
Two Cases of Myofibrillar Myopathies: Genetic and Quality of Life Study



Abstract
:1. Introduction
2. Case 1. Childhood Case of a Thin Girl with Multiple Contractions, Scoliosis and Rigid Spine
2.1. History
2.2. Diagnosis
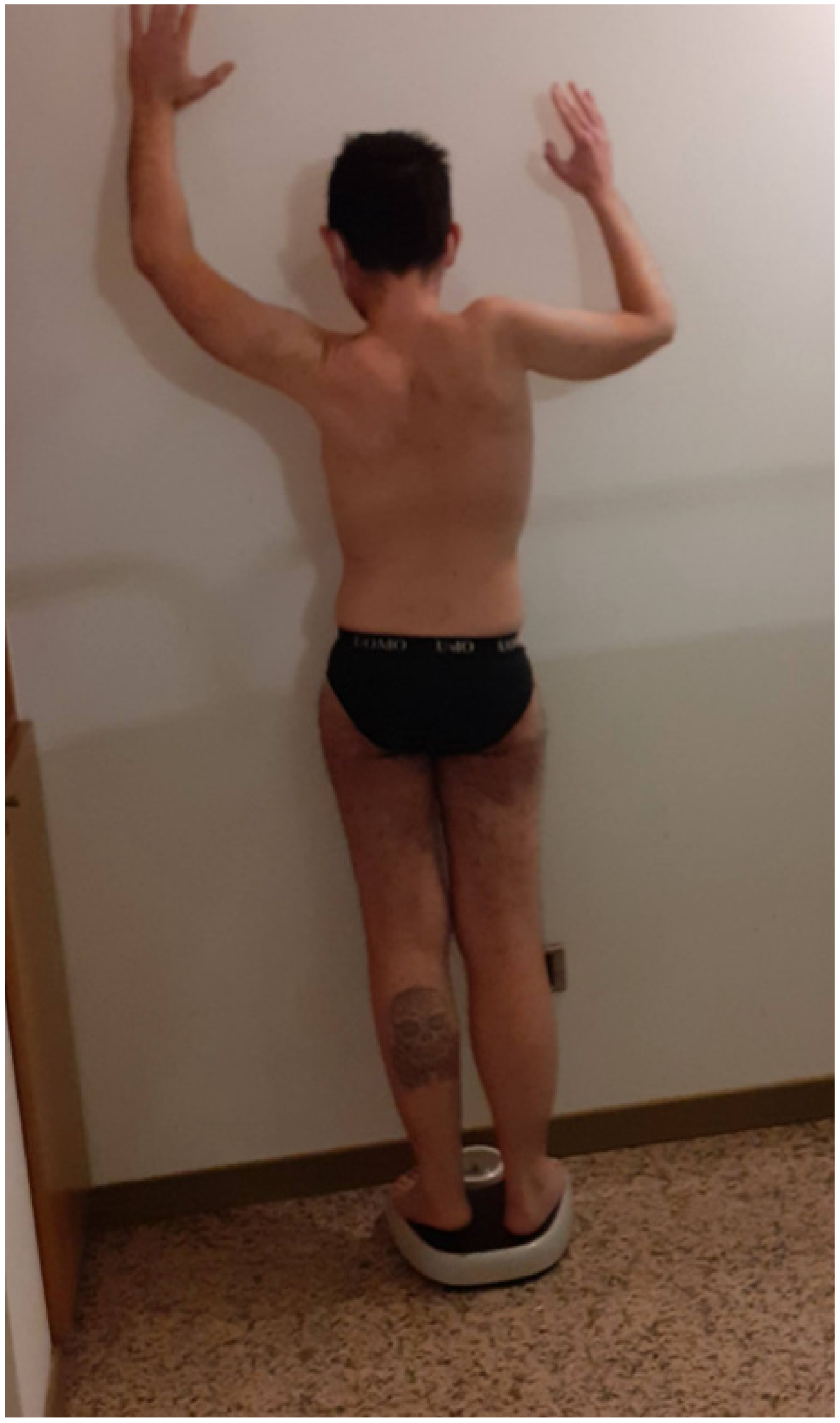
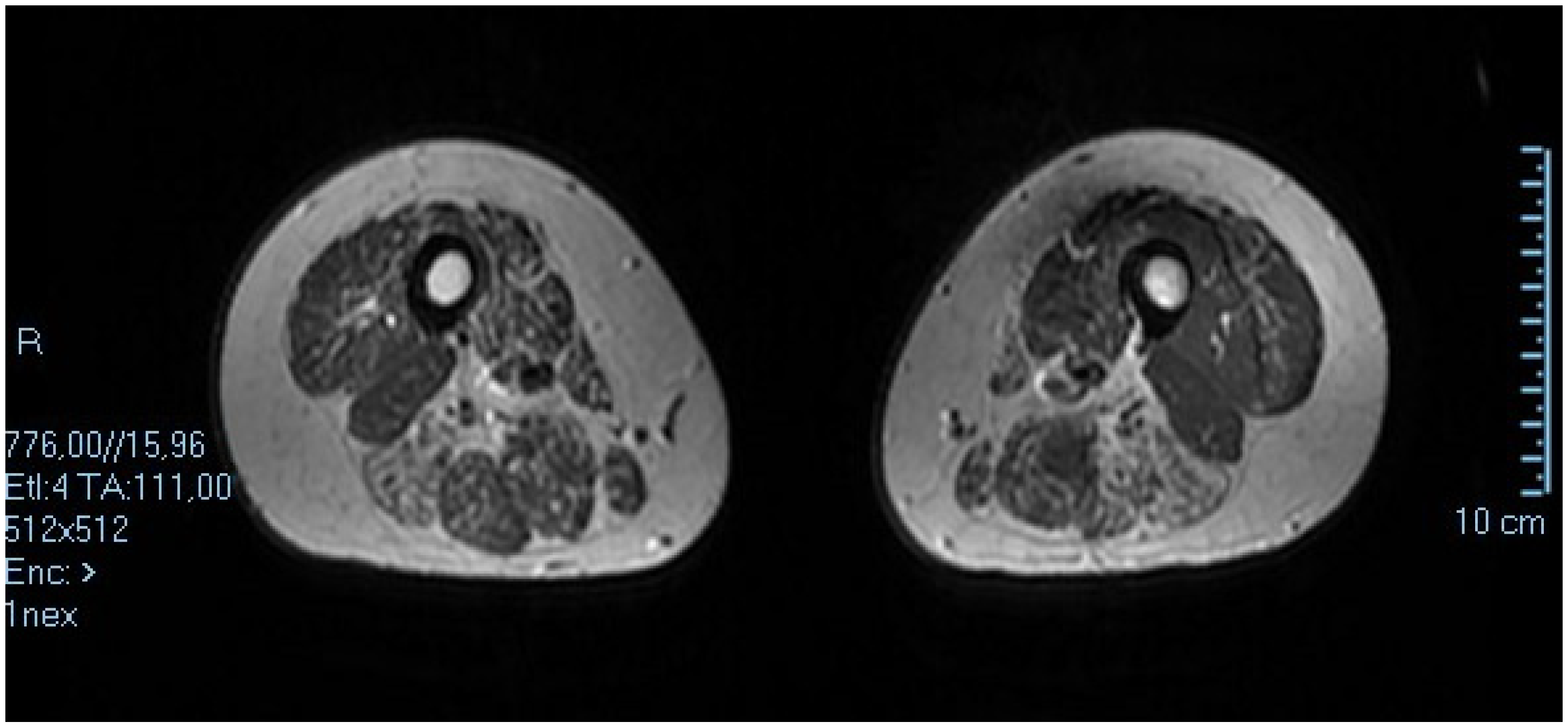
3. Case 2: An Asymmetric Limb-Girdle Myopathy Associated with the LDB3 Variant, Is Found in the Propositus (Figure 2)
3.1. History

3.2. Genetic and Psychosocial Assesment
3.3. Treatment
4. Discussion
4.1. Genetic and Clinical Approach
4.2. Therapeutic Approach
4.3. Psychosocial Issues
4.4. Physical Therapy
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Konersman, C.G.; Bordini, B.J.; Scharer, G.; Lawlor, M.W.; Zangwill, S.; Southern, J.F.; Amos, L.; Geddes, G.C.; Kliegman, R.; Collins, M.P. BAG3 myofibrillar myopathy presenting with cardiomyopathy. Neuromuscul. Disord. 2015, 25, 418–422. [Google Scholar] [CrossRef] [PubMed]
- Odgerel, Z.; Sarkozy, A.; Lee, H.-S.; Mckenna, C.; Rankin, J.; Straub, V.; Lochmüller, H.; Paola, F.; D’Amico, A.; Bertini, E.; et al. Inheritance patterns and phenotypic features of myofibrillar myopathy associated with a BAG3 mutation. Neuromuscul. Disord. 2010, 20, 438–442. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Selcen, D.; Muntoni, F.; Burton, B.K.; Pegoraro, E.; Sewry, C.; Bite, A.V.; Engel, A.G. Mutation in BAG3 causes severe dominant childhood muscular dystrophy. Ann. Neurol. 2009, 65, 83–89. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- D’Avila, F.; Meregalli, M.; Lupoli, S.; Barcella, M.; Orro, A.; De Santis, F.; Sitzia, C.; Farini, A.; D’Ursi, P.; Erratico, S.; et al. Exome sequencing identifies variants in two genes encoding the LIM-proteins NRAP and FHL1 in an Italian patient with BAG3 myofibrillar myopathy. J. Muscle Res. Cell Motil. 2016, 37, 101–115. [Google Scholar] [CrossRef] [Green Version]
- Dubowitz, V.; Brooke, M. Muscle Biopsy: A Modern Approach; Saunders: London, UK, 1973; 220p. [Google Scholar]
- McDermott-Roe, C.; Lv, W.; Maximova, T.; Wada, S.; Bukowy, J.; Marquez, M.; Lai, S.; Shehu, A.; Benjamin, I.; Geurts, A.; et al. Investigation of a dilated cardiomyopathy-associated variant inBAG3 using genome-edited iPSC-derived cardiomyocytes. J. Clin. Investig. 2019, 4, e128799. [Google Scholar] [CrossRef]
- Pfeffer, G.; Povitz, M. Respiratory management of patients with neuromuscular disease: Current perspectives. Degener. Neurol. Neuromuscul. Dis. 2016, 6, 111–118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Angelini, C.; Pegoraro, E.; Turella, E.; Intino, M.T.; Pini, A.; Costa, C. Deflazacort in Duchenne dystrophy: Study of long-term effect. Muscle Nerve Off. J. Am. Assoc. Electrodiagn. Med. 1994, 17, 386–391. [Google Scholar] [CrossRef]
- Lee, H.C.; Cherk, S.W.; Chan, S.K.; Wong, S.; Tong, T.W.; Ho, W.S.; Chan, A.Y.; Lee, K.C.; Mak, C.M. BAG3-related myofibrillar myopathy in a Chinese family. Clin. Genet. 2012, 81, 394–398. [Google Scholar] [CrossRef]
- Jaffer, F.; Murphy, S.M.; Scoto, M.; Healy, E.; Rossor, A.M.; Brandner, S.; Phadke, R.; Selcen, D.; Jungbluth, H.; Muntoni, M. BAG3 mutations: Another cause of giant axonal neuropathy. J. Peripher. Nerv. Syst. 2012, 17, 210–216. [Google Scholar] [CrossRef]
- Kostera-Pruszczyk, A.; Suszek, M.; Płoski, R.; Franaszczyk, M.; Potulska-Chromik, A.; Pruszczyk, P.; Sadurska, E.; Karolczak, J.; Kamińska, A.M.; Rędowicz, M.J. BAG3-related myopathy, polyneuropathy and cardiomyopathy with long QT syndrome. J. Muscle Res. Cell Motil. 2015, 36, 423–432. [Google Scholar] [CrossRef] [Green Version]
- Schänzer, A.; Rupp, S.; Gräf, S.; Zengeler, D.; Jux, C.; Akintürk, H.; Gulatz, L.; Mazhari, N.; Acker, T.; Van Coster, R.; et al. Dysregulated autophagy in re-restrictive cardiomyopathy due to Pro209Leu mutation in BAG3. Mol. Genet. Metab. 2018, 123, 388–399. [Google Scholar] [CrossRef] [PubMed]
- Noury, J.-B.; Maisonobe, T.; Richard, P.; Delague, V.; Malfatti, E.; Stojkovic, T. Rigid spine syndrome associated with sensory-motor axonal neuropathy resembling Charcot-Marie-Tooth disease is characteristic of bcl-2-associated athanogene-3 gene mutations even without cardiac involvement. Muscle Nerve 2018, 57, 330–334. [Google Scholar] [CrossRef] [PubMed]
- Fu, J.; Ma, M.; Song, J.; Pang, M.; Li, G.; Zhang, J. BAG3 p.Pro209Ser mutation identified in a Chinese family with Charcot-Marie-Tooth disease. J. Neurol. 2020, 267, 1080–1085. [Google Scholar] [CrossRef] [PubMed]
- Domínguez, F.; Cuenca, S.; Bilińska, Z.; Toro, R.; Villard, E.; Barriales-Villa, R.; Ochoa, J.P.; Asselbergs, F.; Sammani, A.; Franaszczyk, M. Dilated Cardiomyopathy Due to BLC2-Associated Athanogene 3 (BAG3) Mutations. J. Am. Coll. Cardiol. 2018, 72, 2471–2481. [Google Scholar] [CrossRef] [PubMed]
- Selcen, D. Myofibrillar myopathies. Neuromuscul. Disord. 2011, 21, 161–171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.; Yang, H.; Tan, J.; Fan, Y.; Fan, J.; Xiao, F. A novel LDB3 (c. 1720G > A) mutation causes myofibrillar myopathy. Neurol. Asia 2021, 26, 301–305. [Google Scholar]
- Ruparelia, A.A.; Oorschot, V.; Vaz, R.; Ramm, G.; Bryson-Richardson, R.J. Zebrafish models of BAG3 myofibrillar myopathy suggest a toxic gain of function leading to BAG3 insufficiency. Acta Neuropathol. 2014, 128, 821–833. [Google Scholar] [CrossRef]
- Savarese, M.; Di Fruscio, G.; Mutarelli, M.; Torella, A.; Magri, F.; Santorelli, F.M.; Comi, G.P.; Bruno, C.; Nigro, V. MotorPlex provides accurate variant detection across large muscle genes both in single myopathic patients and in pools of DNA samples. Acta Neuropathol. Commun. 2014, 2, 100. [Google Scholar] [CrossRef]
- Ruparelia, A.A.; McKaige, E.A.; Williams, C.; Schulze, K.E.; Fuchs, M.; Oorschot, V.; Lacene, E.; Meregalli, M.; Lee, C.; Serrano, R.J.; et al. Metformin rescues muscle function in BAG3 myofibrillar myopathy models. Autophagy 2021, 17, 2494–2510. [Google Scholar] [CrossRef]
- Angelis, A.; Tordrup, D.; Kanavos, P. Socio-economic burden of rare diseases: A systematic review of cost of illness evidence. Health Policy 2015, 119, 964–979. [Google Scholar] [CrossRef] [Green Version]
- López-Bastida, J.; Oliva-Moreno, J.; Linertová, R.; Serrano-Aguilar, P. Social/economic costs and health-related quality of life in patients with rare diseases in Europe. Eur. J. Health Econ. 2016, 17 (Suppl. S1), 1–5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fagoaga, J.; Girabent-Farrés, M.; Bagur-Calafat, C. Traducción y validación de la escala Individualized Neuromuscular Quality of Life para la población española: Evaluación de la calidad de vida para personas afectas de enfermedades neuromusculares. Rev. De Neurol. 2017, 194, 200. [Google Scholar] [CrossRef] [Green Version]
- Sansone, V.A.; Panzeri, M.; Montanari, M.; Apolone, G.; Gandossini, S.; Rose, M.R.; Politano, L.; Solimene, C.; Siciliano, G.; Volpi, L.; et al. Italian validation of INQoL, a quality of life questionnaire for adults with muscle diseases. Eur. J. Neurol. 2010, 17, 1178–1187. [Google Scholar] [CrossRef] [PubMed] [Green Version]



{kind=link}
{kind=link}
{kind=link}
| Reference | Age Atonset (Years) | Features Atonset | Cardiomyopathy | Contractures | Weakness | Peripheral Neuropathy | Respiratory Failure |
|---|---|---|---|---|---|---|---|
| Odgerel et al., 2010 [2] | 5 | Not reported | Restrictive- hypertrophic | Not reported | Yes | Axonal neuropathy | Yes |
| Odgerel et al., 2010 [2] | 12 | Not reported | Restrictive- hypertrophic heart transplantation | Not reported | Yes | Axonal neuropathy | Yes |
| Odgerel et al., 2010 [2] | 12 | Pes cavus, weakness, cardiopathy | Restrictive- hypertrophic | Scoliosis and rigid spine | Distal weakness and neck weakness | Axonal neuropathy | Yes |
| Odgerel et al., 2010 [2] | 5 | Gait disturbance | Restrictive- hypertrophic heart transplantation | Not reported | Proximal weakness | Axonal neuropathy | Yes |
| Lee HC et al., 2012 [8] | 6 | Gait disturbance | Restrictive- hypertrophic | Multiple contractures and rigid spine | Mild proximal weakness | Axonal neuropathy | Not reported |
| D’avila et al., 2016 [4] | 11 | Contractures | Hypertrophic and arrhythmias | Rigid spine | Proximal weakness | Axonal neuropathy | Yes |
| Selcen et al., 2009 [3] | Toddler | Toe walker | restrictive heart transplant | Toe walker | Severe weakness | Not reported | Yes |
| Selcen et al., 2009 [3] | 13 | Scoliosis rigid spine, fatigability | hypertrophic | Scoliosis and rigid spine | Distal and proximal weakness | Axonal demyelinating neuropathy | Yes |
| Selcen et al., 2009 [3] | Toddler | Toe walker | Restrictive | Scoliosis, rigid spine and toe walker | Progressive proximal weakness | Not reported | Yes |
| Jaffer et al., 2012 [9] | Toddler | Toe walker | Restrictive-heart transplantation | Multiple contractures and rigid spine | Distal and proximal weakness | Axonal neuropathy | Not reported |
| Jaffer et al., 2012 [9] | Toddler | Toe walker | Restrictive | Multiple contractures, scoliosis and rigid spine | Distal and proximal weakness | Axonal neuropathy | Yes |
| Kostera Pruszczyk et al., 2015 [10] Present Case | 12 3 | Toe walker and foot deformity Foot deformity | Restrictive (subclinical) long QT, transplantation Restrictive Heart Transplantation | Rigid spine multiple contractures Rigid spine Rotatory scoliosis | Subclinical weakness Distal and proximal weakness | Axonal demyelinating neuropathy Axonal neuropathy | No |
| Reference | Age Atonset (Years) | Features at Onset | Cardiomyopathy | Contractures | Weakness | Peripheral Neuropathy | Respiratory Failure |
|---|---|---|---|---|---|---|---|
| Li et al. 2021 [16] | 13 years | Gait difficulties | Left ventricular diastolic dysfunction | No | Distal | No | No |
| Present case | 3 years | Gait difficulties | Atrial dilatation, EF 50% | No | Distal | Axonal neuropathy | No |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Angelini, C.; Ceolin, C.; Rodriguez, A.A.; Nigro, V. Two Cases of Myofibrillar Myopathies: Genetic and Quality of Life Study. Muscles 2023, 2, 177-186. https://doi.org/10.3390/muscles2020013
Angelini C, Ceolin C, Rodriguez AA, Nigro V. Two Cases of Myofibrillar Myopathies: Genetic and Quality of Life Study. Muscles. 2023; 2(2):177-186. https://doi.org/10.3390/muscles2020013
Chicago/Turabian StyleAngelini, Corrado, Chiara Ceolin, Alicia Aurora Rodriguez, and Vincenzo Nigro. 2023. "Two Cases of Myofibrillar Myopathies: Genetic and Quality of Life Study" Muscles 2, no. 2: 177-186. https://doi.org/10.3390/muscles2020013