.; Truscott, R.J. Alpha-crystallin: Molecular chaperone and protein surfactant. Biochim. Biophys. Acta 1994, 1204, 195–206. [Google Scholar] [CrossRef] [PubMed]
- Boelens, W.C.; De Jong, W.W. Alpha-Crystallins, versatile stress-proteins. Mol. Biol. Rep. 1995, 21, 75–80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, Y.; Macrae, T.H. Small heat shock proteins: Molecular structure and chaperone function. Cells Mol. Life Sci. 2005, 62, 2460–2476. [Google Scholar] [CrossRef]
- Armstrong, C.L.; Krueger-Naug, A.M.; Currie, R.W.; Hawkes, R. Constitutive expression of the 25-kDa heat shock protein Hsp25 reveals novel parasagittal bands of Purkinje cells in the adult mouse cerebellar cortex. J. Comp. Neurol. 2000, 416, 383–397. [Google Scholar] [CrossRef]
- Valero, J.; Berciano, M.T.; Weruaga, E.; Lafarga, M.; Alonso, J.R. Pre-neurodegeneration of mitral cells in the pcd mutant mouse is associated with DNA damage, transcriptional repression, and reorganization of nuclear speckles and Cajal bodies. Mol. Cells Neurosci. 2006, 33, 283–295. [Google Scholar] [CrossRef] [PubMed]
- Takata, H.; Hanafusa, T.; Mori, T.; Shimura, M.; Iida, Y.; Ishikawa, K.; Yoshikawa, K.; Yoshikawa, Y.; Maeshima, K. Chromatin Compaction Protects Genomic DNA from Radiation Damage. PLoS ONE 2013, 8, e75622. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cann, K.L.; Dellaire, G. Heterochromatin and the DNA damage response: The need to relaxThis paper is one of a selection of papers in a Special Issue entitled 31st Annual International Asilomar Chromatin and Chromosomes Conference, and has undergone the Journal’s usual peer review process. Biochem. Cell Biol. 2011, 89, 45–60. [Google Scholar] [CrossRef]
- Falk, M.; Lukásová, E.; Kozubek, S. Chromatin structure influences the sensitivity of DNA to gamma-radiation. Biochim. Biophys. Acta 2008, 1783, 2398–2414. [Google Scholar] [CrossRef] [Green Version]
- Friedman, D.; Strowbridge, B.W. Functional Role of NMDA Autoreceptors in Olfactory Mitral Cells. J. Neurophysiol. 2000, 84, 39–50. [Google Scholar] [CrossRef] [Green Version]
- Lowe, G. Electrical signaling in the olfactory bulb. Curr. Opin. Neurobiol. 2003, 13, 476–481. [Google Scholar] [CrossRef]
- Djurisic, M.; Antic, S.; Chen, W.R.; Zecevic, D. Voltage Imaging from Dendrites of Mitral Cells: EPSP Attenuation and Spike Trigger Zones. J. Neurosci. 2004, 24, 6703–6714. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Cerebellum of an adult mouse. (A) dorsal view of a whole-mount cerebellum where its main regions can be easily distinguished: vermis, paravermis, hemispheres, paraflocculi, and flocculi; the cerebellar cortex is also divided into ten lobes, identified using Roman numerals (in this view only lobes from V to IX are shown). (B) A sagittal section of the cerebellar vermis showing the four main transverse domains (in different colors); the complete foliation pattern of the ten lobes can be identified in this section. a, anterior; COP, copula pyramidis; LS, lobulus simplex; p, posterior; and PML, paramedian lobe. Taken from White and Sillitoe [3].
Figure 1.
Cerebellum of an adult mouse. (A) dorsal view of a whole-mount cerebellum where its main regions can be easily distinguished: vermis, paravermis, hemispheres, paraflocculi, and flocculi; the cerebellar cortex is also divided into ten lobes, identified using Roman numerals (in this view only lobes from V to IX are shown). (B) A sagittal section of the cerebellar vermis showing the four main transverse domains (in different colors); the complete foliation pattern of the ten lobes can be identified in this section. a, anterior; COP, copula pyramidis; LS, lobulus simplex; p, posterior; and PML, paramedian lobe. Taken from White and Sillitoe [3].

Figure 2.
Structure of the cerebellar cortex. In the outermost part is the molecular layer, composed of the dendrites of Purkinje cells, the axons of granule cells (parallel fibers), and the climbing fibers; basket and stellate cells are also located in this layer. Deep below is the Purkinje layer, formed by the somas of these neurons. The inner layer of the cerebellar cortex is the granular layer, made up of granule cells and other interneurons, such as Golgi cells.
Figure 2.
Structure of the cerebellar cortex. In the outermost part is the molecular layer, composed of the dendrites of Purkinje cells, the axons of granule cells (parallel fibers), and the climbing fibers; basket and stellate cells are also located in this layer. Deep below is the Purkinje layer, formed by the somas of these neurons. The inner layer of the cerebellar cortex is the granular layer, made up of granule cells and other interneurons, such as Golgi cells.

Figure 3.
Basic diagrams of the main cerebellar circuits. (A) mossy fibers excite granule cells, which also stimulate Purkinje, stellate, and basket cells. At the same time, stellate and basket cells inhibit Purkinje cells; the latter constitutes the only output of the cerebellar cortex, inhibiting deep cerebellar nuclei. (B) climbing fibers and other inputs from either the raphe nucleus, the locus coeruleus, or the pedunculopontine nucleus excite Purkinje cells, which inhibit deep cerebellar nuclei. All cerebellar inputs send excitatory collateral synapses to deep cerebellar nuclei (dotted lines). (C) granule cells, stimulated by mossy fibers, excite Golgi cells, which inhibits the latter in a negative feedback loop. +, excitatory synapsis; −, inhibitory synapsis.
Figure 3.
Basic diagrams of the main cerebellar circuits. (A) mossy fibers excite granule cells, which also stimulate Purkinje, stellate, and basket cells. At the same time, stellate and basket cells inhibit Purkinje cells; the latter constitutes the only output of the cerebellar cortex, inhibiting deep cerebellar nuclei. (B) climbing fibers and other inputs from either the raphe nucleus, the locus coeruleus, or the pedunculopontine nucleus excite Purkinje cells, which inhibit deep cerebellar nuclei. All cerebellar inputs send excitatory collateral synapses to deep cerebellar nuclei (dotted lines). (C) granule cells, stimulated by mossy fibers, excite Golgi cells, which inhibits the latter in a negative feedback loop. +, excitatory synapsis; −, inhibitory synapsis.

Figure 4.
Schema of Zebrin II and HSP25 expression patterns in the posterior part of the adult mouse cerebellum. Only data relative to the vermis are shown, as the lobe X does not extend to the hemispheres. Black bands imply strong labelling and grey bands indicate moderate labelling. Zebrin II expression is practically inexistent in anterior region (lobes I–V); half of Purkinje cells are strongly labeled and the other half, in posterior region, are not (lobe VIII and part of lobe IX); almost all the Purkinje cells are strongly marked in central (lobes V and VI) and nodular regions (part of lobe IX and X). No data of HSP25 in the anterior and posterior regions are shown due to its null expression; some bands can be distinguished in the central (lobes V and VI) and nodular regions (part of lobe IX and X), although expression is less intense in the former. Data were taken from Marzban and Hawkes [31], Armstrong et al. [32] and Sillitoe and Hawkes [33].
Figure 4.
Schema of Zebrin II and HSP25 expression patterns in the posterior part of the adult mouse cerebellum. Only data relative to the vermis are shown, as the lobe X does not extend to the hemispheres. Black bands imply strong labelling and grey bands indicate moderate labelling. Zebrin II expression is practically inexistent in anterior region (lobes I–V); half of Purkinje cells are strongly labeled and the other half, in posterior region, are not (lobe VIII and part of lobe IX); almost all the Purkinje cells are strongly marked in central (lobes V and VI) and nodular regions (part of lobe IX and X). No data of HSP25 in the anterior and posterior regions are shown due to its null expression; some bands can be distinguished in the central (lobes V and VI) and nodular regions (part of lobe IX and X), although expression is less intense in the former. Data were taken from Marzban and Hawkes [31], Armstrong et al. [32] and Sillitoe and Hawkes [33].

Figure 5.
Comparison of the resistance of lobe X in different animal models of cerebellar neurodegeneration. (A) Toppler mouse (taken from Duchala et al.) [45]; (B) Robotic mouse (modified from Isaacs et al.; Copyright (2003) Society for Neuroscience) [46]; (C) Shaker rat (Modified from Tolbert et al.) [47]; (D) Lurcher mouse (modified from Duffin et al.) [58]; (E) NPC1 mouse (modified from Praggastis et al.) [59]; and (F) PCD mouse. In all images, Purkinje cells are stained with different labeling techniques and colors, in parasagittal sections; Roman numbers identify different cerebellar lobes (lobe X is always indicated). Note that lobe X is the most resistant part of the cerebellar cortex, where Purkinje cells continue to remain alive; arrows point to isolated Purkinje cells in (C); a Nissl background staining (blue) can be also observed in (E). Scale bars 500 µm for (A), 1 mm for (C,E,F), and 100 µm for (D).
Figure 5.
Comparison of the resistance of lobe X in different animal models of cerebellar neurodegeneration. (A) Toppler mouse (taken from Duchala et al.) [45]; (B) Robotic mouse (modified from Isaacs et al.; Copyright (2003) Society for Neuroscience) [46]; (C) Shaker rat (Modified from Tolbert et al.) [47]; (D) Lurcher mouse (modified from Duffin et al.) [58]; (E) NPC1 mouse (modified from Praggastis et al.) [59]; and (F) PCD mouse. In all images, Purkinje cells are stained with different labeling techniques and colors, in parasagittal sections; Roman numbers identify different cerebellar lobes (lobe X is always indicated). Note that lobe X is the most resistant part of the cerebellar cortex, where Purkinje cells continue to remain alive; arrows point to isolated Purkinje cells in (C); a Nissl background staining (blue) can be also observed in (E). Scale bars 500 µm for (A), 1 mm for (C,E,F), and 100 µm for (D).


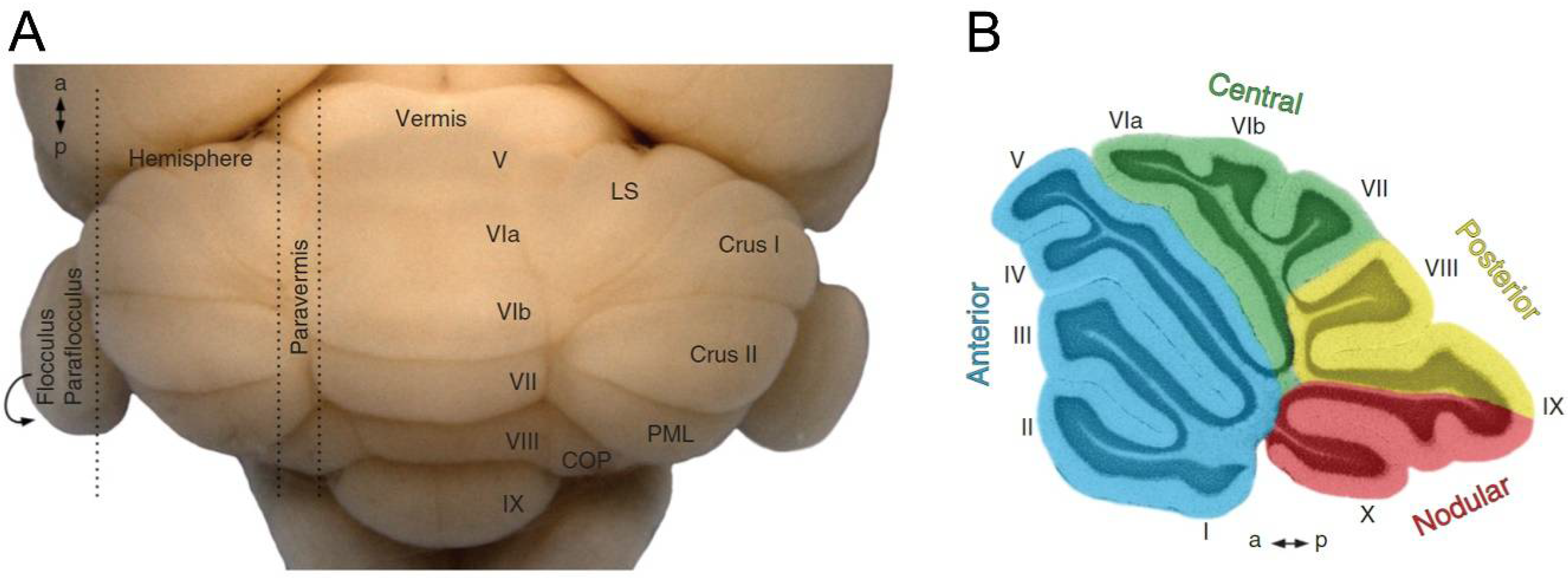{kind=link}
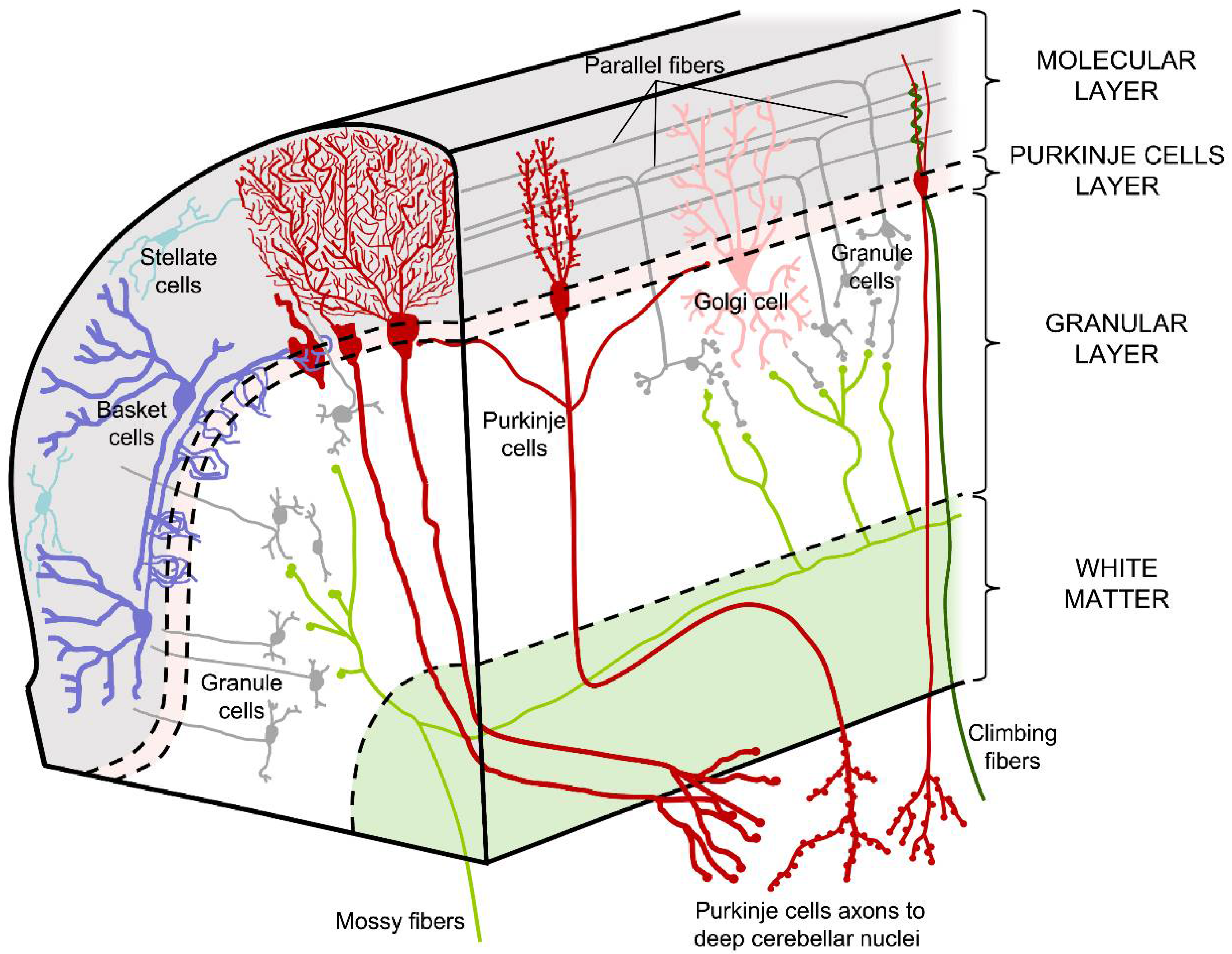{kind=link}
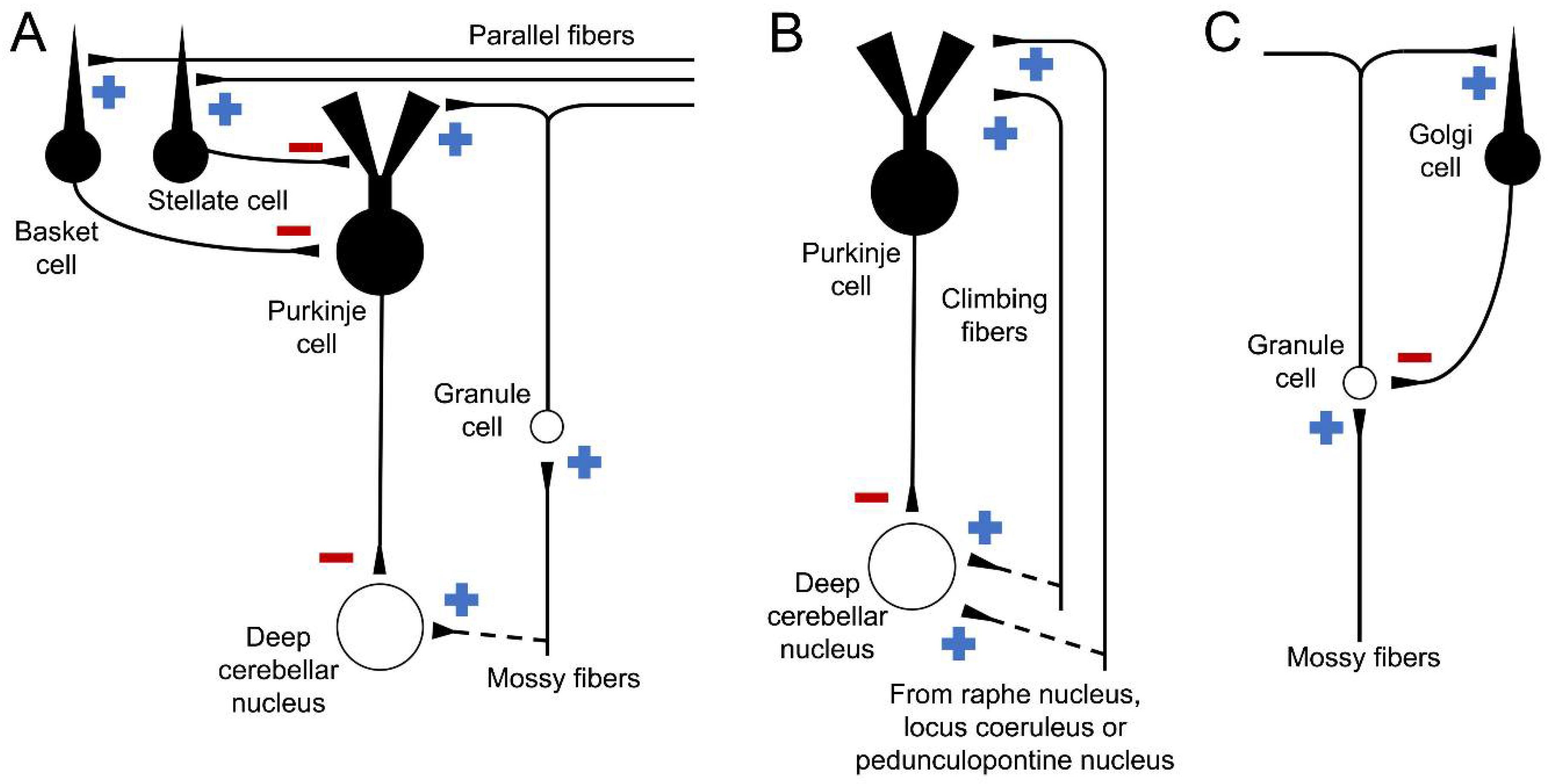{kind=link}
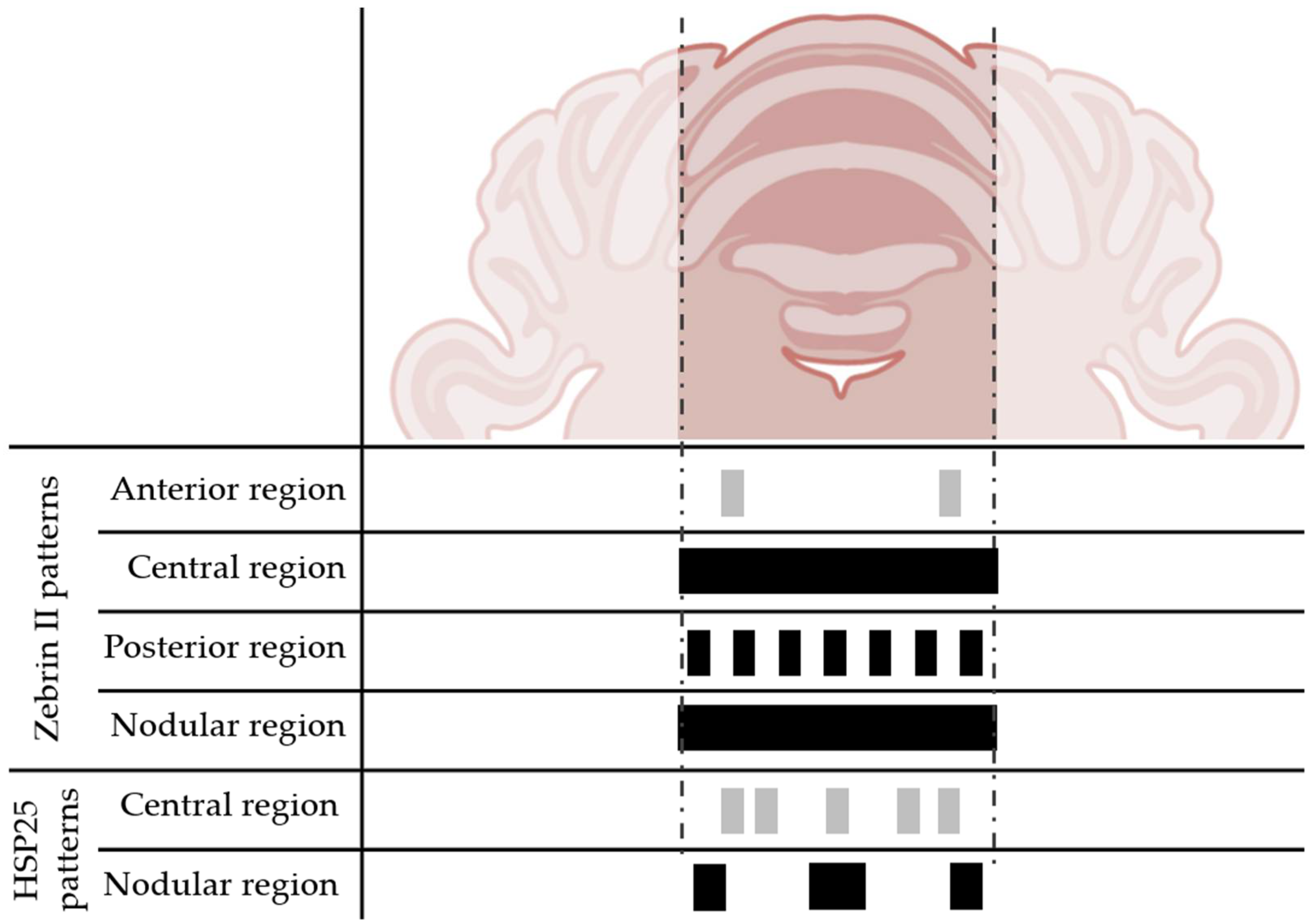{kind=link}
{kind=link}
Table 1.
Summary of the reviewed genetic models.
| Model | Mutation | Cause | Pathology | Degeneration Pattern |
|---|---|---|---|---|
| Leaner | Spontaneous tgla/tgla. | Ca2+ channel subunit damaged. | Ataxia starts at P10 and Purkinje cell loss observed at P40. | Posterior and nodular zones are less vulnerable. |
| Toppler | Spontaneous. Mutation located in chromosome 8. | Unknown. | Ataxia starts at 4–5 weeks of age. Major Purkinje cell death at P14–P30. | Purkinje cells of lobe X survive at P30. |
| Robotic | Induced by N-ethyl-N-nitrosourea. Af4 gene is altered. | Transcription cofactor coded by Af4 is truncated. | Ataxia starts at 3 weeks of age. Purkinje cell death starts at the 8th week. | Antero-posterior neurodegeneration. |
| Shaker (rat) | Spontaneous. Unknown. Two variants described: severe and mild. | Unknown. | Ataxia and body tremors at 3 months of age in the severe variant. Ataxia present only the in mild variant. | The anterior region is the most affected. Lobe X appears unaffected. |
| Lurcher | Spontaneous. +/Lc present ataxia. Lc/Lc is lethal. | δ2 glutamate receptor acts as a Ca2+ channel. | Purkinje cell death occurs from P10 to P65. | Antero-posterior neurodegeneration. Lobe X degeneration is delayed. |
| NPC1 | Spontaneous. Npc1 gene is mutated. | Intracellular cholesterol transport is altered. | Purkinje cell degeneration starts at P40. | Antero-posterior neurodegeneration. Lobe X is the most resistant region. |
| Nervous | Spontaneous. Located in chromosome 8 but unknown. | Unknown | Neurodegeneration starts at birth and slows down after two months. | Zebrin II positive Purkinje cells are more vulnerable. Lobe X shows some surviving cells. |
| PCD | Spontaneous. pcd/pcd. | Ccp1 gene affected. Hyper-glutamylation of microtubules. | Purkinje cell death starts around P18. | Lobe X is the last lobe to degenerate. Some survival cells detected at 9 months. |
| Tambaleante | Spontaneous. Glycine-to-glutamate substitution. | E3 ubiquitin ligase protein HERC1 overexpressed. | Some Purkinje cells remain alive at 2.5 months. | Random. Lobe X cells remain alive a few days longer. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Hernández-Pérez, C.; Weruaga, E.; Díaz, D. Lobe X of the Cerebellum: A Natural Neuro-Resistant Region. Anatomia 2023, 2, 43-62. https://doi.org/10.3390/anatomia2010005
AMA Style
Hernández-Pérez C, Weruaga E, Díaz D. Lobe X of the Cerebellum: A Natural Neuro-Resistant Region. Anatomia. 2023; 2(1):43-62. https://doi.org/10.3390/anatomia2010005
Chicago/Turabian StyleHernández-Pérez, Carlos, Eduardo Weruaga, and David Díaz. 2023. "Lobe X of the Cerebellum: A Natural Neuro-Resistant Region" Anatomia 2, no. 1: 43-62. https://doi.org/10.3390/anatomia2010005