
Distantly Related Homologue of UhpT in Pseudomonas aeruginosa



Abstract
:1. Introduction
- 4 TM helices are positioned in the center of the transporter, which contains the majority of residues important for substrate binding site and creates a central pore;
- 4 TM helices are positioned outside of the previous TM helices and are important for structural integrity;
- 4 TM helices are positioned on the side of the proteins and are important for interdomain contacts.
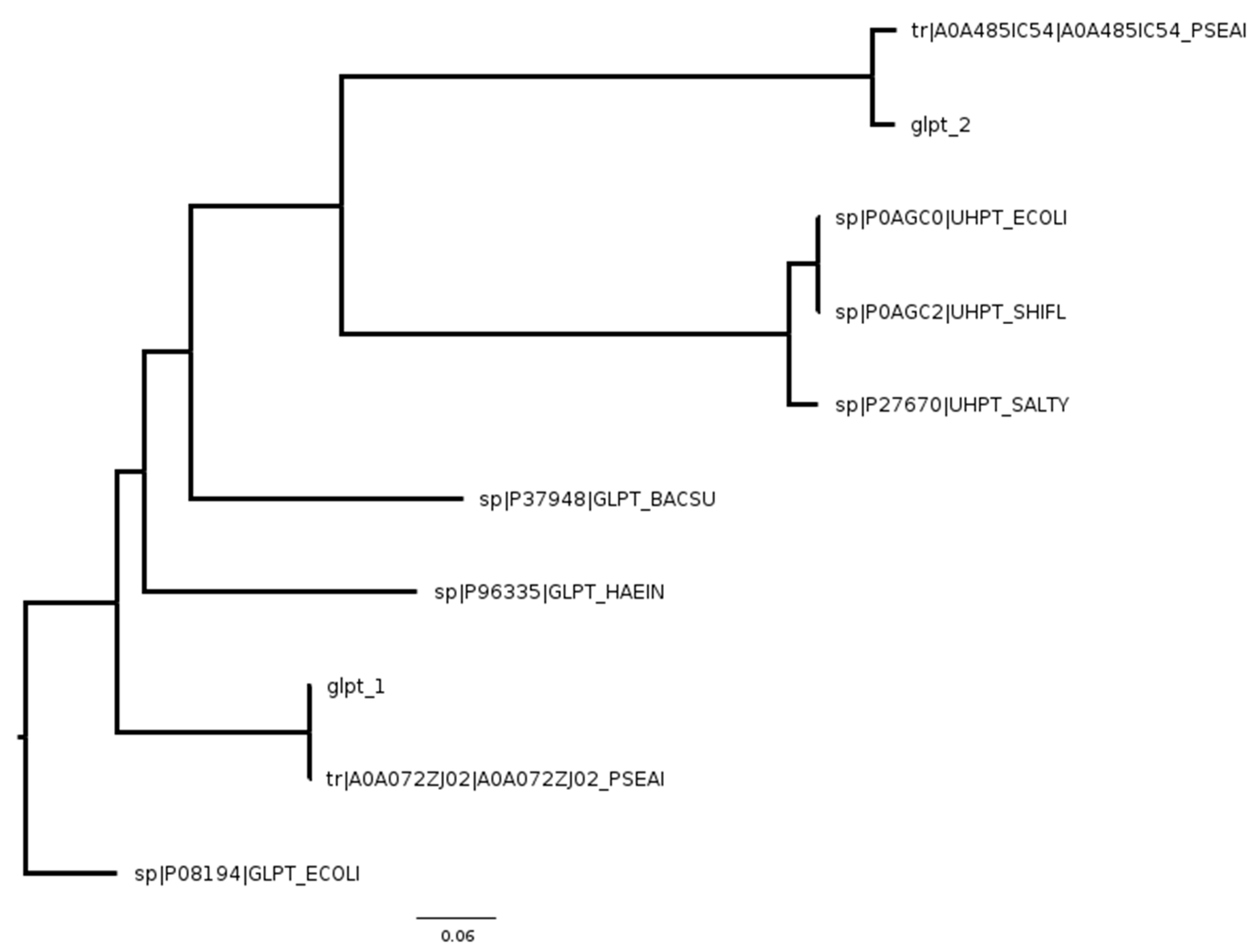
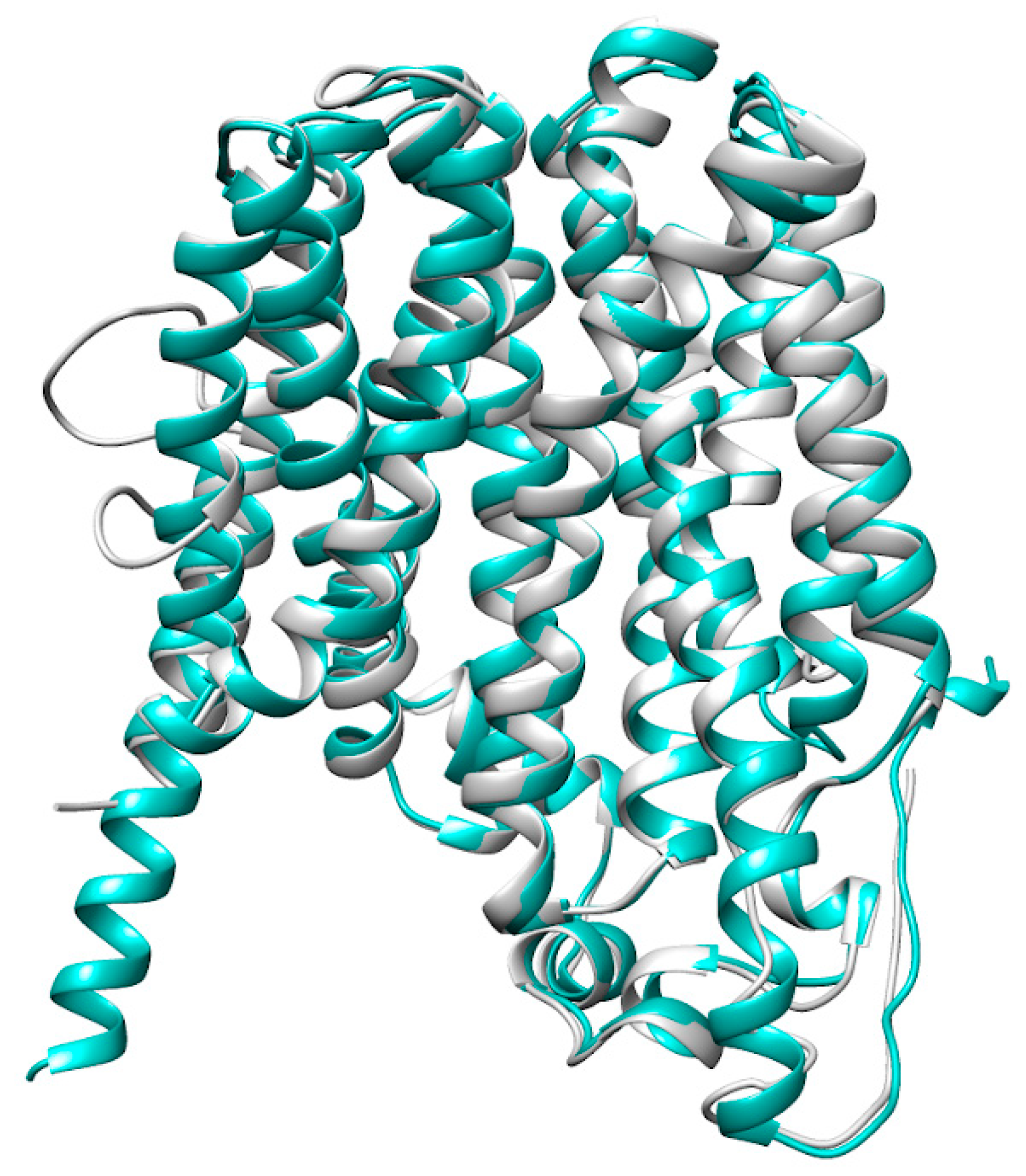
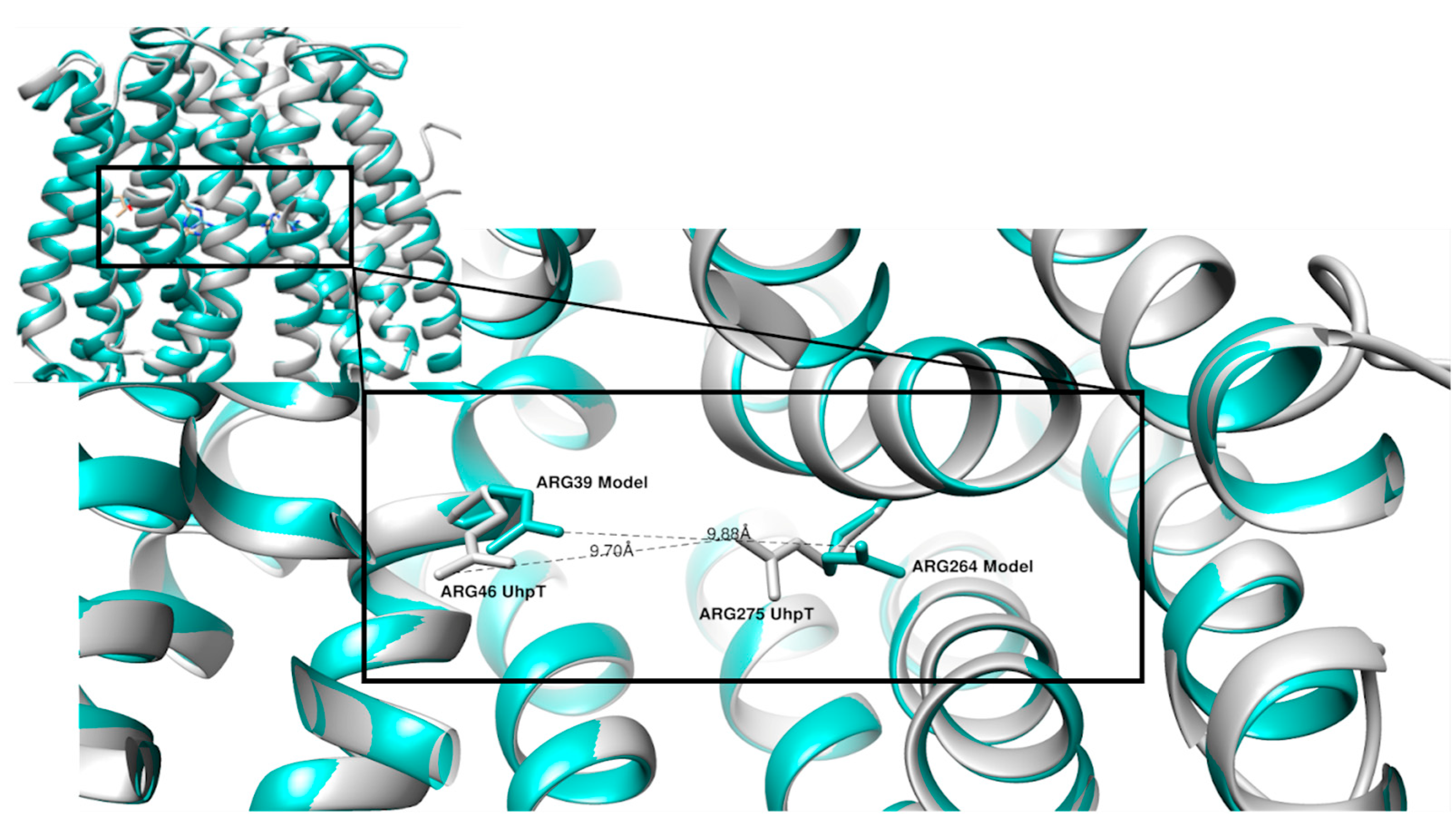
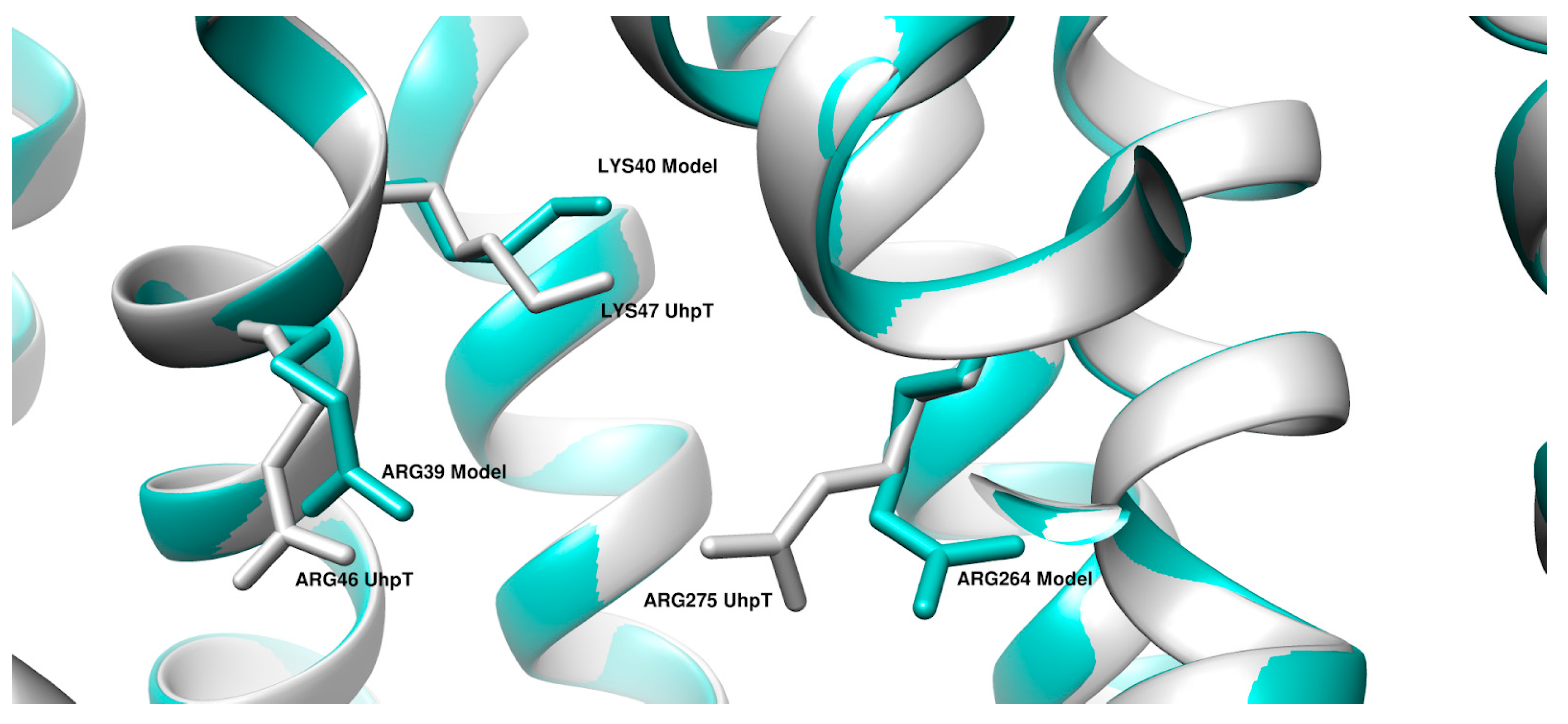
2. Results
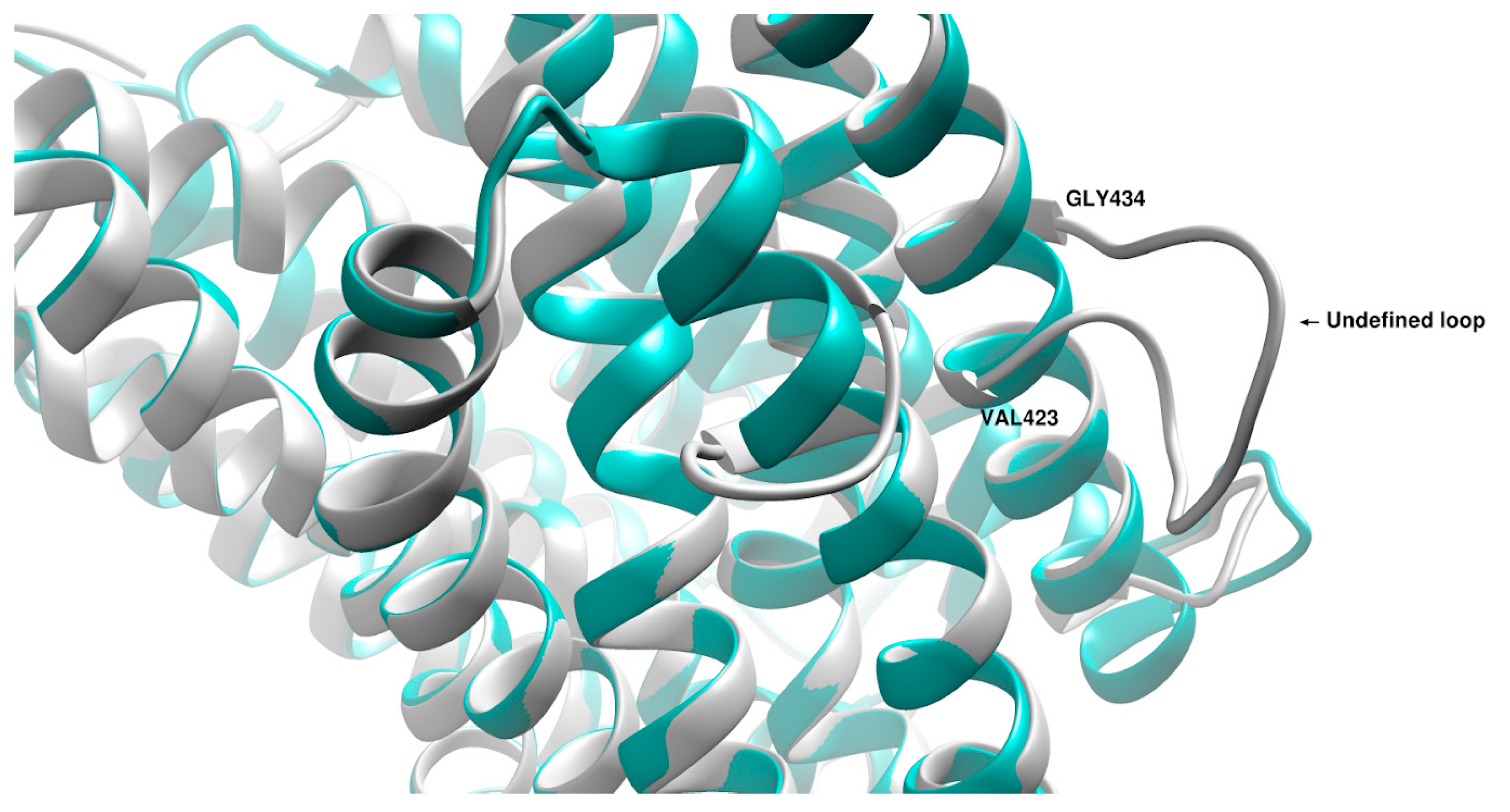
3. Discussion
4. Materials and Methods
4.1. PA Collection and Sequencing Pipeline
4.2. Building Hidden Markov Model (HMM)
- the proteins have to be an X-ray experiment;
- resolution must be ≤3.5 Å;
- the proteins have to be wild-type;
- no mutations in the sequence.
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Lyczak, J.B.; Cannon, C.L.; Pier, G.B. Lung Infections Associated with Cystic Fibrosis. Clin. Microbiol. Rev. 2002, 15, 194–222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stover, C.K.; Pham, X.Q.; Erwin, A.L.; Mizoguchi, S.D.; Warrener, P.; Hickey, M.J.; Brinkman, F.S.L.; Hufnagle, W.O.; Kowalik, D.J.; Lagrou, M.; et al. Complete genome sequence of Pseudomonas aeruginosa PAO1, an opportunistic pathogen. Nature 2000, 406, 959–964. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poole, K. Outer Membranes and Efflux: The Path to Multidrug Resistance in Gram- Negative Bacteria. Curr. Pharm. Biotechnol. 2002, 3, 77–98. [Google Scholar] [CrossRef] [PubMed]
- Ferrara, A.M. Potentially multidrug-resistant non-fermentative Gram-negative pathogens causing nosocomial pneumonia. Int. J. Antimicrob. Agents 2006, 27, 183–195. [Google Scholar] [CrossRef] [PubMed]
- Rossolini, G.M.; Mantengoli, E. Treatment and control of severe infections caused by multiresistant Pseudomonas aeruginosa. Clin. Microbiol. Infect. 2005, 11, 17–32. [Google Scholar] [CrossRef] [Green Version]
- Gupta, V. Metallo beta lactamases in Pseudomonas aeruginosaand Acinetobacter species. Expert Opin. Investig. Drugs 2008, 17, 131–143. [Google Scholar] [CrossRef]
- Zhao, W.-H.; Hu, Z.-Q. β-Lactamases identified in clinical isolates of Pseudomonas aeruginosa. Crit. Rev. Microbiol. 2010, 36, 245–258. [Google Scholar] [CrossRef]
- Ramirez, M.S.; Tolmasky, M.E. Aminoglycoside modifying enzymes. Drug Resist. Update 2010, 13, 151–171. [Google Scholar] [CrossRef] [Green Version]
- Strateva, T.; Yordanov, D. Pseudomonas aeruginosa—A phenomenon of bacterial resistance. J. Med. Microbiol. 2009, 58, 1133–1148. [Google Scholar] [CrossRef] [Green Version]
- Kahan, F.M.; Kahan, J.S.; Cassidy, P.J.; Kropp, H. The mechanism of action of fosfomycin (phosphonomycin). Ann. N. Y. Acad. Sci. 1974, 235, 364–386. [Google Scholar] [CrossRef]
- Falagas, M.E.; Giannopoulou, K.P.; Kokolakis, G.N.; Rafailidis, P.I. Fosfomycin: Use Beyond Urinary Tract and Gastrointestinal Infections. Clin. Infect. Dis. 2008, 46, 1069–1077. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oliver, A.; Levin, B.R.; Juan, C.; Baquero, F.; Blázquez, J. Hypermutation and the Preexistence of Antibiotic-Resistant Pseudomonas aeruginosa Mutants: Implications for Susceptibility Testing and Treatment of Chronic Infections. Antimicrob. Agents Chemother. 2004, 48, 4226–4233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Falagas, M.E.; Kastoris, A.C.; Karageorgopoulos, D.; Rafailidis, P.I. Fosfomycin for the treatment of infections caused by multidrug-resistant non-fermenting Gram-negative bacilli: A systematic review of microbiological, animal and clinical studies. Int. J. Antimicrob. Agents 2009, 34, 111–120. [Google Scholar] [CrossRef] [Green Version]
- Alper, M.D.; Ames, B.N. Transport of antibiotics and metabolite analogs by systems under cyclic AMP control: Positive selection of Salmonella typhimurium cya and crp mutants. J. Bacteriol. 1978, 133, 149–157. [Google Scholar] [CrossRef] [Green Version]
- Castañeda-García, A.; Rodríguez-Rojas, A.; Guelfo, J.R.; Blázquez, J. The Glycerol-3-Phosphate Permease GlpT Is the Only Fosfomycin Transporter in Pseudomonas aeruginosa. J. Bacteriol. 2009, 191, 6968–6974. [Google Scholar] [CrossRef] [Green Version]
- Olekhnovich, I.; Dahl, J.L.; Kadner, R.J. Separate contributions of UhpA and CAP to activation of transcription of the uhpT promoter of Escherichia coli. J. Mol. Biol. 1999, 292, 973–986. [Google Scholar] [CrossRef] [PubMed]
- Karageorgopoulos, D.; Wang, R.; Yu, X.-H.; Falagas, M.E. Fosfomycin: Evaluation of the published evidence on the emergence of antimicrobial resistance in Gram-negative pathogens. J. Antimicrob. Chemother. 2012, 67, 255–268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nikolaidis, I.; Favini-Stabile, S.; Dessen, A. Resistance to antibiotics targeted to the bacterial cell wall. Protein Sci. 2014, 23, 243–259. [Google Scholar] [CrossRef]
- Mirakhur, A.; Gallagher, M.; Ledson, M.; Hart, C.; Walshaw, M. Fosfomycin therapy for multiresistant Pseudomonas aeruginosa in cystic fibrosis. J. Cyst. Fibros. 2003, 2, 19–24. [Google Scholar] [CrossRef] [Green Version]
- Okazaki, M.; Suzuki, K.; Asano, N.; Araki, K.; Shukuya, N.; Egami, T.; Uchimura, H.; Watanabe, T.; Higurashi, Y.; Morita, K. Effectiveness of fosfomycin combined with other antimicrobial agents against multidrug-resistant Pseudomonas aeruginosa isolates using the efficacy time index assay. J. Infect. Chemother. 2002, 8, 37–42. [Google Scholar] [CrossRef]
- Pao, S.S.; Paulsen, I.T.; Saier, M.H., Jr. Major Facilitator Superfamily. Microbiol. Mol. Biol. Rev. 1998, 62, 1–34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marger, M.D.; Saier, M.H., Jr. A major superfamily of transmembrane facilitators that catalyse uniport, symport and antiport. Trends Biochem. Sci. 1993, 18, 13–20. [Google Scholar] [CrossRef]
- Maiden, M.C.J.; Davis, E.O.; Baldwin, S.A.; Moore, D.C.M.; Henderson, P.J.F. Mammalian and bacterial sugar transport proteins are homologous. Nature 1987, 325, 641–643. [Google Scholar] [CrossRef]
- Yan, N. Structural advances for the major facilitator superfamily (MFS) transporters. Trends Biochem. Sci. 2013, 38, 151–159. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Jiang, X.; Kaback, H.R. Role of the irreplaceable residues in the LacY alternating access mechanism. Proc. Natl. Acad. Sci. USA 2012, 109, 12438–12442. [Google Scholar] [CrossRef] [Green Version]
- Dang, S.; Sun, L.; Huang, Y.; Lu, F.; Liu, Y.; Gong, H.; Wang, J.; Yan, N. Structure of a fucose transporter in an outward-open conformation. Nature 2010, 467, 734–738. [Google Scholar] [CrossRef]
- Solcan, N.; Kwok, J.; Fowler, P.W.; Cameron, A.D.; Drew, D.; Iwata, S.; Newstead, S. Alternating access mechanism in the POT family of oligopeptide transporters. EMBO J. 2012, 31, 3411–3421. [Google Scholar] [CrossRef] [Green Version]
- Huang, Y.; Lemieux, M.J.; Song, J.; Auer, M.; Wang, D.N. Structure and Mechanism of the Glycerol-3-Phosphate Transporter from Escherichia coli. Science 2003, 301, 616–620. [Google Scholar] [CrossRef] [Green Version]
- Hall, J.A.; Maloney, P.C. Altered Oxyanion Selectivity in Mutants of UhpT, the Pi -linked Sugar Phosphate Carrier of Escherichia coli. J. Biol. Chem. 2005, 280, 3376–3381. [Google Scholar] [CrossRef] [Green Version]
- Chen, S.-Y.; Pan, C.-J.; Lee, S.; Peng, W.; Chou, J.Y. Functional analysis of mutations in the glucose-6-phosphate transporter that cause glycogen storage disease type Ib. Mol. Genet. Metab. 2008, 95, 220–223. [Google Scholar] [CrossRef]
- Rice, P.; Longden, I.; Bleasby, A. EMBOSS: The European Molecular Biology Open Software Suite. Trends Genet. 2000, 16, 276–277. [Google Scholar] [CrossRef]
- Sievers, F.; Wilm, A.; Dineen, D.; Gibson, T.J.; Karplus, K.; Li, W.; Lopez, R.; McWilliam, H.; Remmert, M.; Söding, J.; et al. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol. 2011, 7, 539. [Google Scholar] [CrossRef] [PubMed]
- Martí-Renom, M.A.; Stuart, A.C.; Fiser, A.; Sánchez, R.; Melo, F.; Šali, A. Comparative Protein Structure Modeling of Genes and Genomes. Annu. Rev. Biophys. Biomol. Struct. 2000, 29, 291–325. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Webb, B.; Sali, A. Comparative Protein Structure Modeling Using MODELLER. Curr. Protoc. Bioinform. 2016, 54, 5–6. [Google Scholar] [CrossRef] [Green Version]
- Fiser, A.; Do, R.K.; SSali, A. Modeling of loops in protein structures. Protein Sci. 2000, 9, 1753–1773. [Google Scholar] [CrossRef] [Green Version]
- Šali, A.; Blundell, T.L. Comparative Protein Modelling by Satisfaction of Spatial Restraints. J. Mol. Biol. 1993, 234, 779–815. [Google Scholar] [CrossRef]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef]
- Varadi, M.; Anyango, S.; Deshpande, M.; Nair, S.; Natassia, C.; Yordanova, G.; Yuan, D.; Stroe, O.; Wood, G.; Laydon, A.; et al. AlphaFold Protein Structure Database: Massively expanding the structural coverage of protein-sequence space with high-accuracy models. Nucleic Acids Res. 2022, 50, D439–D444. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera-a visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [Green Version]
- Jessen-Marshall, A.E.; Brooker, R.J. Evidence That Transmembrane Segment 2 of the Lactose Permease Is Part of a Conformationally Sensitive Interface between the Two Halves of the Protein. J. Biol. Chem. 1996, 271, 1400–1404. [Google Scholar] [CrossRef]
- Fann, M.-C.; Busch, A.; Maloney, P.C. Functional Characterization of Cysteine Residues in GlpT, the Glycerol 3-Phosphate Transporter of Escherichia coli. J. Bacteriol. 2003, 185, 3863–3870. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Available online: https://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 1 May 2022).
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [Green Version]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D.; et al. SPAdes: A new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seemann, T. Prokka: Rapid Prokaryotic Genome Annotation. Bioinformatics 2014, 30, 2068–2069. [Google Scholar] [CrossRef] [Green Version]
- Eddy, S.R. Hidden Markov models. Curr. Opin. Struct. Biol. 1996, 6, 361–365. [Google Scholar] [CrossRef]
- Eddy, S.R. What is a hidden Markov model? Nat. Biotechnol. 2004, 22, 1315–1316. [Google Scholar] [CrossRef] [Green Version]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krissinel, E.; Henrick, K. Multiple Alignment of Protein Structure in Three Dimensions. In Computational Life Sciences; Springer: Berlin/Heidelberg, Germany, 2005; pp. 67–68. [Google Scholar]
- Needleman, S.B.; Wunsch, C.D. A general method applicable to the search for similarities in the amino acid sequence of two proteins. J. Mol. Biol. 1970, 48, 443–453. [Google Scholar] [CrossRef]
- HMMER. Available online: http://hmmer.org/ (accessed on 1 May 2022).
- The UniProt Consortium. UniProt: The universal protein knowledgebase in 2021. Nucleic Acids Res. 2021, 49, D480–D489. [Google Scholar] [CrossRef]
- Suzek, B.; Wang, Y.; Huang, H.; McGarvey, P.B.; Wu, C.H. The UniProt Consortium UniRef clusters: A comprehensive and scalable alternative for improving sequence similarity searches. Bioinformatics 2015, 31, 926–932. [Google Scholar] [CrossRef]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Frimmersdorf, E.; Horatzek, S.; Pelnikevich, A.; Wiehlmann, L.; Schomburg, D. How Pseudomonas aeruginosa adapts to various environments: A metabolomic approach. Environ. Microbiol. 2010, 12, 1734–1747. [Google Scholar] [CrossRef] [PubMed]
- Ng, F.M.-W.; Dawes, E.A. Chemostat studies on the regulation of glucose metabolism in Pseudomonas aeruginosa by citrate. Biochem. J. 1973, 132, 129–140. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| UniProt ID of UhpT | Organism | UniProt ID of GlpT | Organism |
|---|---|---|---|
| P0AGC0 | E. coli | P37948 | B. subtilis |
| P0AGC2 | S. flexneri | P96335 | H. influenzae |
| P27670 | S. typhi | P08194 | E. coli |
| A0A485IC54 * | P. aeruginosa | A0A072Z * | P. aeruginosa |
| PDB id and Chain | Name of the Protein | Organism |
|---|---|---|
| 1pw4:A | Crystal Structure of the Glycerol-3-Phosphate Transporter | Escherichia coli |
| 6e9n:A | D-galactonate:proton symporter in the inward open form | Escherichia coli |
| 7cko:A | Human MCT1/Basigin-2 complex in the presence of anti-cancer drug candidate 7ACC2 in the inward-open conformation | Homo sapiens |
| 6kki:A | Drug:Proton Antiporter-1 (DHA1) Family SotB, in the inward-occluded conformation | Escherichia coli K-12 |
| 6kkj:B | Drug:Proton Antiporter-1 (DHA1) Family SotB, in the inward open conformation | Escherichia coli K-12 |
| 4zp2:A | Multidrug transporter MdfA in complex with n-dodecyl-N, N-dimethylamine-N-oxide | Escherichia coli K-12 |
| 6oop:A | Protein B | Escherichia coli |
| 4zow:A | Multidrug transporter MdfA in complex with chloramphenicol | Escherichia coli K-12 |
| 6vs0:A | Protein B | Escherichia coli |
| Predicted MFS Domain | |||
|---|---|---|---|
| Positive | Negative | ||
| Protein with MFS Domain | Positive | 288 (TP) | 53 (FN) |
| Negative | 54 (FP) | 55,204 (TN) | |
| Scoring Index | Value |
|---|---|
| TPR | 0.84 |
| TNR | 0.99 |
| ACC | 0.99 |
| MCC | 0.84 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Orioli, T.; Dolce, D. Distantly Related Homologue of UhpT in Pseudomonas aeruginosa. Bacteria 2022, 1, 266-278. https://doi.org/10.3390/bacteria1040020
Orioli T, Dolce D. Distantly Related Homologue of UhpT in Pseudomonas aeruginosa. Bacteria. 2022; 1(4):266-278. https://doi.org/10.3390/bacteria1040020
Chicago/Turabian StyleOrioli, Tommaso, and Daniela Dolce. 2022. "Distantly Related Homologue of UhpT in Pseudomonas aeruginosa" Bacteria 1, no. 4: 266-278. https://doi.org/10.3390/bacteria1040020