
Heat Shock Proteins: Important Helpers for the Development, Maintenance and Regeneration of Skeletal Muscles
 , and
, and 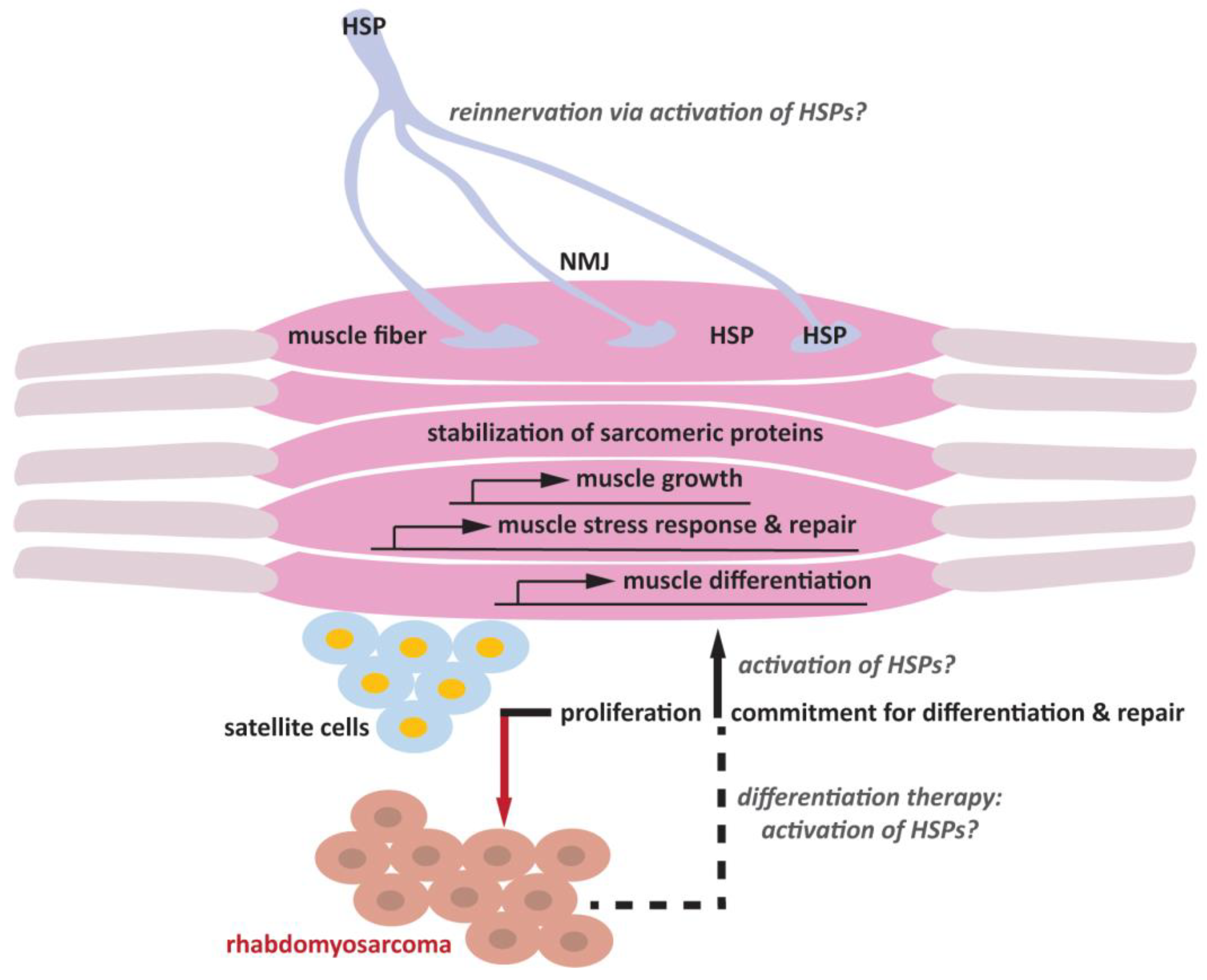{kind=link}
Abstract
:1. Introduction
2. Heat Shock Proteins
3. Heat Shock Proteins and Skeletal Muscle Differentiation
4. Heat Shock Proteins, Muscle Activity and Resistance Training
5. Heat Shock Proteins and Muscle Disease
6. Heat Shock Protein Muscle-Specific Targets
7. Future Research Direction: Regeneration of Muscle Fibers and Neuromuscular Junction
8. Skeletal Muscle Differentiation as a Potential Therapeutic Avenue for Rhabdomyosarcoma
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Taetzsch, T.; Valdez, G. NMJ maintenance and repair in aging. Curr. Opin. Physiol. 2018, 4, 57–64. [Google Scholar] [CrossRef]
- Dumont, N.A.; Wang, Y.X.; Rudnicki, M.A. Intrinsic and extrinsic mechanisms regulating satellite cell function. Development 2015, 142, 1572–1581. [Google Scholar] [CrossRef] [PubMed]
- Le Grand, F.; Rudnicki, M.A. Skeletal muscle satellite cells and adult myogenesis. Curr. Opin. Cell Biol. 2007, 19, 628–633. [Google Scholar] [CrossRef] [PubMed]
- Emery, A.E. The muscular dystrophies. Lancet 2002, 359, 687–695. [Google Scholar] [CrossRef] [PubMed]
- Skapek, S.X.; Ferrari, A.; Gupta, A.A.; Lupo, P.J.; Butler, E.; Shipley, J.; Barr, F.G.; Hawkins, D.S. Rhabdomyosarcoma. Nat. Rev. Dis. Prim. 2019, 5, 1. [Google Scholar] [CrossRef]
- Chal, J.; Pourquié, O. Making muscle: Skeletal myogenesis in vivo and in vitro. Development 2017, 144, 2104–2122. [Google Scholar] [CrossRef] [PubMed]
- Snijders, T.; Nederveen, J.P.; McKay, B.R.; Joanisse, S.; Verdijk, L.B.; van Loon, L.J.C.; Parise, G. Satellite cells in human skeletal muscle plasticity. Front. Physiol. 2015, 6, 283. [Google Scholar] [CrossRef]
- Murphy, M.M.; Lawson, J.A.; Mathew, S.J.; Hutcheson, D.A.; Kardon, G. Satellite cells, connective tissue fibroblasts and their interactions are crucial for muscle regeneration. Development 2011, 138, 3625–3637. [Google Scholar] [CrossRef]
- Kim, Y.E.; Hipp, M.S.; Bracher, A.; Hayer-Hartl, M.; Ulrich Hartl, F. Molecular Chaperone Functions in Protein Folding and Proteostasis. Annu. Rev. Biochem. 2013, 82, 323–355. [Google Scholar] [CrossRef]
- Jayaraj, G.G.; Hipp, M.S.; Hartl, F.U. Functional Modules of the Proteostasis Network. Cold Spring Harb. Perspect. Biol. 2020, 12, a033951. [Google Scholar] [CrossRef]
- Craig, E.A.; Weissman, J.S.; Horwich, A.L. Heat shock proteins and molecular chaperones: Mediators of protein conformation and turnover in the cell. Cell 1994, 78, 365–372. [Google Scholar] [CrossRef]
- Morimoto, R.I. Regulation of the heat shock transcriptional response: Cross talk between a family of heat shock factors, molecular chaperones, and negative regulators. Genes Dev. 1998, 12, 3788–3796. [Google Scholar] [CrossRef] [PubMed]
- Brehme, M.; Voisine, C.; Rolland, T.; Wachi, S.; Soper, J.H.; Zhu, Y.; Orton, K.; Villella, A.; Garza, D.; Vidal, M.; et al. A Chaperome Subnetwork Safeguards Proteostasis in Aging and Neurodegenerative Disease. Cell Rep. 2014, 9, 1135–1150. [Google Scholar] [CrossRef]
- Calderwood, S.K.; Gong, J. Heat Shock Proteins Promote Cancer: It’s a Protection Racket. Trends Biochem. Sci. 2016, 41, 311–323. [Google Scholar] [CrossRef]
- Haslbeck, M.; Weinkauf, S.; Buchner, J. Small heat shock proteins: Simplicity meets complexity. J. Biol. Chem. 2019, 294, 2121–2132. [Google Scholar] [CrossRef] [PubMed]
- Kappé, G.; Franck, E.; Verschuure, P.; Boelens, W.C.; Leunissen, J.A.M.; De Jong, W.W. The human genome encodes 10 α-crystallin–related small heat shock proteins: HspB1–10. Cell Stress Chaperones 2003, 8, 53–61. [Google Scholar] [CrossRef]
- Fontaine, J.-M.; Rest, J.S.; Welsh, M.J.; Benndorf, R. The sperm outer dense fiber protein is the 10th member of the superfamily of mammalian small stress proteins. Cell Stress Chaperones 2003, 8, 62–69. [Google Scholar] [CrossRef] [PubMed]
- Tiago, T.; Hummel, B.; Morelli, F.F.; Basile, V.; Vinet, J.; Galli, V.; Mediani, L.; Antoniani, F.; Pomella, S.; Cassandri, M.; et al. Small heat-shock protein HSPB3 promotes myogenesis by regulating the lamin B receptor. Cell Death Dis. 2021, 12, 452. [Google Scholar] [CrossRef]
- Sugiyama, Y.; Suzuki, A.; Kishikawa, M.; Akutsu, R.; Hirose, T.; Waye, M.M.Y.; Tsui, S.K.W.; Yoshida, S.; Ohno, S. Muscle Develops a Specific Form of Small Heat Shock Protein Complex Composed of MKBP/HSPB2 and HSPB3 during Myogenic Differentiation. J. Biol. Chem. 2000, 275, 1095–1104. [Google Scholar] [CrossRef]
- Ritossa, F. Discovery of the heat shock response. Cell Stress Chaperones 1996, 1, 97–98. [Google Scholar] [CrossRef]
- Åkerfelt, M.; Morimoto, R.I.; Sistonen, L. Heat shock factors: Integrators of cell stress, development and lifespan. Nat. Rev. Mol. Cell Biol. 2010, 11, 545–555. [Google Scholar] [CrossRef] [PubMed]
- Åkerfelt, M.; Trouillet, D.; Mezger, V.; Sistonen, L. Heat Shock Factors at a Crossroad between Stress and Development. Ann. N. Y. Acad. Sci. 2007, 1113, 15–27. [Google Scholar] [CrossRef]
- Le Breton, L.; Mayer, M.P. A model for handling cell stress. Elife 2016, 5, e22850. [Google Scholar] [CrossRef]
- Fehrenbach, E.; Niess, A.M. Role of heat shock proteins in the exercise response. Exerc. Immunol. Rev. 1999, 5, 57–77. [Google Scholar]
- Weihl, C.C.; Töpf, A.; Bengoechea, R.; Duff, J.; Charlton, R.; Garcia, S.K.; Domínguez-González, C.; Alsaman, A.; Hernández-Laín, A.; Franco, L.V.; et al. Loss of function variants in DNAJB4 cause a myopathy with early respiratory failure. Acta Neuropathol. 2023, 145, 127–143. [Google Scholar] [CrossRef] [PubMed]
- Irobi, J.; Van Impe, K.; Seeman, P.; Jordanova, A.; Dierick, I.; Verpoorten, N.; Michalik, A.; De Vriendt, E.; Jacobs, A.; Van Gerwen, V.; et al. Hot-spot residue in small heat-shock protein 22 causes distal motor neuropathy. Nat. Genet. 2004, 36, 597–601. [Google Scholar] [CrossRef]
- Evgrafov, O.V.; Mersiyanova, I.; Irobi, J.; Van Den Bosch, L.; Dierick, I.; Leung, C.L.; Schagina, O.; Verpoorten, N.; Van Impe, K.; Fedotov, V.; et al. Mutant small heat-shock protein 27 causes axonal Charcot-Marie-Tooth disease and distal hereditary motor neuropathy. Nat. Genet. 2004, 36, 602–606. [Google Scholar] [CrossRef]
- Selcen, D.; Muntoni, F.; Burton, B.K.; Pegoraro, E.; Sewry, C.; Bite, A.V.; Engel, A.G. Mutation in BAG3 causes severe dominant childhood muscular dystrophy. Ann. Neurol. 2009, 65, 83–89. [Google Scholar] [CrossRef] [PubMed]
- Saveri, P.; Magri, S.; Maderna, E.; Balistreri, F.; Lombardi, R.; Ciano, C.; Moda, F.; Garavaglia, B.; Reale, C.; Pinter, G.L.; et al. DNAJB2-related Charcot-Marie-Tooth disease type 2: Pathomechanism insights and phenotypic spectrum widening. Eur. J. Neurol. 2022, 29, 2056–2065. [Google Scholar] [CrossRef]
- Sarparanta, J.; Jonson, P.H.; Kawan, S.; Udd, B. Neuromuscular Diseases Due to Chaperone Mutations: A Review and Some New Results. Int. J. Mol. Sci. 2020, 21, 1409. [Google Scholar] [CrossRef]
- Harms, M.B.; Sommerville, R.B.; Allred, P.; Bell, S.; Ma, D.; Cooper, P.; Lopate, G.; Pestronk, A.; Weihl, C.C.; Baloh, R.H. Exome sequencing reveals DNAJB6 mutations in dominantly-inherited myopathy. Ann. Neurol. 2012, 71, 407–416. [Google Scholar] [CrossRef]
- Ghaoui, R.; Palmio, J.; Brewer, J.; Lek, M.; Needham, M.; Evilä, A.; Hackman, P.; Jonson, P.-H.; Penttilä, S.; Vihola, A.; et al. Mutations in HSPB8 causing a new phenotype of distal myopathy and motor neuropathy. Neurology 2015, 86, 391–398. [Google Scholar] [CrossRef] [PubMed]
- Molkentin, J.D.; Olson, E.N. Combinatorial control of muscle development by basic helix-loop-helix and MADS-box transcription factors. Proc. Natl. Acad. Sci. USA 1996, 93, 9366–9373. [Google Scholar] [CrossRef] [PubMed]
- Gopal-Srivastava, R.; Piatigorsky, J. The murine alpha B-crystallin/small heat shock protein enhancer: Identification of alpha BE-1, alpha BE-2, alpha BE-3, and MRF control elements. Mol. Cell. Biol. 1993, 13, 7144–7152. [Google Scholar] [CrossRef]
- Echeverría, P.C.; Briand, P.-A.; Picard, D. A Remodeled Hsp90 Molecular Chaperone Ensemble with the Novel Cochaperone Aarsd1 Is Required for Muscle Differentiation. Mol. Cell. Biol. 2016, 36, 1310–1321. [Google Scholar] [CrossRef] [PubMed]
- Wagatsuma, A.; Shiozuka, M.; Kotake, N.; Takayuki, K.; Yusuke, H.; Mabuchi, K.; Matsuda, R.; Yamada, S. Pharmacological inhibition of HSP90 activity negatively modulates myogenic differentiation and cell survival in C2C12 cells. Mol. Cell. Biochem. 2011, 358, 265–280. [Google Scholar] [CrossRef] [PubMed]
- Bar-Lavan, Y.; Shemesh, N.; Dror, S.; Ofir, R.; Yeger-Lotem, E.; Ben-Zvi, A. A Differentiation Transcription Factor Establishes Muscle-Specific Proteostasis in Caenorhabditis elegans. PLOS Genet. 2016, 12, e1006531. [Google Scholar] [CrossRef] [PubMed]
- Tucker, N.R.; Ustyugov, A.; Bryantsev, A.L.; Konkel, M.E.; Shelden, E.A. Hsp27 is persistently expressed in zebrafish skeletal and cardiac muscle tissues but dispensable for their morphogenesis. Cell Stress Chaperones 2009, 14, 521–533. [Google Scholar] [CrossRef]
- Middleton, R.C.; Shelden, E.A. Small heat shock protein HSPB1 regulates growth of embryonic zebrafish craniofacial muscles. Exp. Cell Res. 2013, 319, 860–874. [Google Scholar] [CrossRef]
- Thorsteinsdóttir, S.; Deries, M.; Cachaço, A.S.; Bajanca, F. The extracellular matrix dimension of skeletal muscle development. Dev. Biol. 2011, 354, 191–207. [Google Scholar] [CrossRef]
- Goody, M.F.; Sher, R.B.; Henry, C.A. Hanging on for the ride: Adhesion to the extracellular matrix mediates cellular responses in skeletal muscle morphogenesis and disease. Dev. Biol. 2015, 401, 75–91. [Google Scholar] [CrossRef] [PubMed]
- Csapo, R.; Gumpenberger, M.; Wessner, B. Skeletal Muscle Extracellular Matrix—What Do We Know About Its Composition, Regulation, and Physiological Roles? A Narrative Review. Front. Physiol. 2020, 11, 253. [Google Scholar] [CrossRef] [PubMed]
- Shemesh, N.; Jubran, J.; Dror, S.; Simonovsky, E.; Basha, O.; Argov, C.; Hekselman, I.; Abu-Qarn, M.; Vinogradov, E.; Mauer, O.; et al. The landscape of molecular chaperones across human tissues reveals a layered architecture of core and variable chaperones. Nat. Commun. 2021, 12, 2180. [Google Scholar] [CrossRef] [PubMed]
- Obi, S.; Nakajima, T.; Hasegawa, T.; Nakamura, F.; Sakuma, M.; Toyoda, S.; Tei, C.; Inoue, T. Heat induces myogenic transcription factors of myoblast cells via transient receptor potential vanilloid 1 (Trpv1). FEBS Open Bio 2018, 9, 101–113. [Google Scholar] [CrossRef]
- Noble, E.G.; Milne, K.J.; Melling, C.J. Heat shock proteins and exercise: A primer. Appl. Physiol. Nutr. Metab. 2008, 33, 1050–1075. [Google Scholar] [CrossRef]
- Milne, K.J.; Noble, E.G. Exercise-induced elevation of HSP70 is intensity dependent. J. Appl. Physiol. 2002, 93, 561–568. [Google Scholar] [CrossRef]
- Kampinga, H.H.; Hageman, J.; Vos, M.J.; Kubota, H.; Tanguay, R.M.; Bruford, E.A.; Cheetham, M.E.; Chen, B.; Hightower, L.E. Guidelines for the nomenclature of the human heat shock proteins. Cell Stress Chaperones 2009, 14, 105–111. [Google Scholar] [CrossRef]
- Krüger, K.; Reichel, T.; Zeilinger, C. Role of heat shock proteins 70/90 in exercise physiology and exercise immunology and their diagnostic potential in sports. J. Appl. Physiol. 2019, 126, 916–927. [Google Scholar] [CrossRef]
- Senf, S.M. Skeletal muscle heat shock protein 70: Diverse functions and therapeutic potential for wasting disorders. Front. Physiol. 2013, 4, 330. [Google Scholar] [CrossRef]
- Senf, S.M.; Dodd, S.L.; Judge, S. FOXO signaling is required for disuse muscle atrophy and is directly regulated by Hsp70. Am. J. Physiol. Physiol. 2010, 298, C38–C45. [Google Scholar] [CrossRef]
- Senf, S.M.; Howard, T.M.; Ahn, B.; Ferreira, L.F.; Judge, S. Loss of the Inducible Hsp70 Delays the Inflammatory Response to Skeletal Muscle Injury and Severely Impairs Muscle Regeneration. PLoS ONE 2013, 8, e62687. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Gampert, L.; Nething, K.; Steinacker, J.M. Response and function of skeletal muscle heat shock protein 70. Front. Biosci. 2006, 11, 2802–2827. [Google Scholar] [CrossRef]
- Gaiser, A.M.; Kaiser, C.J.O.; Haslbeck, V.; Richter, K. Downregulation of the Hsp90 System Causes Defects in Muscle Cells of Caenorhabditis Elegans. PLoS ONE 2011, 6, e25485. [Google Scholar] [CrossRef] [PubMed]
- Paulsen, G.; Vissing, K.; Kalhovde, J.M.; Ugelstad, I.; Bayer, M.L.; Kadi, F.; Schjerling, P.; Hallén, J.; Raastad, T. Maximal eccentric exercise induces a rapid accumulation of small heat shock proteins on myofibrils and a delayed HSP70 response in humans. Am. J. Physiol. Integr. Comp. Physiol. 2007, 293, R844–R853. [Google Scholar] [CrossRef] [PubMed]
- Thompson, H.S.; Maynard, E.B.; Morales, E.R.; Scordilis, S.P. Exercise-induced HSP27, HSP70 and MAPK responses in human skeletal muscle. Acta Physiol. Scand. 2003, 178, 61–72. [Google Scholar] [CrossRef]
- Frankenberg, N.T.; Lamb, G.D.; Overgaard, K.; Murphy, R.M.; Vissing, K. Small heat shock proteins translocate to the cytoskeleton in human skeletal muscle following eccentric exercise independently of phosphorylation. J. Appl. Physiol. 2014, 116, 1463–1472. [Google Scholar] [CrossRef] [PubMed]
- Dodd, S.L.; Hain, B.; Senf, S.M.; Judge, A.R. Hsp27 inhibits IKKβ-induced NF-κΕ activity and skeletal muscle atrophy. FASEB J. 2009, 23, 3415–3423. [Google Scholar] [CrossRef]
- Ubaida-Mohien, C.; Gonzalez-Freire, M.; Lyashkov, A.; Moaddel, R.; Chia, C.W.; Simonsick, E.M.; Sen, R.; Ferrucci, L. Physical Activity Associated Proteomics of Skeletal Muscle: Being Physically Active in Daily Life May Protect Skeletal Muscle from Aging. Front. Physiol. 2019, 10, 312. [Google Scholar] [CrossRef] [PubMed]
- Sarparanta, J.; Jonson, P.H.; Golzio, C.; Sandell, S.; Luque, H.; Screen, M.; McDonald, K.; Stajich, J.M.; Mahjneh, I.; Vihola, A.; et al. Mutations affecting the cytoplasmic functions of the co-chaperone DNAJB6 cause limb-girdle muscular dystrophy. Nat. Genet. 2012, 44, 450–455. [Google Scholar] [CrossRef]
- Qian, F.-Y.; Guo, Y.-D.; Zu, J.; Zhang, J.-H.; Zheng, Y.-M.; Abdoulaye, I.A.; Pan, Z.-H.; Xie, C.-M.; Gao, H.-C.; Zhang, Z.-J. A novel recessive mutation affecting DNAJB6a causes myofibrillar myopathy. Acta Neuropathol. Commun. 2021, 9, 23. [Google Scholar] [CrossRef]
- Ji, G.; Wang, N.; Han, X.; Wang, Y.; Zhang, J.; Wu, Y.; Wu, H.; Ma, S.; Song, X. Case Report: A Novel Splice-Site Mutation in DNAJB6 Associated with Juvenile-Onset Proximal–Distal Myopathy in a Chinese Patient. Front. Genet. 2022, 13, 925926. [Google Scholar] [CrossRef] [PubMed]
- Stürner, E.; Behl, C. The Role of the Multifunctional BAG3 Protein in Cellular Protein Quality Control and in Disease. Front. Mol. Neurosci. 2017, 10, 177. [Google Scholar] [CrossRef] [PubMed]
- Takayama, S.; Reed, J.C. Molecular chaperone targeting and regulation by BAG family proteins. Nature 2001, 3, E237–E241. [Google Scholar] [CrossRef] [PubMed]
- Jaffer, F.; Murphy, S.; Scoto, M.; Healy, E.; Rossor, A.; Brandner, S.; Phadke, R.; Selcen, D.; Jungbluth, H.; Muntoni, F.; et al. BAG3 mutations: Another cause of giant axonal neuropathy. J. Peripher. Nerv. Syst. 2012, 17, 210–216. [Google Scholar] [CrossRef] [PubMed]
- Al-Tahan, S.; Weiss, L.; Yu, H.; Tang, S.; Saporta, M.; Vihola, A.; Mozaffar, T.; Udd, B.; Kimonis, V. New family with HSPB8-associated autosomal dominant rimmed vacuolar myopathy. Neurol. Genet. 2019, 5, e349. [Google Scholar] [CrossRef]
- Vicart, P.; Caron, A.; Guicheney, P.; Li, Z.; Prevost, M.C.; Faure, A.; Chateau, D.; Chapon, F.; Tome, F.; Dupret, J.M.; et al. A Missense Mutation in the αB-Crystallin Chaperone Gene Causes a Desmin-Related Myopathy. Nat. Genet. 1998, 20, 92–95. [Google Scholar] [CrossRef]
- Vendredy, L.; Adriaenssens, E.; Timmerman, V. Small heat shock proteins in neurodegenerative diseases. Cell Stress Chaperones 2020, 25, 679–699. [Google Scholar] [CrossRef]
- Scarlino, S.; Domi, T.; Pozzi, L.; Romano, A.; Pipitone, G.B.; Falzone, Y.M.; Mosca, L.; Penco, S.; Lunetta, C.; Sansone, V.; et al. Burden of Rare Variants in ALS and Axonal Hereditary Neuropathy Genes Influence Survival in ALS: Insights from a Next Generation Sequencing Study of an Italian ALS Cohort. Int. J. Mol. Sci. 2020, 21, 3346. [Google Scholar] [CrossRef]
- Chen, J.; Liu, X.; Xu, Y.; Fan, D. Rare variants of HSPB1 are probably associated with amyotrophic lateral sclerosis. Nan Fang Yi Ke Da Xue Xue Bao 2021, 41, 75–78. [Google Scholar] [CrossRef]
- Thakur, S.S.; Swiderski, K.; Ryall, J.; Lynch, G.S. Therapeutic potential of heat shock protein induction for muscular dystrophy and other muscle wasting conditions. Philos. Trans. R. Soc. B Biol. Sci. 2017, 373, 20160528. [Google Scholar] [CrossRef]
- Miyabara, E.; Martin, J.L.; Griffin, T.M.; Moriscot, A.S.; Mestril, R. Overexpression of inducible 70-kDa heat shock protein in mouse attenuates skeletal muscle damage induced by cryolesioning. Am. J. Physiol. Physiol. 2006, 290, C1128–C1138. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.-C.; Lin, C.-H.; Lin, C.-Y.; Lee, C.-C.; Lin, M.-T.; Wen, H.-C. Transgenic overexpression of heat shock protein 72 in mouse muscle protects against exhaustive exercise-induced skeletal muscle damage. J. Formos. Med. Assoc. 2013, 112, 24–30. [Google Scholar] [CrossRef] [PubMed]
- Miyabara, E.H.; Nascimento, T.L.; Rodrigues, D.C.; Moriscot, A.S.; Davila, W.F.; AitMou, Y.; Detombe, P.P.; Mestril, R. Overexpression of inducible 70-kDa heat shock protein in mouse improves structural and functional recovery of skeletal muscles from atrophy. Pflügers Arch.-Eur. J. Physiol. 2012, 463, 733–741. [Google Scholar] [CrossRef] [PubMed]
- Kayani, A.C.; Close, G.L.; Dillmann, W.H.; Mestril, R.; Jackson, M.J.; McArdle, A. Overexpression of HSP10 in skeletal muscle of transgenic mice prevents the age-related fall in maximum tetanic force generation and muscle cross-sectional area. Am. J. Physiol. Integr. Comp. Physiol. 2010, 299, R268–R276. [Google Scholar] [CrossRef] [PubMed]
- Herzog, W. The multiple roles of titin in muscle contraction and force production. Biophys. Rev. 2018, 10, 1187–1199. [Google Scholar] [CrossRef]
- Squire, J. Special Issue: The Actin-Myosin Interaction in Muscle: Background and Overview. Int. J. Mol. Sci. 2019, 20, 5715. [Google Scholar] [CrossRef]
- Gigli, M.; Begay, R.L.; Morea, G.; Graw, S.L.; Sinagra, G.; Taylor, M.R.G.; Granzier, H.; Mestroni, L. A Review of the Giant Protein Titin in Clinical Molecular Diagnostics of Cardiomyopathies. Front. Cardiovasc. Med. 2016, 3, 21. [Google Scholar] [CrossRef]
- Minajeva, A.; Kulke, M.; Fernandez, J.M.; Linke, W.A. Unfolding of Titin Domains Explains the Viscoelastic Behavior of Skeletal Myofibrils. Biophys. J. 2001, 80, 1442–1451. [Google Scholar] [CrossRef]
- Bullard, B.; Ferguson, C.; Minajeva, A.; Leake, M.C.; Gautel, M.; Labeit, D.; Ding, L.; Labeit, S.; Horwitz, J.; Leonard, K.R.; et al. Association of the Chaperone αB-Crystallin with Titin in Heart Muscle. J. Biol. Chem. 2004, 279, 7917–7924. [Google Scholar] [CrossRef]
- Kötter, S.; Unger, A.; Hamdani, N.; Lang, P.; Vorgerd, M.; Nagel-Steger, L.; Linke, W.A. Human myocytes are protected from titin aggregation-induced stiffening by small heat shock proteins. J. Cell Biol. 2014, 204, 187–202. [Google Scholar] [CrossRef]
- Unger, A.; Beckendorf, L.; Böhme, P.; Kley, R.; Von Frieling-Salewsky, M.; Lochmüller, H.; Schröder, R.; Fürst, D.O.; Vorgerd, M.; Linke, W.A. Translocation of molecular chaperones to the titin springs is common in skeletal myopathy patients and affects sarcomere function. Acta Neuropathol. Commun. 2017, 5, 72. [Google Scholar] [CrossRef] [PubMed]
- Voelkel, T.; Andresen, C.; Unger, A.; Just, S.; Rottbauer, W.; Linke, W.A. Lysine methyltransferase Smyd2 regulates Hsp90-mediated protection of the sarcomeric titin springs and cardiac function. Biochim. Biophys. Acta (BBA)—Mol. Cell Res. 2013, 1833, 812–822. [Google Scholar] [CrossRef] [PubMed]
- Arndt, V.; Dick, N.; Tawo, R.; Dreiseidler, M.; Wenzel, D.; Hesse, M.; Fürst, D.O.; Saftig, P.; Saint, R.; Fleischmann, B.K.; et al. Chaperone-Assisted Selective Autophagy Is Essential for Muscle Maintenance. Curr. Biol. 2010, 20, 143–148. [Google Scholar] [CrossRef] [PubMed]
- González-Morales, N.; Holenka, T.K.; Schöck, F. Filamin actin-binding and titin-binding fulfill distinct functions in Z-disc cohesion. PLOS Genet. 2017, 13, e1006880. [Google Scholar] [CrossRef]
- Collier, M.P.; Alderson, T.R.; de Villiers, C.P.; Nicholls, D.; Gastall, H.Y.; Allison, T.M.; Degiacomi, M.T.; Jiang, H.; Mlynek, G.; Fürst, D.O.; et al. HspB1 phosphorylation regulates its intramolecular dynamics and mechanosensitive molecular chaperone interaction with filamin C. Sci. Adv. 2019, 5, eaav8421. [Google Scholar] [CrossRef]
- Juo, L.-Y.; Liao, W.-C.; Shih, Y.-L.; Yang, B.-Y.; Liu, A.-B.; Yan, Y.-T. HSPB7 interacts with dimerized FLNC and its absence results in progressive myopathy in skeletal muscles. J. Cell Sci. 2016, 129, 1661–1670. [Google Scholar] [CrossRef] [PubMed]
- Homma, S.; Iwasaki, M.; Shelton, G.D.; Engvall, E.; Reed, J.C.; Takayama, S. BAG3 Deficiency Results in Fulminant Myopathy and Early Lethality. Am. J. Pathol. 2006, 169, 761–773. [Google Scholar] [CrossRef]
- Lavoie, J.; Gingras-Breton, G.; Tanguay, R.; Landry, J. Induction of Chinese hamster HSP27 gene expression in mouse cells confers resistance to heat shock. HSP27 stabilization of the microfilament organization. J. Biol. Chem. 1993, 268, 3420–3429. [Google Scholar] [CrossRef]
- Lavoie, J.; Hickey, E.; Weber, L.; Landry, J. Modulation of actin microfilament dynamics and fluid phase pinocytosis by phosphorylation of heat shock protein 27. J. Biol. Chem. 1993, 268, 24210–24214. [Google Scholar] [CrossRef]
- Guay, J.; Lambert, H.; Gingras-Breton, G.; Lavoie, J.; Huot, J.; Landry, J. Regulation of actin filament dynamics by p38 map kinase-mediated phosphorylation of heat shock protein 27. J. Cell Sci. 1997, 110 Pt 3, 357–368. [Google Scholar] [CrossRef]
- Lavoie, J.N.; Lambert, H.; Hickey, E.; Weber, L.A.; Landry, J. Modulation of cellular thermoresistance and actin filament stability accompanies phosphorylation-induced changes in the oligomeric structure of heat shock protein 27. Mol. Cell. Biol. 1995, 15, 505–516. [Google Scholar] [CrossRef] [PubMed]
- Perng, M.D.; Cairns, L.; van den IJssel, P.; Prescott, A.; Hutcheson, A.M.; Quinlan, R.A. Intermediate Filament Interactions can be Altered by HSP27 and alphaB-Crystallin. J. Cell Sci. 1999, 112 Pt 13, 2099–2112. [Google Scholar] [CrossRef] [PubMed]
- Djabali, K.; Piron, G.; de Néchaud, B.; Portier, M.-M. αB-Crystallin Interacts with Cytoplasmic Intermediate Filament Bundles during Mitosis. Exp. Cell Res. 1999, 253, 649–662. [Google Scholar] [CrossRef]
- Wu, T.; Mu, Y.; Bogomolovas, J.; Fang, X.; Veevers, J.; Nowak, R.B.; Pappas, C.T.; Gregorio, C.C.; Evans, S.M.; Fowler, V.M.; et al. HSPB7 is indispensable for heart development by modulating actin filament assembly. Proc. Natl. Acad. Sci. USA 2017, 114, 11956–11961. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.; Conover, G.M.; Elliott, J.L.; Der Perng, M.; Herrmann, H.; Quinlan, R.A. αB-crystallin is a sensor for assembly intermediates and for the subunit topology of desmin intermediate filaments. Cell Stress Chaperones 2017, 22, 613–626. [Google Scholar] [CrossRef] [PubMed]
- Elliott, J.L.; Der Perng, M.; Prescott, A.R.; Jansen, K.A.; Koenderink, G.H.; Quinlan, R.A. The Specificity of the Interaction between αB-Crystallin and Desmin Filaments and its Impact on Filament Aggregation and Cell Viability. Philos. Trans. R. Soc. Lond. B. Biol. Sci. 2013, 368, 20120375. [Google Scholar] [CrossRef]
- Ojima, K.; Ichimura, E.; Suzuki, T.; Oe, M.; Muroya, S.; Nishimura, T. HSP90 modulates the myosin replacement rate in myofibrils. Am. J. Physiol. Physiol. 2018, 315, C104–C114. [Google Scholar] [CrossRef]
- Fennel, Z.J.; Amorim, F.T.; Deyhle, M.R.; Hafen, P.S.; Mermier, C.M. The Heat Shock Connection: Skeletal Muscle Hypertrophy and Atrophy. Am. J. Physiol. Integr. Comp. Physiol. 2022, 323, R133–R148. [Google Scholar] [CrossRef]
- Bodine, S.C.; Stitt, T.N.; Gonzalez, M.; Kline, W.O.; Stover, G.L.; Bauerlein, R.; Zlotchenko, E.; Scrimgeour, A.; Lawrence, J.C.; Glass, D.J.; et al. Akt/mTOR pathway is a crucial regulator of skeletal muscle hypertrophy and can prevent muscle atrophy in vivo. Nature 2001, 3, 1014–1019. [Google Scholar] [CrossRef]
- Fan, W.; Gao, X.K.; Rao, X.S.; Shi, Y.P.; Liu, X.C.; Wang, F.Y.; Liu, Y.F.; Cong, X.X.; He, M.Y.; Xu, S.B.; et al. Hsp70 Interacts with Mitogen-Activated Protein Kinase (MAPK)-Activated Protein Kinase 2 To Regulate p38MAPK Stability and Myoblast Differentiation during Skeletal Muscle Regeneration. Mol. Cell. Biol. 2018, 38, e00211-18. [Google Scholar] [CrossRef]
- Shaknovich, R.; Shue, G.; Kohtz, D.S. Conformational Activation of a Basic Helix-Loop-Helix Protein (MyoDl) by the C-Terminal Region of Murine HSP90 (HSP84). Mol. Cell. Biol. 1992, 12, 288–299. [Google Scholar] [CrossRef] [PubMed]
- Singh, B.N.; Rao, K.S.; Rao, C.M. Ubiquitin–proteasome-mediated degradation and synthesis of MyoD is modulated by αB-crystallin, a small heat shock protein, during muscle differentiation. Biochim. Biophys. Acta (BBA)—Mol. Cell Res. 2010, 1803, 288–299. [Google Scholar] [CrossRef] [PubMed]
- Clarkson, P.M.; Tremblay, I. Exercise-induced muscle damage, repair, and adaptation in humans. J. Appl. Physiol. 1988, 65, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Deschenes, M.R.; Li, S.; Adan, M.A.; Oh, J.J.; Ramsey, H.C. Muscle fibers and their synapses differentially adapt to aging and endurance training. Exp. Gerontol. 2018, 106, 183–191. [Google Scholar] [CrossRef] [PubMed]
- Deschenes, M.; Tenny, K.; Wilson, M. Increased and decreased activity elicits specific morphological adaptations of the neuromuscular junction. Neuroscience 2006, 137, 1277–1283. [Google Scholar] [CrossRef]
- Ebiressi, S. The quasi-parallel lives of satellite cells and atrophying muscle. Front. Aging Neurosci. 2015, 7, 140. [Google Scholar] [CrossRef]
- Jensen, L.; Jørgensen, L.H.; Bech, R.D.; Frandsen, U.; Schrøder, H.D. Skeletal Muscle Remodelling as a Function of Disease Progression in Amyotrophic Lateral Sclerosis. BioMed Res. Int. 2016, 2016, 5930621. [Google Scholar] [CrossRef]
- Dadon-Nachum, M.; Melamed, E.; Offen, D. The “Dying-Back” Phenomenon of Motor Neurons in ALS. J. Mol. Neurosci. 2011, 43, 470–477. [Google Scholar] [CrossRef]
- Fischer, L.R.; Culver, D.G.; Tennant, P.; Davis, A.A.; Wang, M.; Castellano-Sanchez, A.; Khan, J.; Polak, M.A.; Glass, J.D. Amyotrophic lateral sclerosis is a distal axonopathy: Evidence in mice and man. Exp. Neurol. 2004, 185, 232–240. [Google Scholar] [CrossRef]
- Gentile, F.; Scarlino, S.; Falzone, Y.M.; Lunetta, C.; Tremolizzo, L.; Quattrini, A.; Riva, N. The Peripheral Nervous System in Amyotrophic Lateral Sclerosis: Opportunities for Translational Research. Front. Neurosci. 2019, 13, 601. [Google Scholar] [CrossRef]
- Picchiarelli, G.; Demestre, M.; Zuko, A.; Been, M.; Higelin, J.; Dieterlé, S.; Goy, M.-A.; Mallik, M.; Sellier, C.; Scekic-Zahirovic, J.; et al. FUS-mediated regulation of acetylcholine receptor transcription at neuromuscular junctions is compromised in amyotrophic lateral sclerosis. Nat. Neurosci. 2019, 22, 1793–1805. [Google Scholar] [CrossRef] [PubMed]
- Spaulding, E.L.; Sleigh, J.N.; Morelli, K.H.; Pinter, M.J.; Burgess, R.W.; Seburn, K.L. Synaptic Deficits at Neuromuscular Junctions in Two Mouse Models of Charcot–Marie–Tooth Type 2d. J. Neurosci. 2016, 36, 3254–3267. [Google Scholar] [CrossRef] [PubMed]
- Calabrese, V.; Scapagnini, G.; Ravagna, A.; Giuffrida Stella, A.M.; Butterfield, D.A. Molecular chaperones and their roles in neural cell differentiation. Dev. Neurosci. 2002, 24, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Brown, I.R.; Rush, S.J. Expression of heat shock genes (hsp70) in the mammalian brain: Distinguishing constitutively expressed and hyperthermia-inducible mRNA species. J. Neurosci. Res. 1990, 25, 14–19. [Google Scholar] [CrossRef] [PubMed]
- D’Souza, S.M.; Brown, I.R. Constitutive expression of heat shock proteins Hsp90, Hsc70, Hsp70 and Hsp60 in neural and non-neural tissues of the rat during postnatal development. Cell Stress Chaperones 1998, 3, 188–199. [Google Scholar] [CrossRef]
- Casas-Tintó, S.; Reyes, T.D.L. Neural functions of small heat shock proteins. Neural Regen. Res. 2022, 17, 512. [Google Scholar] [CrossRef]
- McArdle, A.; Dillmann, W.H.; Mestril, R.; Faulkner, J.A.; Jackson, M.J. Overexpression of HSP70 in mouse skeletal muscle protects against muscle damage and age-related muscle dysfunction. FASEB J. 2003, 18, 355–357. [Google Scholar] [CrossRef]
- Selsby, J.T.; Rother, S.; Tsuda, S.; Pracash, O.; Quindry, J.; Dodd, S.L. Intermittent hyperthermia enhances skeletal muscle regrowth and attenuates oxidative damage following reloading. J. Appl. Physiol. 2007, 102, 1702–1707. [Google Scholar] [CrossRef]
- Asthana, P.; Zhang, G.; Sheikh, K.A.; Ma, C.H.E. Heat shock protein is a key therapeutic target for nerve repair in autoimmune peripheral neuropathy and severe peripheral nerve injury. Brain Behav. Immun. 2020, 91, 48–64. [Google Scholar] [CrossRef]
- Luo, S.; Zhang, B.; Dong, X.-P.; Tao, Y.; Ting, A.; Zhou, Z.; Meixiong, J.; Luo, J.; Chiu, F.A.; Xiong, W.C.; et al. HSP90β Regulates Rapsyn Turnover and Subsequent AChR Cluster Formation and Maintenance. Neuron 2008, 60, 97–110. [Google Scholar] [CrossRef]
- Chan, Z.C.-K.; Deng, L.; Lee, C.W. Grp94 Regulates the Recruitment of Aneural AChR Clusters for the Assembly of Postsynaptic Specializations by Modulating ADF/Cofilin Activity and Turnover. eNeuro 2020, 7, ENEURO.0025-20.2020. [Google Scholar] [CrossRef] [PubMed]
- Gess, B.; Auer-Grumbach, M.; Schirmacher, A.; Strom, T.; Zitzelsberger, M.; Rudnik-Schöneborn, S.; Röhr, D.; Halfter, H.; Young, P.; Senderek, J. HSJ1-related hereditary neuropathies: Novel mutations and extended clinical spectrum. Neurology 2014, 83, 1726–1732. [Google Scholar] [CrossRef] [PubMed]
- Claeys, K.G.; Sozanska, M.; Martin, J.-J.; Lacene, E.; Vignaud, L.; Stockholm, D.; Laforêt, P.; Eymard, B.; Kichler, A.; Scherman, D.; et al. DNAJB2 Expression in Normal and Diseased Human and Mouse Skeletal Muscle. Am. J. Pathol. 2010, 176, 2901–2910. [Google Scholar] [CrossRef] [PubMed]
- Raut, S.; Mallik, B.; Parichha, A.; Amrutha, V.; Sahi, C.; Kumar, V. RNAi-Mediated Reverse Genetic Screen Identified Drosophila Chaperones Regulating Eye and Neuromuscular Junction Morphology. G3 Genes Genomes Genet. 2017, 7, 2023–2038. [Google Scholar] [CrossRef]
- Santana, E.; Reyes, T.D.L.; Casas-Tintó, S. Small heat shock proteins determine synapse number and neuronal activity during development. PLoS ONE 2020, 15, e0233231. [Google Scholar] [CrossRef]
- Poon, A.; Zhang, Y.; Chandrasekaran, A.; Phanthong, P.; Schmid, B.; Nielsen, T.T.; Freude, K.K. Modeling neurodegenerative diseases with patient-derived induced pluripotent cells: Possibilities and challenges. New Biotechnol. 2017, 39, 190–198. [Google Scholar] [CrossRef]
- Martins, J.-M.F.; Fischer, C.; Urzi, A.; Vidal, R.; Kunz, S.; Ruffault, P.-L.; Kabuss, L.; Hube, I.; Gazzerro, E.; Birchmeier, C.; et al. Self-Organizing 3D Human Trunk Neuromuscular Organoids. Cell Stem Cell 2020, 27, 498. [Google Scholar] [CrossRef]
- Williamson, D.; Missiaglia, E.; Pritchard-Jones, K.; Oberlin, O.; Shipley, J.; Delattre, O.; De Reyniès, A.; Pierron, G.; Thuille, B.; Palenzuela, G.; et al. Fusion Gene-Negative Alveolar Rhabdomyosarcoma is Clinically and Molecularly Indistinguishable from Embryonal Rhabdomyosarcoma. J. Clin. Oncol. 2010, 28, 2151–2158. [Google Scholar] [CrossRef]
- Shern, J.F.; Selfe, J.; Izquierdo, E.; Patidar, R.; Chou, H.-C.; Song, Y.K.; Yohe, M.E.; Sindiri, S.; Wei, J.; Wen, X.; et al. Genomic Classification and Clinical Outcome in Rhabdomyosarcoma: A Report from an International Consortium. J. Clin. Oncol. 2021, 39, 2859–2871. [Google Scholar] [CrossRef]
- Oberlin, O.; Rey, A.; Lyden, E.; Bisogno, G.; Stevens, M.; Meyer, W.H.; Carli, M.; Anderson, J.R. Prognostic Factors in Metastatic Rhabdomyosarcomas: Results of a Pooled Analysis from United States and European Cooperative Groups. J. Clin. Oncol. 2008, 26, 2384–2389. [Google Scholar] [CrossRef]
- MacQuarrie, K.L.; Yao, Z.; Fong, A.P.; Diede, S.J.; Rudzinski, E.R.; Hawkins, D.S.; Tapscott, S.J. Comparison of Genome-Wide Binding of MyoD in Normal Human Myogenic Cells and Rhabdomyosarcomas Identifies Regional and Local Suppression of Promyogenic Transcription Factors. Mol. Cell. Biol. 2013, 33, 773–784. [Google Scholar] [CrossRef] [PubMed]
- Tenente, I.M.; Hayes, M.N.; Ignatius, M.S.; McCarthy, K.; Yohe, M.; Sindiri, S.; Gryder, B.; Oliveira, M.L.; Ramakrishnan, A.; Tang, Q.; et al. Myogenic regulatory transcription factors regulate growth in rhabdomyosarcoma. Elife 2017, 6, e19214. [Google Scholar] [CrossRef] [PubMed]
- Yohe, M.E.; Gryder, B.E.; Shern, J.F.; Song, Y.K.; Chou, H.-C.; Sindiri, S.; Mendoza, A.; Patidar, R.; Zhang, X.; Guha, R.; et al. MEK inhibition induces MYOG and remodels super-enhancers in RAS-driven rhabdomyosarcoma. Sci. Transl. Med. 2018, 10, eaan4470. [Google Scholar] [CrossRef] [PubMed]
- Ciarapica, R.; Carcarino, E.; Adesso, L.; De Salvo, M.; Bracaglia, G.; Leoncini, P.P.; Dall’agnese, A.; Verginelli, F.; Milano, G.M.; Boldrini, R.; et al. Pharmacological inhibition of EZH2 as a promising differentiation therapy in embryonal RMS. BMC Cancer 2014, 14, 139. [Google Scholar] [CrossRef]
- Vella, S.; Pomella, S.; Leoncini, P.P.; Colletti, M.; Conti, B.; Marquez, V.E.; Strillacci, A.; Roma, J.; Gallego, S.; Milano, G.M.; et al. MicroRNA-101 is repressed by EZH2 and its restoration inhibits tumorigenic features in embryonal rhabdomyosarcoma. Clin. Epigenet. 2015, 7, 82. [Google Scholar] [CrossRef]
- Laubscher, D.; Gryder, B.E.; Sunkel, B.D.; Andresson, T.; Wachtel, M.; Das, S.; Roschitzki, B.; Wolski, W.; Wu, X.S.; Chou, H.-C.; et al. BAF complexes drive proliferation and block myogenic differentiation in fusion-positive rhabdomyosarcoma. Nat. Commun. 2021, 12, 6924. [Google Scholar] [CrossRef]
- Phelps, M.P.; Bailey, J.N.; Vleeshouwer-Neumann, T.; Chen, E.Y. CRISPR screen identifies the NCOR/HDAC3 complex as a major suppressor of differentiation in rhabdomyosarcoma. Proc. Natl. Acad. Sci. USA 2016, 113, 15090–15095. [Google Scholar] [CrossRef]
- Pomella, S.; Sreenivas, P.; Gryder, B.E.; Wang, L.; Milewski, D.; Cassandri, M.; Baxi, K.; Hensch, N.R.; Carcarino, E.; Song, Y.; et al. Interaction between SNAI2 and MYOD enhances oncogenesis and suppresses differentiation in Fusion Negative Rhabdomyosarcoma. Nat. Commun. 2021, 12, 192. [Google Scholar] [CrossRef]
- Li, J.; Labbadia, J.; Morimoto, R.I. Rethinking HSF1 in Stress, Development, and Organismal Health. Trends Cell Biol. 2017, 27, 895–905. [Google Scholar] [CrossRef]
- He, H.; Chen, C.; Xie, Y.; Asea, A.; Calderwood, S.K. HSP70 and heat shock factor 1 cooperate to repress Ras-induced transcriptional activation of the c-fos gene. Cell Stress Chaperones 2000, 5, 406–411. [Google Scholar] [CrossRef]
- Lassar, A.B.; Thayer, M.J.; Overell, R.W.; Weintraub, H. Transformation by activated ras or fos prevents myogenesis by inhibiting expression of MyoD1. Cell 1989, 58, 659–667. [Google Scholar] [CrossRef] [PubMed]
- Gvozdenov, Z.; Bendix, L.D.; Kolhe, J.; Freeman, B.C. The Hsp90 Molecular Chaperone Regulates the Transcription Factor Network Controlling Chromatin Accessibility. J. Mol. Biol. 2019, 431, 4993–5003. [Google Scholar] [CrossRef] [PubMed]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pomella, S.; Cassandri, M.; Antoniani, F.; Crotti, S.; Mediani, L.; Silvestri, B.; Medici, M.; Rota, R.; Rosa, A.; Carra, S. Heat Shock Proteins: Important Helpers for the Development, Maintenance and Regeneration of Skeletal Muscles. Muscles 2023, 2, 187-203. https://doi.org/10.3390/muscles2020014
Pomella S, Cassandri M, Antoniani F, Crotti S, Mediani L, Silvestri B, Medici M, Rota R, Rosa A, Carra S. Heat Shock Proteins: Important Helpers for the Development, Maintenance and Regeneration of Skeletal Muscles. Muscles. 2023; 2(2):187-203. https://doi.org/10.3390/muscles2020014
Chicago/Turabian StylePomella, Silvia, Matteo Cassandri, Francesco Antoniani, Samuele Crotti, Laura Mediani, Beatrice Silvestri, Margherita Medici, Rossella Rota, Alessandro Rosa, and Serena Carra. 2023. "Heat Shock Proteins: Important Helpers for the Development, Maintenance and Regeneration of Skeletal Muscles" Muscles 2, no. 2: 187-203. https://doi.org/10.3390/muscles2020014