
Decellularization of Mouse Kidneys to Generate an Extracellular Matrix Gel for Human Induced Pluripotent Stem Cell Derived Renal Organoids

1
Research Department of Surgical Biotechnology, UCL Division of Surgery & Interventional Science, Royal Free Hospital, Rowland Hill Street, London NW3 2QG, UK
2
UCL Institute of Immunity & Transplantation, The Pears Building, Pond Street, London NW3 2PP, UK
*
Author to whom correspondence should be addressed.
Organoids 2023, 2(1), 66-78; https://doi.org/10.3390/organoids2010005
Submission received: 24 December 2022
/
Revised: 12 February 2023
/
Accepted: 24 February 2023
/
Published: 22 March 2023
(This article belongs to the Special Issue Advances in Organoid Technology—Selected Papers from "Organoids Are Us 2022")
{kind=link}
{kind=link}
{kind=link}
Abstract
:Chronic Kidney Disease (CKD) is a major cause of morbidity and mortality characterized by progressive renal fibrosis, and in extreme cases, renal failure. Human CKD models that replicate the biological complexity of the kidney and CKD are lacking and will be invaluable in identifying drugs to revert and/or prevent fibrosis. To address this unmet need, we developed 3D renal organoids where human induced pluripotent stem cells (hiPSCs) were differentiated to renal progenitors within a renal extracellular matrix (rECM) gel, based on the premise that an rECM could recreate the renal niche to facilitate hiPSC-derived renal progenitor generation. We used mouse kidneys as a source of rECM and identified that superior detergent-mediated decellularization of mouse kidneys was achieved with a combination of 0.5% w/v Sodium Dodecyl Sulphate and 1% v/v Triton-X and mechanical agitation for 60 h. HiPSCs that underwent specification to become metanephric mesenchyme (MM) were subsequently cultured within the rECM gel and, notably, mesenchymal to epithelial transition (MET) was observed, as judged by expression of nephron markers K-cadherin, Nephrin and WT1. These data demonstrate a role for rECM gel in developing human renal organoids from hiPSCs, which will aid the further development of a human disease model for renal fibrosis.
1. Introduction
The kidneys develop from two primitive tissues in the embryo, called the metanephric mesenchyme (MM) and the ureteric bud (UB), which reciprocally act upon signals from the other to form the mature kidneys [1]. Kidneys play a vital role in many bodily functions including filtering the blood of metabolic waste, urine production, controlling blood pressure and production of hormones, including erythropoietin and the metabolically active form of Vitamin-D.
Chronic Kidney Disease (CKD) arises when renal structure and/or function is chronically impaired; it affects over 9% of the population globally and is responsible for high morbidity and mortality worldwide [2,3,4]. Developing in response to conditions including Diabetes, Glomerulonephritis and untreated renal calculi, CKD often progresses into End-Stage-Renal-Disease (ESRD) resulting in renal failure. Dialysis can help replace abnormal renal filtration but fails to aid selective reabsorption or renal endocrine function, which dramatically limits quality of life for patients. At present, there are no effective drugs to directly target or reverse renal fibrosis [5]. Thus, for patients who develop ESRD, organ transplantation is their last chance to regain proper renal functionality. However, aside from the shortage of transplant donors, which dramatically limits the impact of this treatment, additional drawbacks of kidney transplantation include the need for lifelong immunosuppressive treatment to prevent organ rejection and, importantly, because transplantation fails to address the root pathology of fibrosis, newly transplanted kidneys may also be jeopardized by fibrotic damage. Thus, new therapies are required to tackle this mounting clinically unmet need. However, a significant impediment is the absence of valid human disease models as many promising pre-clinical drug candidates fall short in clinical trials [5]. At present, animal models and 2D primary cell cultures are common platforms for pre-clinical drug screening. The 2D cultures do not produce satisfactorily relatable data with clinical trials, and interspecies variation is often responsible for significant disparities between findings in pre-clinical animal research and human clinical trials [6].
Human induced pluripotent stem cells (iPSCs)—somatic cells ‘reprogrammed’ to revert to a pluripotent state through the introduction of key stem cell factors—have boosted disease modeling efforts [7,8]. iPSCs recapitulate development at the cellular level, are capable of self-renewal and differentiation into any cell of the embryonic germ layers and are particularly useful as they can be directly generated from patients—both healthy and from those bearing disease-relevant genetic polymorphisms [9]. Early attempts to develop protocols for the targeted differentiation of renal cells from human pluripotent stem cells (hPSCs) met with limited success, attributable to the sheer anatomical, physiological and developmental complexity of the kidney. More recently, however, there have been multiple improved reports using human PSCs to generate morphologically and functionally coherent kidney podocytes [10], ureteric bud progenitors [11], metanephric mesenchyme [12] and self-organizing 3D structures called kidney organoids from embryonic stem cells (ESC) [13] and iPSCs [14,15,16]. Organoids enable physiological cell differentiation, replicate tissue- and organ-level physiology and have been proposed as a basis for a human disease model for fibrotic kidney disease. Crucially, where primary cell cultures sourced from already lineage-committed cells are unable to maintain phenotypic stability for long, iPSC-derived kidney organoids display a more physiologically relevant phenotype in response to nephrotoxic drugs including chemotherapeutic agent cisplatin and the antibiotic gentamycin, which makes them a tractable model for drug-screening assays [15,16].
While current efforts to generate renal organoids from hiPSCs have advanced considerably in recent years, maturation of the resultant organoid renal lineage cells requires further improvement and optimization [17]. One potential avenue under study attempts to better understand and exploit the role of the extracellular matrix (ECM) in providing the required niche factors, e.g., proteins (collagen, laminin, fibronectin), glycosaminoglycans and other secreted factors which could direct the differentiation of hiPSCs to mature lineage-specific cell types [18,19]. Via a process called decellularization, tissues are treated with a combination of detergents, digestive enzymes and/or subjected to mechanically disruptive processes such as freeze-thawing, in order to achieve cell death whilst leaving the residual tissue ECM architecture intact [19]. As such, decellularization techniques of various organs including the lungs [20], pancreas [21,22], heart [23] and liver [24,25] are being investigated as potential tissue scaffolds that closely replicate the characteristics of in vivo tissue.
In this report, we explore the role of the renal ECM in directing differentiation of hiPSCs derivatives towards the renal lineage. Using mouse kidneys, we sought to develop a suitable protocol for kidney decellularization. Four decellularization methods (decellularization protocols 1–4, DP1–4 hereafter) were investigated, and tissues subjected to them were characterized to determine whether they could maintain the kidney ECM whilst depleting the tissue of cellular material. DP1, the protocol that was found to best preserve the renal architecture, was taken forward to create a rudimentary renal ECM hydrogel as a substrate in which to generate human iPSC renal derivatives.
2. Materials and Methods
2.1. Animals and Tissue Preparation
All experimental animals were sacrificed by Schedule 1 methods ratified by the UK Home Office under the Animals (Scientific Procedures) Act (ASPA), 1986. A total of 25 C57 BL/6 mice in the age range of three months to one year were dissected per-abdominally to obtain 50 kidneys. These organs were stored separately in 30 mL phosphate-buffered saline (PBS) (Sigma Aldrich, Gillingham, Dorset, UK) briefly to cleanse blood from the surface. All perinephric fat was dissected out with a scalpel, the kidneys were washed in PBS for a second time and each kidney was longitudinally sliced into three sections of approximately 2 mm thickness each. This yielded a total of 150 kidney slices. Additionally, three kidneys were cryo-preserved to be used as a negative control for decellularization procedures. Standards were available for quantitative assays (collagen and glycosaminoglycan assays) along with the respective kits to serve as positive control.
2.2. Kidney Decellularization
Freshly dissected and cleaned kidneys were washed in PBS within 30 min of harvest and prepared as mentioned above. The remaining slices were transferred to 60 mm petri-dishes containing 30 mL double distilled water (ddH2O) for cellular lysis for 4 h. The first batch of sliced kidneys (60 slices) were divided into four parts. Each part was subjected to one of four immersion decellularization protocols: Decellularization Protocol 1—dH2O-treated slices were transferred to a bottle containing 0.5% w/v Sodium Dodecyl Sulphate (SDS) (Sigma Aldrich) in 150 mL PBS and set up on continuous magnetic stirring for 36 h. Next, the samples were immersed in 1% v/v Triton-X (Sigma Aldrich) in 20 mL PBS for 24 h with continuous orbital shaking. At 64 h, slices were harvested and stored at 4 °C in 30 mL fresh PBS. Decellularization Protocol 2—ddH2O-treated slices were immersed in 0.5% w/v Sodium dodecyl sulphate (SDS) in 30 mL PBS for 48 hrs. Slices were harvested at 52 h and stored in 30 mL fresh PBS at 4 °C. Decellularization Protocol 3—ddH2O-treated slices were exposed to 1% v/v Triton-X in 30 mL PBS for 48 h followed by 0.5% w/v SDS in 30 mL PBS for the next 24 h. Treated scaffolds were harvested at 76 h and stored at 4 °C in 30 mL fresh PBS. Decellularization Protocol 4—ddH2O-lysed slices were treated with 1% v/v Triton-X in 30 mL PBS for 96 h followed by fresh medium for 48 h. Slices were harvested at 100 h and stored at 4 °C in 30 mL PBS.
2.3. Nuclease Treatment of Scaffolds
Decellularized renal scaffolds were treated with Benzonase® nuclease (Sigma Aldrich) to remove any traces of residual DNA or RNA adhering to the scaffolds post-decellularization as per supplied protocol and stored for further processing.
2.4. Sterilization of Treated Scaffolds
Nuclease-treated scaffolds were sterilized with 10× Penicillin/Streptomycin/Amphotericin-B (PSA) (Sigma Aldrich) solution in PBS for 3 h with continuous orbital shaking. The slices were washed in PBS three times for 10 min per wash; samples were then retained for cryo-embedding in tissue-freezing medium for subsequent qualitative analysis and the remainder were stored in fresh PBS at 4 °C.
2.5. Preparing Decellularized Murine Kidneys for Histology
Two scaffolds from each DP were embedded in cryomolds onto optimal cutting temperature (OCT) tissue-freezing medium (Triangle Biomedical Sciences, Durham, NC, USA), frozen in a bath of isopropyl alcohol (Sigma Aldrich) on dry ice, labeled, wrapped in aluminum foil and stored at −80 °C for cryosectioning to 10µm slices to be immunostained for ECM proteins collagen I, collagen IV, laminin and fibronectin in addition to testing for any residual nucleic acids using 4′,6-diamidino2-phenylindole (DAPI, Sigma Aldrich).
2.6. ECM Component Quantification
Collagen (soluble) and sulphated glycosaminoglycan (sGAG) were quantified using the SIRCOL soluble collagen assay kit (Biocolor, Carrickfergus, Co., Antrim, UK) and sGAG assay kit (Biocolor), respectively, following supplied protocols. The mean weight of decellularized murine kidneys used in these assays was 17.3 mg.
2.7. Total Residual DNA Analysis
Total residual DNA content in decellularized scaffolds was assessed on sections of kidney slices that had previously undergone papain digestion as part of the sGAG assay, but which were not utilized for S-GAG quantification. The DNA concentration was estimated using a Nanodrop Lyte spectrophotometer (ThermoFisher Scientific, Inchinnan, Scotland, UK) for absorbances at 260 nm and 280 nm. Purity of the samples was assessed by the ratio of absorbances at 260 nm and 280 nm (A260/A280), while the DNA yield was quantified using absorbance at 280 nm (A280). Three observations per sample were recorded and analyzed.
2.8. Immuno-Histochemistry of Decellularized Mouse Kidney Slices
The cryo-embedded kidney scaffolds were sectioned using a cryostat (R.Wolf Electronics, London, UK) into 10 µm slices, applied electrostatically to adhesion slides (VWR International, Lutterworth, UK), air-dried for 10 min and stored in a slide holder at −80 °C for subsequent immunofluorescence staining. The tissue sections were processed as per standard protocols for fixation with Acetone (Sigma Aldrich) (15 min), blocking with 3% Goat serum (Sigma Aldrich) and 0.2% Triton-X in PBS (1 h, room temperature), staining with 1:100 dilutions of primary antibodies (in blocking serum) for collagen I, collagen IV, fibronectin and laminin (Abcam) (overnight incubation, 4 °C) followed by incubation with phyco-erythrin (PE) conjugated Goat anti-Rabbit secondary antibodies (Invitrogen, ThermoFisher Scientific, Inchinnan, Scotland, UK) (1:100) (PE 594 nm emission) (1 h, room temperature) protected from light with necessary intervening lavage with PBS. Sections were further stained with 0.02% DAPI (Sigma Aldrich) (30 min), DPX mounting media (Sigma Aldrich) was added and the sections were visualized under an EVOS® fluorescent microscope (Life Technologies ThermoFisher Scientific, Inchinnan, Scotland, UK).
2.9. Preparation of Renal ECM (rECM) Homogenate Gel with Decellularized Mouse Kidneys
Decellularized murine kidney scaffolds prepared by the protocol yielding the best combination of ECM protein preservation and native murine cellular remnant clearance (as depicted in results section) were chosen to generate the murine rECM homogenate gel. Briefly, following Benzonase nuclease treatment and sterilization, each decellularized murine kidney scaffold with a mean weight of 17.25 mg was crushed and homogenized using a 2 mL Dulce® tissue grinder (Sigma Aldrich) in 200 uL of 10% w/v Gelatin (Sigma Aldrich) as a solvent. This homogenized scaffold–gelatin mix, denoted the rECM gel, was transferred to individual wells of a 48-well plate, one scaffold-rECM gel mix per well. Each well was later seeded with 3–4 hiPSC-derived embryoid bodies (EBs) as outlined below.
2.10. Formation of hiPSC Embryoid Bodies
Episomally-reprogrammed hiPSCs (ThermoFisher Scientific), grown in feeder-free conditions in 6-well tissue culture plates (Corning Costar®, High Wycombe, Buckinghamshire, UK) in the presence of E8 medium (Life Technologies) and rho-kinase inhibitor Y27632, were passaged using Accutase (Stemcell Technologies, Cambridge, UK). A total of 3.6 × 10⁶ cells in suspension were seeded into a well of an Aggrewell-400 plate (Stemcell Technologies); each Aggrewell well contained 1200 microwells per well. The cells were cultured in Basal Media composed of DMEM/F-12 (Sigma Aldrich), non-essential amino acids (Minimum essential medium (Sigma Aldrich)), L-glutamine (Glutamax, Sigma Aldrich), Beta-mercaptoethanol (Life Technologies), B-27 supplement (Life Technologies) and Insulin-transferrin-selenium (Life Technologies) and further supplemented with 10 uM Y27632 (Sigma Aldrich) and 0.5 ng/mL human BMP4 (Peprotech, Altrincham, Cheshire, UK). The Aggrewell-400 plate was centrifuged at 100× g for 3 min to allow for uniform distribution of hiPSCs and culture media into each microwell. This set-up was incubated at 37 °C (5% CO2, 95% humidity) for 48 h. The plate was observed using an EVOS cell imaging system (Life Technologies) to look for generation of embryoid bodies (EBs).
2.11. Directed Differentiation of hiPSC EBs to Metanephric Mesenchyme
To generate metanephric mesenchyme, hiPSC-derived EBs were subjected to a differentiation protocol adapted from Taguchi et al. [12]. At Day 2 (48 h after seeding the Aggrewell-400 plate), hiPSC EBs were harvested and transferred to one well of a 6-well tissue culture plate. Under microscopic guidance (EVOS® cell imaging system (Life Technologies) at 4× magnification) EBs were gently aspirated with a 1000 uL pipette and transferred to an ultra-low attachment 24-well plate (Corning Costar®) at a density of 50 EBs per well. Each well contained basal media supplemented with 20 ng/mL bFGF (Peprotech) and 1 ng/mL human Activin-A (Peprotech) prior to adding EBs. As negative control, 2 wells were conditioned only with Basal Media without growth factors. The above setup was incubated at 37 °C (5% CO2 95% humidity) for 2 days for Epiblast generation. On Day 4, the EBs were transferred to media containing 10 uL CHIR99021 (Stemgent, Reprocell, Durham, UK) and 1 ng/mL hBMP4 to allow for Posterior Nascent Mesoderm differentiation. The medium was refreshed on alternate days for 6 days. On Day 10, the EBs were transferred to media supplemented with 3 uL CHIR99021, 3 ng/mL hBMP4, 10 uM Y27632, 10 ng/mL Activin-A and 0.1 mM Retinoic Acid (Sigma Aldrich) to facilitate generation of Posterior Intermediate Mesoderm. Finally, on Day 12, the EBs were transferred to media containing 5 ng/mL human FGF-9 (Peprotech), 1 uM CHIR99021 and 10 uM Y27632 to allow for differentiation to Metanephric Mesenchyme. All steps involving EB transfer between stages require EBs to be centrifuged at 800 rpm for 2 min to allow for separation of the media from the previous stage, discarding supernatant, aspirating the EB pellet and re-suspending it in fresh medium for the next stage of differentiation onto a separate ultra-low attachment 24-well plate.
2.12. Incubation of Renal Lineage Committed EBs in Murine (rECM) Homogenate Gel
Lineage committed EBs at Metanephric Mesenchyme stage were harvested from the differentiation medium and transferred onto the murine rECM gel already applied to wells of a 48-well plate on ice. After 30 min, the rECM gel gelatinized around the EBs creating a 3D conditioned environment. This EB–rECM gel construct was cultured with basal media, supplemented with 5 ng/mL FGF9, 10 uM Y27632 and 1 uM CHIR99021, applied on top of the gel layer. The plate was then incubated at 37 °C (5% CO2, 95% humidity) for 12 days to assess if culture within the rECM gel could provide a 3D environment for Mesenchymal to Epithelial transition of Metanephric Mesenchyme progenitors.
2.13. Renal Phenotypic Marker Expression
On Day 12 of gel embedding, the EBs within the rECM gel in the 48-well plate were imaged with an EVOS cell imaging system at 10× magnification. The EBs were next processed for immunofluorescence staining following standard protocols for fixation with Methanol–Acetone (7:3, Sigma Aldrich) for 5 minutes, blocking with 1% Tween-20 (Sigma Aldrich) and 5% Donkey serum (Sigma Aldrich) in PBS (15 min), staining with Primary antibodies (goat anti-human WT1, mouse anti-human K-cadherin and sheep anti-human Nephrin; all Abcam) at a dilution of 1:100 (in Blocking solution) (30 min), followed by incubation with secondary antibodies (Donkey anti-goat Alexa Fluor 488 nm, Donkey anti-mouse Alexa Fluor 594 nm and Donkey anti-sheep 637 nm; all Abcam) at 1:100 dilution (60 min). Following this, DPX mounting medium was added to each well. Samples were imaged with a Confocal microscope (488 nm and 543 nm excitation laser). EBs grown in basal media without the rECM gel or supplementary growth factors were used as negative controls. Unprocessed mouse kidneys sectioned and stained for renal progenitor and podocyte marker WT1, podocyte marker Nephrin and renal epithelial cell marker K-cadherin were used as positive controls.
2.14. Statistical Analyses
Statistical analyses were carried out using SPSS software version 21. Variance and significance levels were analyzed using one way ANOVA with significance established at p < 0.05. Homogeneity of Variance was tested using Levine’s statistic. Significant differences within and between groups of data were assessed using the post hoc Bonferroni test. Normality of data distribution was assessed using a QQ plot. Graphs have been generated with Microsoft Excel 2019 and the data are expressed as the Mean +/− Standard Error.
3. Results
Herein, we sought to develop an efficient protocol for murine renal decellularization that could be performed with basic equipment and would balance maximal cellular removal with optimal preservation of ECM components. Our overall aim was to use decellularized kidney tissue to create a renal ECM gel (henceforth rECM) that could be used to promote terminal differentiation and maturation of renal lineage-committed hiPSC derivatives. The first step towards this aim was to achieve renal decellularization. Given that it is difficult to procure human kidneys, and there is a lack of universal access to specialist equipment for tissue perfusion, we opted to develop an immersion decellularization method using mouse kidneys. We elected to use mouse kidneys because they are of mammalian origin, available with relative ease and are suitable for immersion decellularization by virtue of their dimensions. Four different decellularization methods, denoted as decellularization protocols (DP) 1–4, combining varying concentrations and incubation times in ionic (0.5% w/v SDS) and/or non-ionic (1% v/v Triton-X100) detergents, in addition to mechanical methods and enzymatic treatment, were tested. As experimental controls, we included kidneys which were not subjected to a decellularization process (denoted native ‘untreated’ kidney) and kidneys which underwent an initial lysis stage comprising 4 h of immersion in ddH2O with no further decellularization (‘Lysed’ sample).
3.1. Qualitative Analysis of Decellularized Murine Renal Scaffolds
Post-decellularization, samples from each of the four treatment protocols were preserved for qualitative assessment with respect to their gross morphological appearance, in addition to observation by light microscopy and immunohistochemistry. Samples from the Native (untreated) kidney slices and Lysed samples were assessed as controls.
3.1.1. Appearance of Kidneys Following Decellularization
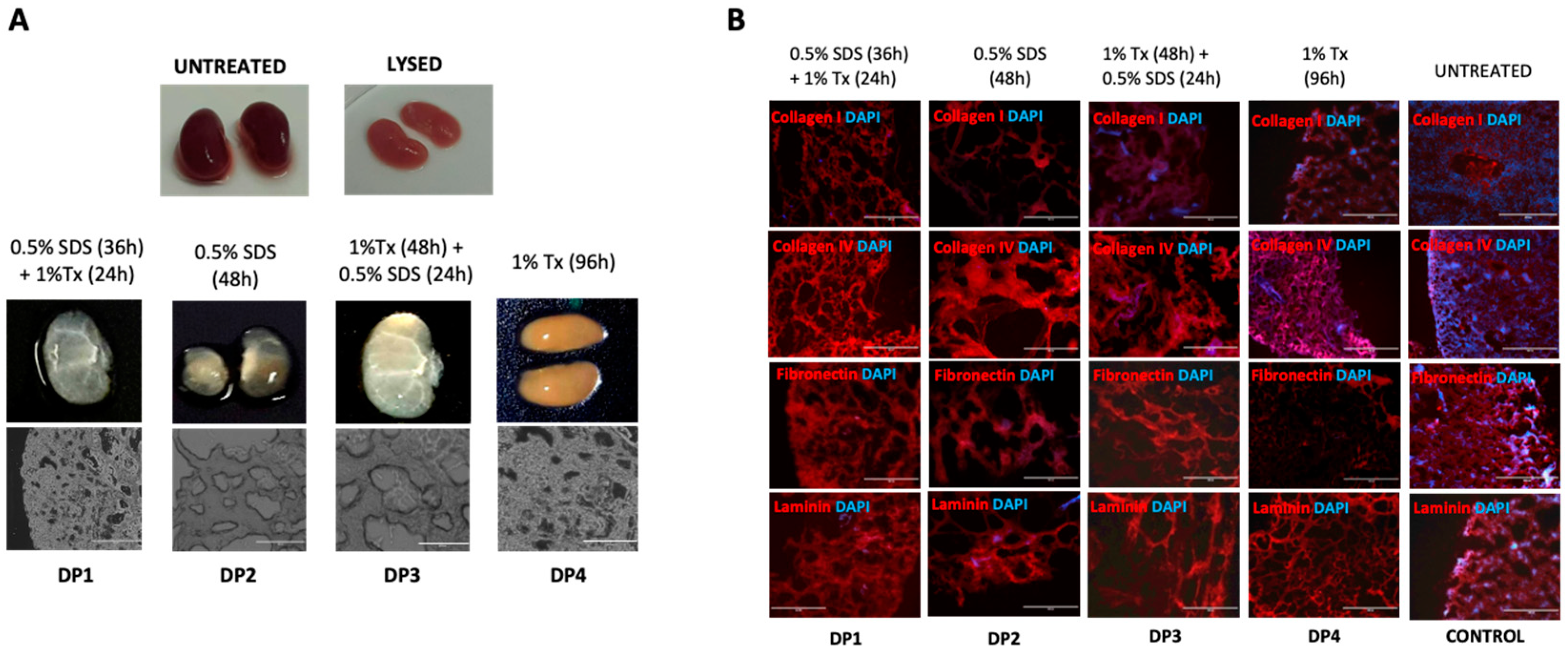
Figure 1A shows the gross macroscopic appearance of kidney tissues that underwent decellularization using one of the four different decellularization protocols: DP1- 0.5% SDS (36 h) + 1% Tx (24 h); DP2- 0.5% SDS (48 h); DP3- 1% Tx (48 h) + 0.5% SDS (24 h) and DP4- 1% Tx (96 h) in comparison with the controls. Shown are macroscopic images of whole (Untreated and Lysed), and sliced kidneys (DP1–4), and additionally for DP1–4 samples, light micrographs of the decellularized kidney tissues (bottom panels). In these transmitted light images, samples treated with DP1 and DP4 appeared to better preserve the tissue architecture in comparison to samples treated with DP2 and DP3 protocols where wider lacunar spaces are evident in the tissue sections (Figure 1A, bottom panels). Furthermore, as a potential indicator of enhanced cellular removal, tissue scaffolds treated with DP1 appeared to be the most visibly transparent when compared to scaffolds treated with the other protocols.
3.1.2. Microscopic Analysis of Decellularized Scaffolds for ECM Component Preservation
The biochemical characteristics of decellularized tissues were next assessed to determine the efficiency of the decellularization protocols. Decellularized and control tissues were sectioned, fixed and immunostained to examine their expression of the ECM proteins collagen I, collagen IV, laminin and fibronectin, to determine which, if any, protocols were able to best preserve the ECM following decellularization (Figure 1B). Sections were also counterstained with DAPI to visualize nuclear material in the sections as a qualitative measure to assess the extent of cellular removal (Figure 1B). The immunofluorescence images revealed superior protein preservation in DP1- and DP4-treated samples. However, DP4-treated samples also showed a high DAPI signal, indicating significant persistence of residual nuclear material even after decellularization. DP2 and DP3 protocols, while appearing to remove cells better than that of DP4, simultaneously appeared to have had a harsher effect on the tissue samples, as was evident with the staining pattern when compared to both DP1- and DP4-treated samples.
3.2. Quantitative Analysis of Scaffolds for ECM Component Preservation and Cell Clearance
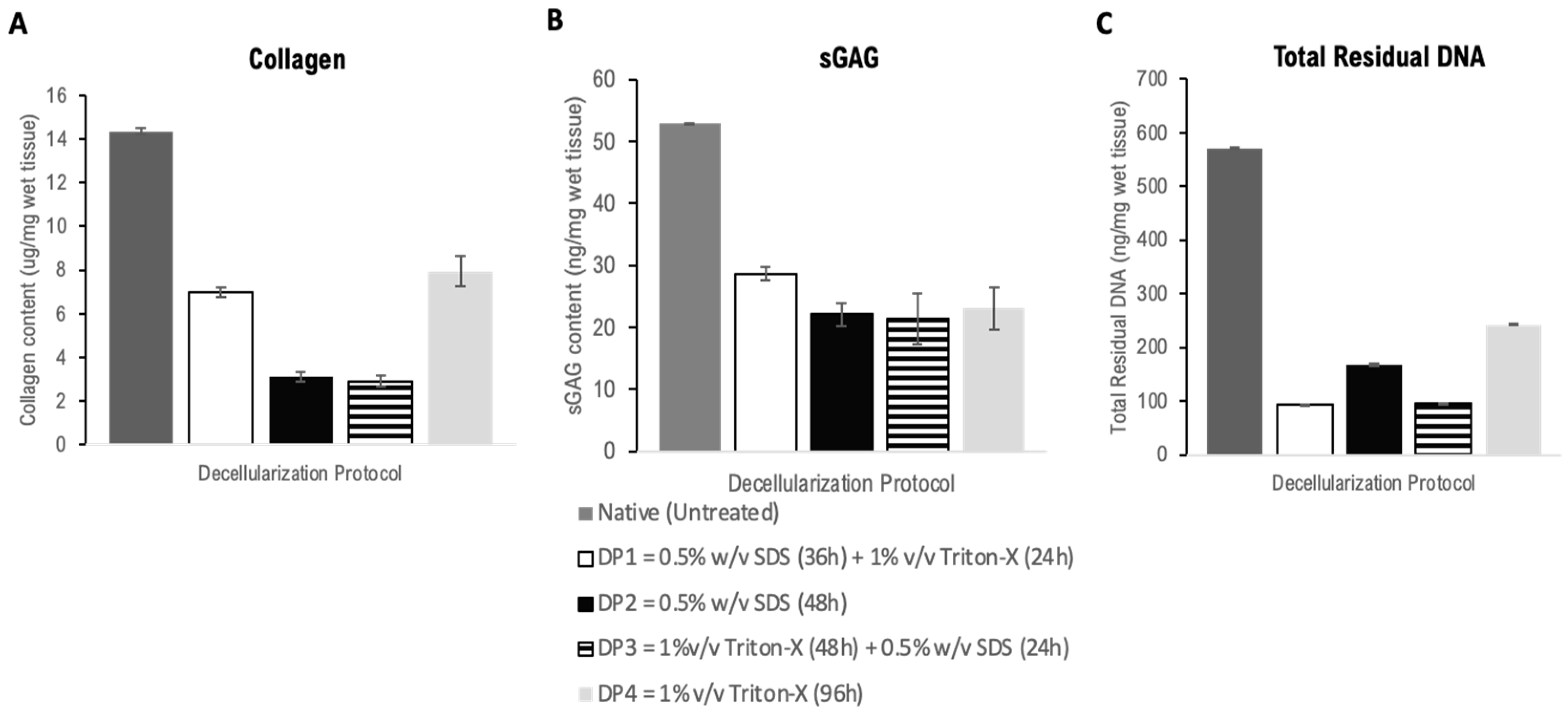
Following qualitative assessment of ECM protein expression, we formally quantified the soluble collagen content, glycosaminoglycan (GAG) and residual DNA content of the decellularized scaffolds (Figure 2). Scaffolds from each DP treatment group were quantified separately to identify the treatment which best preservation these components, which could be used to decellularize subsequent batches of murine kidneys for the remainder of the study.
3.2.1. Collagen Quantification
Collagen quantification was carried out on three independent samples from each decellularization protocol (Figure 2A). DP4 and DP1 samples preserved significantly higher quantities of soluble collagen than DP2- and DP3-treated samples whose collagen content was less than half the value obtained from samples treated with DP1 and 4. However, when compared with the control native (untreated) tissue samples, each of the decellularization treatment protocols significantly leached out soluble collagen during the treatment procedure.
3.2.2. Sulphated Glycosaminoglycan (sGAG) Quantification
sGAG content quantification was conducted using the Blyscan sGAG quantification kit on papain extracts from three independent samples from each DP treatment group (Figure 2B). There was no significant difference in preserved sGAG content between any of the decellularized cohorts. Nonetheless, samples treated with DP1 retained the highest sGAG content post-decellularization followed by DP4, DP2 and DP3 in descending order. Each of these values were significantly lower when compared to the control native (untreated) tissue samples following normalization for initial tissue weight.
3.2.3. Total DNA Content
Samples preserved from each decellularization protocol were quantified for DNA content as a measure of cellular clearance (Figure 2C). Nanodrop analysis revealed that the lowest residual DNA contents were from samples in the DP1 group, followed by DP3 and DP2, with DP4 having the highest DNA content. In contrast, native (untreated) tissue samples were found to have a significantly higher DNA content following normalization compared to all decellularization protocols.
Taking the results from the qualitative analysis, collagen quantification, sGAG quantification, and total residual DNA estimation together, we found that DP1 decellularized mouse kidneys most efficiently among all decellularized cohorts. Hence, DP1 was utilized for subsequent batches of mouse kidney decellularizations.
3.3. Generating a Renal Precursor Population from hiPSCs
To test whether the renal ECM gel could support the mesenchymal to epithelial transition of differentiating hiPSC derived renal progenitors, we next took hiPSC-derived Embryoid Bodies (EBs) and directed them to the metanephric mesenchyme lineage in accordance with a protocol developed by Taguchi et al. [12]. Using reverse transcriptase Polymerase Chain Reaction (Supplementary Methods, Supplementary Table S1), stage specific renal gene expression was confirmed by expression of FGF5 (for the epiblast stage), WT1 and HOX11 (for posterior intermediate mesenchyme) and PAX2 and SIX2 (for metanephric mesenchyme) (Supplementary Figure S1).
3.4. Examination of Markers Indicating Renal Epithelial Transition in Renal Lineage-Committed Embryoid Bodies Cultured in rECM Gel
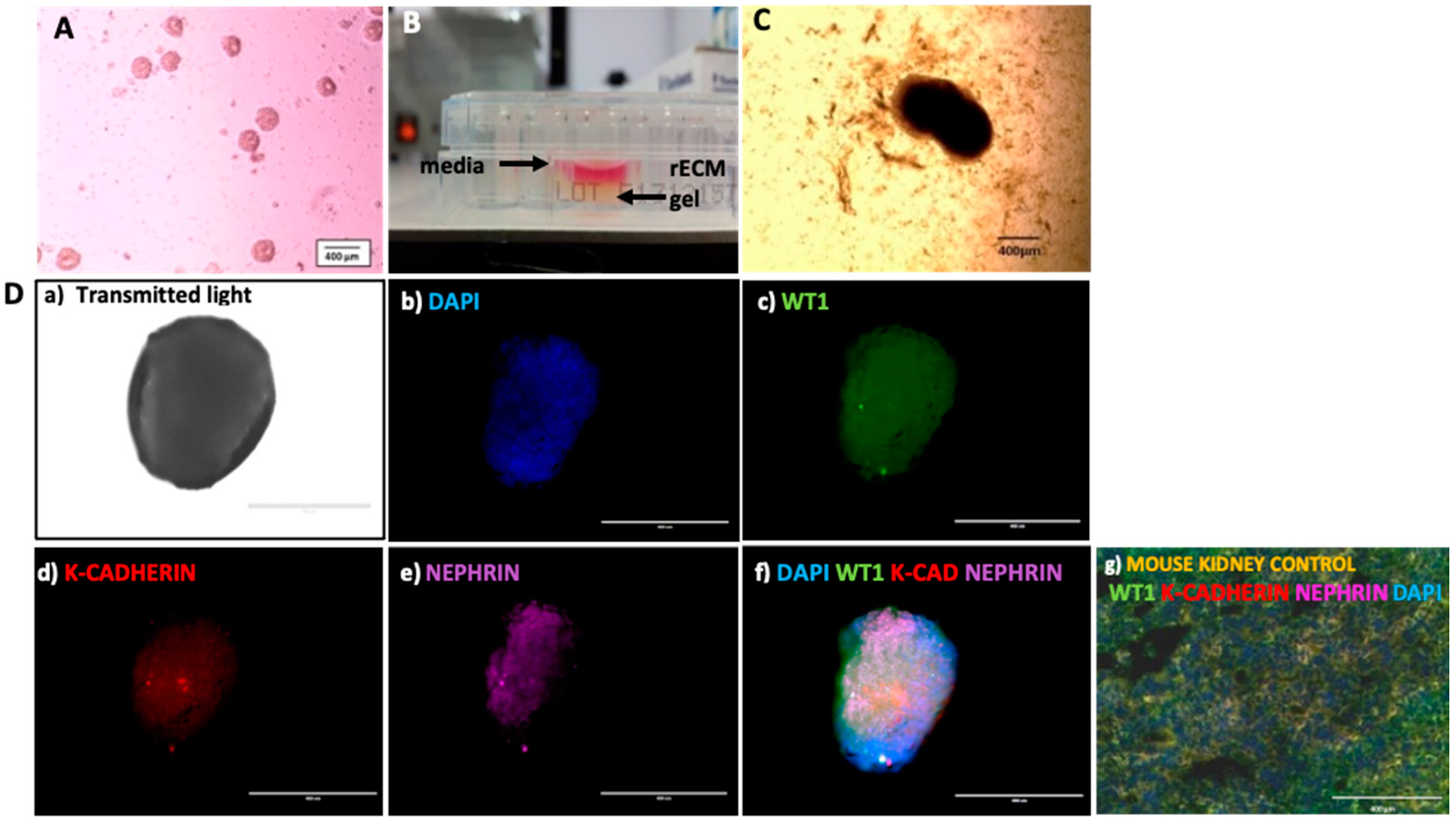
Metanephric mesenchyme EBs (MM-EB, Figure 3A) were then cultured in rECM gel, overlaid with renal culture media (Figure 3B). These EB–rECM constructs were cultured for 12 days. Figure 3C shows an EB embedded within the rECM at day 3 post-embedding. RECM-embedded lineage committed EBs at Day 12 were harvested and assessed by immunofluorescence for markers concomitant with renal MET and maturation (Figure 3D).
Figure 3D(a) shows an rECM embedded EB under transmitted light. Staining with DAPI nuclear counterstain (Figure 3D(b)) was undertaken in conjunction with expression of renal markers including nephron progenitor cell and podocyte marker WT1 (Figure 3D(c)), tubular epithelial marker K-cadherin (Figure 3D(d)) and podocyte marker Nephrin (Figure 3D(e)), which revealed similar expression of renal markers in rECM differentiated EBs (Figure 3D(f)) compared to native (untreated) mouse kidney control sections (Figure 3D(g)). These data suggest that culturing renal lineage committed MM-EBs within a 3D renal ECM gel can induce the mesenchymal to epithelial transition.
4. Discussion
In this report, we devised an efficient and effective method for kidney tissue decellularization that could balance cellular removal with optimal preservation of ECM components. Our overall objective was to take the resulting decellularized renal scaffold to generate an ECM-based renal hydrogel (rECM gel) for developing a 3D hiPSC renal organoid model. Toward that aim, we observed hiPSC-derived EBs from the renal metanephric mesenchyme lineage develop into maturing renal epithelial cells, demonstrating that our rECM gel may provide a 3D environment conducive for the renal mesenchymal to epithelial transition and subsequent progenitor maturation.
Four protocols (DP1–4) were devised to achieve decellularization of mouse kidney sections. The protocols utilized varying concentrations of, and incubation periods in, the detergents SDS, Triton-X or both, with subsequent nuclease treatment, and tissues subjected to each protocol were characterized to determine their retention of the key ECM proteins. DP1—which used a combination of 0.5% w/v Sodium Dodecyl Sulphate and 1% v/v Triton-X and mechanical agitation for 60 h—provided the best-preserved renal ECM, as judged by biochemical analysis of ECM components and DNA content to measure efficient cell removal. Conversely, samples treated with DP1 had the lowest levels of DNA retention. This was higher than expected, particularly when there was a lack of cellular debris observed by light microscopy and low or no DAPI staining observed in DP1 immunostained sections [26,27]. A possible reason for this disparity could be attributed to DNA content being measured using papain extracts (obtained while processing decellularized kidney slices for the sGAG quantification assay) which may have interfered with the spectrophotometer readings yielding disproportionately high values. Since our aim was to utilize the renal ECM to make an rECM homogenate gel for cell culture rather than using it in vivo, this was not a major concern. However, to avoid this issue in future in vivo applications it will be prudent to perform DNA quantitation analysis with an alternative method.
Kidneys subjected to the DP1 protocol were then used to create a renal ECM-derived gel matrix to investigate whether culture within the rECM would enhance renal lineage specification by enabling the mesenchymal to epithelial transition of renal progenitors. The importance of a niche-specific culture environment is well established [28]. A case in point is the Ott lab who developed a bioengineered kidney which produced rudimentary urine, using human umbilical vein endothelial cells (HUVEC) and neonatal rat kidney epithelial cells seeded onto perfusion-decellularized kidney scaffolds [29]. We rationalized that, as a naturally derived non-synthetic gelating solvent, gelatin would represent a useful vehicle in which to disperse the homogenized murine renal ECM to create the rECM gel. Tronci et al extensively studied gelatin-based hydrogels and found that crosslinking improved their thermostability [30]. However, we opted not to incorporate synthetic chemicals into the rECM gel to avoid downstream cytotoxicity.
We tested the rECM gel for its potential to induce terminal mesenchymal to epithelial transition in renal lineage-committed MM stage EBs as discussed below. Accordingly, we first generated a population of renal progenitor cells by directed differentiation of hiPSC-derived EBs, by adapting a method by Taguchi et al. [12], to the desired renal Metanephric Mesenchyme (MM) stage. To affirm progression of renal differentiation, we checked for expression of key renal genes including FGF5, expressed by Epiblast, prior to gastrulation; WT1 and HOX11, markers of Posterior Intermediate Mesoderm, the intermediate stage of renal development; and finally, the expression of PAX2 and SIX2, expressed by MM cells [31]. These genes were all expressed, confirming successful generation of the desired renal MM progenitor population. Following culture in rECM gel, the MM precursors gave rise to a rudimentary renal organoid expressing podocyte, nephron and tubular epithelium markers including K-cadherin, Nephrin and WT1. While WT1 is a common marker for both renal progenitor and resident mesenchymal stem cells, expression of K-cadherin is indicative of mesenchymal to epithelial transition in the MM-EBs [32]. Moreover, the MM-EBs expressed both SIX2 and PAX2 prior to rECM gel culture, further confirming renal lineage commitment. Together, these data suggest that MM precursors cultured in rECM gel gave rise to a rudimentary renal organoid with mesenchymal to epithelial transition and possible podocyte development.
Normal renal development relies on the interactions of the MM and the ureteric bud (UB) in conjunction with renal stromal cells, some of which arise from within the kidney and others migrate in from other sites [31,33]. The UB was notably absent from our study, hence, we did not attempt to demonstrate, or expect to observe definitive tubulogenesis with renal epithelial cells acquiring the appropriate spatial geometry as seen in the in vivo developing kidney. Taguchi et al. were able to derive Nephrin+ and E-cadherin+ tubule cells from MM cells but only following co-culture with spinal cord cells, which they suggest provided Wnt signals normally produced by the UB [12]. Thus, it appears that signals typically provided by the UB in directing the MM to develop into nephron lineage cells may have been provided to our differentiated renal cells by the rECM gel. In support of this theory, several studies have reported a role for the ECM in influencing renal cell development and fate [34]. Linton et al. observed that knockout of the ECM protein nephronectin, a ligand for α8β1 integrin, causes a dramatic reduction in GDNF expression in the MM, leading to renal agenesis and hypoplasia [34]. In line with these observations, since our MM stage EBs exposed to renal niche-specific rECM developed expression of both K-cadherin and Nephrin post-culture without any UB element, perhaps the rECM had a role to play in the Wnt signaling required to bring about these changes. However, this is speculation until further experimentation can enable a definitive conclusion to be drawn. A further elegant study from Taguchi et al. was the first to demonstrate branching morphogenesis of mouse embryonic stem cells (mESCs) generating an intricate network of interconnected segmented tubular nephrons [35], but relied on provision of MM and UB progenitors in addition to stromal cell populations. Perhaps if the epithelial–mesenchymal interaction of the MM and UB progenitor populations were established within a renal ECM gel niche, addition of the rECM could enable enhanced in vitro generation of functionally coherent renal organoids even in the absence of supportive renal stromal cells, which are difficult to derive/obtain in the human setting. The rudimentary renal organoid generated here with support of the rECM gel, while lacking segmented nephrons typical of kidney architecture, nonetheless shows promise for developing this co-culture process. Our study provides proof of concept for the use of renal tissue ECM gels to support hiPSC renal derivatives in culture. However, use of a human, rather than mouse, renal ECM would be preferable [6], and work to generate a human renal ECM gel for 3D culture of renal organoids as models of renal development and disease is underway in our laboratory. Future work should examine the mechanical characteristics of the ECM gel including rheological analysis and an examination of the gel ultrastructure by scanning electron microscopy.
In closing, ECM-derived gels could be used in conjunction with stem-cell-derived or primary renal progenitors to better recapitulate the renal niche in the quest to generate a definitive nephron population that could act as a 3D platform for CKD modeling. Indeed, novel human disease models for CKD are urgently required since effective drugs to treat or reverse the impact of renal fibrosis are currently lacking [5]. A renal organoid with morphological and functional relevance to fibrotic renal disease could be used to screen potential anti-fibrotic drug candidates for efficacy and toxicity and serve as the nexus between pre-clinical research and translation to new therapies in CKD.
Supplementary Materials
The following supporting information can be downloaded at https://www.mdpi.com/article/10.3390/organoids2010005/s1, Supplementary Methodology and Supplementary Table S1: Primer information; Supplementary Figure S1: Expression of renal genes measured by reverse transcriptase polymerase chain reaction (RT-PCR).
Author Contributions
Conceptualization, S.N. and A.S.B.; methodology, S.N. and A.S.B.; formal analysis, S.N. and A.S.B.; investigation, S.N.; data curation, S.N. and A.S.B.; writing—original draft preparation, S.N. and A.S.B.; writing—review and editing, S.N. and A.S.B.; visualization, S.N. and A.S.B.; supervision, A.S.B.; project administration, A.S.B.; funding acquisition, A.S.B. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the UCL Division of Surgery & Interventional Science (A.S.B.).
Institutional Review Board Statement
Mice used in this study were humanely sacrificed by Schedule 1 methods in accordance with the UK Home Office, which governs and protects the use of animals for scientific experimentation and research under the Animals (Scientific Procedures) Act 1986. As non-licensed procedures, Schedule 1 procedures do not require institutional ethical approval. Use of commercial hiPSC lines does not fall under the remit of the Human Tissues Act 2004 or require UK institutional ethical approval for their use.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript; or in the decision to publish the results.
References
- Stuart, R.O.; Nigam, S.K. Development of the tubular nephron. Semin. Nephrol. 1995, 15, 315–326. [Google Scholar] [PubMed]
- Bikbov, B.; Purcell, C.A.; Levey, A.S.; Smith, M.; Abdoli, A.; Abebe, M.; Adebayo, O.M.; Afarideh, M.; Agarwal, S.K.; Agudelo-Botero, M.; et al. Global, regional, and national burden of chronic kidney disease, 1990–2017: A systematic analysis for the Global Burden of Disease Study 2017. Lancet 2020, 395, 709–733. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stevens, P.; O’Donoghue, D.; de Lusignan, S.; Van Vlymen, J.; Klebe, B.; Middleton, R.; Hague, N.; New, J.; Farmer, C. Chronic kidney disease management in the United Kingdom: NEOERICA project results. Kidney Int. 2007, 72, 92–99. [Google Scholar] [CrossRef] [Green Version]
- Mills, K.T.; Xu, Y.; Zhang, W.; Bundy, J.D.; Chen, C.-S.; Kelly, T.N.; Chen, J.; He, J. A systematic analysis of worldwide population-based data on the global burden of chronic kidney disease in 2010. Kidney Int. 2015, 88, 950–957. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruiz-Ortega, M.; Lamas, S.; Ortiz, A. Antifibrotic Agents for the Management of CKD: A Review. Am. J. Kidney Dis. 2022, 80, 251–263. [Google Scholar] [CrossRef] [PubMed]
- Cheval, L.; Pierrat, F.; Rajerison, R.; Piquemal, D.; Doucet, A. Of Mice and Men: Divergence of Gene Expression Patterns in Kidney. PLoS ONE 2012, 7, e46876. [Google Scholar] [CrossRef] [Green Version]
- Takahashi, K.; Yamanaka, S. Induction of Pluripotent Stem Cells from Mouse Embryonic and Adult Fibroblast Cultures by Defined Factors. Cell 2006, 126, 663–676. [Google Scholar] [CrossRef] [Green Version]
- Takahashi, K.; Tanabe, K.; Ohnuki, M.; Narita, M.; Ichisaka, T.; Tomoda, K.; Yamanaka, S. Induction of Pluripotent Stem Cells from Adult Human Fibroblasts by Defined Factors. Cell 2007, 131, 861–872. [Google Scholar] [CrossRef] [Green Version]
- Park, I.-H.; Arora, N.; Huo, H.; Maherali, N.; Ahfeldt, T.; Shimamura, A.; Lensch, M.W.; Cowan, C.; Hochedlinger, K.; Daley, G.Q. Disease-Specific Induced Pluripotent Stem Cells. Cell 2008, 134, 877–886. [Google Scholar] [CrossRef] [Green Version]
- Song, B.; Smink, A.M.; Jones, C.V.; Callaghan, J.M.; Firth, S.D.; Bernard, C.C.A.; Laslett, A.; Kerr, P.G.; Ricardo, S.D. The Directed Differentiation of Human iPS Cells into Kidney Podocytes. PLoS ONE 2012, 7, e46453. [Google Scholar] [CrossRef] [Green Version]
- Xia, Y.; Nivet, E.; Sancho-Martinez, I.; Gallegos, T.F.; Suzuki, K.; Okamura, D.; Wu, M.-Z.; Dubova, I.; Esteban, C.R.; Montserrat, N.; et al. Directed differentiation of human pluripotent cells to ureteric bud kidney progenitor-like cells. Nat. Cell Biol. 2013, 15, 1507–1515. [Google Scholar] [CrossRef] [PubMed]
- Taguchi, A.; Kaku, Y.; Ohmori, T.; Sharmin, S.; Ogawa, M.; Sasaki, H.; Nishinakamura, R. Redefining the In Vivo Origin of Metanephric Nephron Progenitors Enables Generation of Complex Kidney Structures from Pluripotent Stem Cells. Cell Stem Cell 2014, 14, 53–67. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takasato, M.; Er, P.X.; Becroft, M.; Vanslambrouck, J.M.; Stanley, E.G.; Elefanty, A.G.; Little, M.H. Directing human embryonic stem cell differentiation towards a renal lineage generates a self-organizing kidney. Nat. Cell Biol. 2013, 16, 118–126. [Google Scholar] [CrossRef] [PubMed]
- Takasato, M.; Er, P.X.; Chiu, H.S.; Maier, B.; Baillie, G.J.; Ferguson, C.; Parton, R.G.; Wolvetang, E.J.; Roost, M.S.; Chuva de Sousa Lopes, S.M.; et al. Kidney organoids from human iPS cells contain multiple lineages and model human nephrogenesis. Nature 2015, 526, 564–568. [Google Scholar] [CrossRef] [PubMed]
- Freedman, B.S.; Brooks, C.R.; Lam, A.Q.; Fu, H.; Morizane, R.; Agrawal, V.; Saad, A.F.; Li, M.K.; Hughes, M.R.; Werff, R.V.; et al. Modelling kidney disease with CRISPR-mutant kidney organoids derived from human pluripotent epiblast spheroids. Nat. Commun. 2015, 6, 8715. [Google Scholar] [CrossRef] [Green Version]
- Morizane, R.; Lam, A.Q.; Freedman, B.S.; Kishi, S.; Valerius, M.T.; Bonventre, J.V. Nephron organoids derived from human pluripotent stem cells model kidney development and injury. Nat. Biotechnol. 2015, 33, 1193–1200. [Google Scholar] [CrossRef] [Green Version]
- Wu, H.; Uchimura, K.; Donnelly, E.L.; Kirita, Y.; Morris, S.A.; Humphreys, B.D. Comparative Analysis and Refinement of Human PSC-Derived Kidney Organoid Differentiation with Single-Cell Transcriptomics. Cell Stem Cell 2018, 23, 869–881.e8. [Google Scholar] [CrossRef] [Green Version]
- Robb, K.P.; Shridhar, A.; Flynn, L.E. Decellularized Matrices As Cell-Instructive Scaffolds to Guide Tissue-Specific Regeneration. ACS Biomater. Sci. Eng. 2017, 4, 3627–3643. [Google Scholar] [CrossRef]
- Brown, B.N.; Badylak, S.F. Extracellular matrix as an inductive scaffold for functional tissue reconstruction. Transl. Res. 2014, 163, 268–285. [Google Scholar] [CrossRef] [Green Version]
- Gilpin, S.E.; Guyette, J.P.; Gonzalez, G.; Ren, X.; Asara, J.M.; Mathisen, D.J.; Vacanti, J.P.; Ott, H.C. Perfusion decellularization of human and porcine lungs: Bringing the matrix to clinical scale. J. Heart Lung Transplant. 2014, 33, 298–308. [Google Scholar] [CrossRef]
- Elebring, E.; Kuna, V.K.; Kvarnström, N.; Sumitran-Holgersson, S. Cold-perfusion decellularization of whole-organ porcine pancreas supports human fetal pancreatic cell attachment and expression of endocrine and exocrine markers. J. Tissue Eng. 2017, 8, 2041731417738145. [Google Scholar] [CrossRef] [PubMed]
- Kuna, V.K.; Kvarnström, N.; Elebring, E.; Holgersson, S.S. Isolation and Decellularization of a Whole Porcine Pancreas. J. Vis. Exp. 2018, 140, e58302. [Google Scholar]
- Guyette, J.P.; Gilpin, S.E.; Charest, J.M.; Tapias, L.F.; Ren, X.; Ott, H.C. Perfusion decellularization of whole organs. Nat. Protoc. 2014, 9, 1451–1468. [Google Scholar] [CrossRef]
- Maghsoudlou, P.; Georgiades, F.; Smith, H.; Milan, A.; Shangaris, P.; Urbani, L.; Loukogeorgakis, S.P.; Lombardi, B.; Mazza, G.; Hagen, C.; et al. Optimization of Liver Decellularization Maintains Extracellular Matrix Micro-Architecture and Composition Predisposing to Effective Cell Seeding. PLoS ONE 2016, 11, e0155324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Bao, J.; Wu, Q.; Zhou, Y.; Li, Y.; Wu, X.; Shi, Y.; Li, L.; Bu, H. Method for perfusion decellularization of porcine whole liver and kidney for use as a scaffold for clinical-scale bioengineering engrafts. Xenotransplantation 2014, 22, 48–61. [Google Scholar] [CrossRef]
- Gilbert, T.W.; Sellaro, T.L.; Badylak, S.F. Decellularization of tissues and organs. Biomaterials 2006, 27, 3675–3683. [Google Scholar] [CrossRef]
- Gilbert, T.; Freund, J.M.; Badylak, S.F. Quantification of DNA in Biologic Scaffold Materials. J. Surg. Res. 2009, 152, 135–139. [Google Scholar] [CrossRef] [Green Version]
- Sellaro, T.L.; Ranade, A.; Faulk, D.M.; McCabe, G.P.; Dorko, K.; Badylak, S.F.; Strom, S.C. Maintenance of human hepatocyte function in vitro by liver-derived extracellular matrix gels. Tissue Eng. Part A 2010, 16, 1075–1082. [Google Scholar] [CrossRef] [Green Version]
- Song, J.J.; Guyette, J.P.; Gilpin, S.; Gonzalez, G.; Vacanti, J.P.; Ott, H.C. Regeneration and experimental orthotopic transplantation of a bioengineered kidney. Nat. Med. 2013, 19, 646–651. [Google Scholar] [CrossRef]
- Tronci, G.; Neffe, A.T.; Pierce, B.F.; Lendlein, A. An entropy–elastic gelatin-based hydrogel system. J. Mater. Chem. 2010, 20, 8875–8884. [Google Scholar] [CrossRef]
- Kobayashi, A.; Valerius, M.T.; Mugford, J.W.; Carroll, T.J.; Self, M.; Oliver, G.; McMahon, A.P. Six2 Defines and Regulates a Multipotent Self-Renewing Nephron Progenitor Population throughout Mammalian Kidney Development. Cell Stem Cell 2008, 3, 169–181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martin, C.E.; Jones, N. Nephrin Signaling in the Podocyte: An Updated View of Signal Regulation at the Slit Diaphragm and Beyond. Front. Endocrinol. 2018, 9, 302. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, A.; Mugford, J.W.; Krautzberger, A.M.; Naiman, N.; Liao, J.; McMahon, A.P. Identification of a Multipotent Self-Renewing Stromal Progenitor Population during Mammalian Kidney Organogenesis. Stem Cell Rep. 2014, 3, 650–662. [Google Scholar] [CrossRef] [Green Version]
- Linton, J.M.; Martin, G.R.; Reichardt, L.F. The ECM protein nephronectin promotes kidney development via integrin alpha8beta1-mediated stimulation of Gdnf expression. Development 2007, 134, 2501–2509. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taguchi, A.; Nishinakamura, R. Higher-Order Kidney Organogenesis from Pluripotent Stem Cells. Cell Stem Cell 2017, 21, 730–746. [Google Scholar] [CrossRef] [Green Version]
Figure 1.
(A) Optical and light microscopy of native and decellularized murine kidneys. Four different decellularization conditions using combinations of Sodium Dodecyl Sulphate (SDS) and Triton-X100 (Tx) followed by DNase (Benzonase® nuclease) digestion (decellularization protocols (DP) 1–4) were used to decellularize mouse kidneys compared with non-decellularized kidney (UNTREATED) and kidneys subjected to a cellular lysis treatment involving ddH2O immersion for 4 h (LYSED). The conditions used in each DP were as follows: DP1—0.5% SDS (36 h) + 1% Tx (24 h); DP2—0.5% SDS (48 h); DP3—1% Tx (48 h) + 0.5% SDS (24 h) and DP4—1% Tx (96 h). Shown are macroscopic images of whole (UNTREATED and LYSED) and sliced decellularized kidneys (DP1–4), and additionally for DP1–4 samples, light micrographs of the decellularized kidney tissues (lower panels). (n > 3 per experiment), Scale bar: 400 µm; Magnification: 10×; (B) Immunofluorescence of extra-cellular matrix proteins post-decellularization. Preservation of collagen I, collagen IV, fibronectin and laminin following decellularization of murine kidney slices by varied combinations of SDS and/or Triton-X100 was assessed. Untreated murine kidneys served as negative controls. (n > 3 per experiment).
Figure 1.
(A) Optical and light microscopy of native and decellularized murine kidneys. Four different decellularization conditions using combinations of Sodium Dodecyl Sulphate (SDS) and Triton-X100 (Tx) followed by DNase (Benzonase® nuclease) digestion (decellularization protocols (DP) 1–4) were used to decellularize mouse kidneys compared with non-decellularized kidney (UNTREATED) and kidneys subjected to a cellular lysis treatment involving ddH2O immersion for 4 h (LYSED). The conditions used in each DP were as follows: DP1—0.5% SDS (36 h) + 1% Tx (24 h); DP2—0.5% SDS (48 h); DP3—1% Tx (48 h) + 0.5% SDS (24 h) and DP4—1% Tx (96 h). Shown are macroscopic images of whole (UNTREATED and LYSED) and sliced decellularized kidneys (DP1–4), and additionally for DP1–4 samples, light micrographs of the decellularized kidney tissues (lower panels). (n > 3 per experiment), Scale bar: 400 µm; Magnification: 10×; (B) Immunofluorescence of extra-cellular matrix proteins post-decellularization. Preservation of collagen I, collagen IV, fibronectin and laminin following decellularization of murine kidney slices by varied combinations of SDS and/or Triton-X100 was assessed. Untreated murine kidneys served as negative controls. (n > 3 per experiment).

Figure 2.
Characterization of the murine renal ECM: (A) Quantification of soluble collagen retained post-decellularization with 4 decellularization protocols (DP1–4) using the SIRCOL Soluble Collagen Assay Kit (Biocolor, UK). Data presented as Mean +/− Standard error. Variance analyzed using one-way ANOVA in SPSS version 21. Significance established at p < 0.05. Post hoc Bonferroni test shows significant difference in preserved collagen content between each treatment group from each DP analyzed except for DP1 compared with DP4, and DP2 compared with DP3. (B) Sulphated glycosaminoglycan (sGAG) content in samples post-decellularization with 4 separate decellularization protocols (DP1–4) using the sGAG quantification assay (Blyscan, Biocolor, UK) compared to untreated kidney. Data presented as Mean +/− Standard error. Variance analyzed using one-way ANOVA in SPSS version 21. Significance established at p < 0.05. Post hoc Bonferroni test showed no significant difference between preserved sGAG content among the treatment groups (p = 1.000). (C) DNA content as a measure of cellular clearance with 4 decellularization protocols (DP1–4) compared to untreated kidney. Data presented as Mean +/− Standard error. Variance analyzed using one-way ANOVA in SPSS version 21. Significance established at p < 0.05. Post hoc Bonferroni analysis revealed statistically significant differences (p < 0.0001) between all decellularized cohort combinations, except for between DP2 and DP3 where statistical significance was p = 0.01. Statistically significant differences were observed between all the decellularized cohorts with the native (untreated) controls. Abbreviations: ECM = Extracellular Matrix; hiPSC = Human Induced Pluripotent Stem Cells; sGAG = Sulphated Glycosaminoglycan; ANOVA = Analysis of Variance; SPSS = Statistical Package for Social Sciences; DP = Decellularization Protocol; DP1 = 0.5% w/v SDS (36 h) + 1% v/v Triton-X (24 h); DP2 = 0.5% w/v SDS (48 h); DP3 = 1% v/v Triton-X (48 h) + 0.5% w/v SDS (24 h); DP4 = 1% v/v Triton-X (96 h).
Figure 2.
Characterization of the murine renal ECM: (A) Quantification of soluble collagen retained post-decellularization with 4 decellularization protocols (DP1–4) using the SIRCOL Soluble Collagen Assay Kit (Biocolor, UK). Data presented as Mean +/− Standard error. Variance analyzed using one-way ANOVA in SPSS version 21. Significance established at p < 0.05. Post hoc Bonferroni test shows significant difference in preserved collagen content between each treatment group from each DP analyzed except for DP1 compared with DP4, and DP2 compared with DP3. (B) Sulphated glycosaminoglycan (sGAG) content in samples post-decellularization with 4 separate decellularization protocols (DP1–4) using the sGAG quantification assay (Blyscan, Biocolor, UK) compared to untreated kidney. Data presented as Mean +/− Standard error. Variance analyzed using one-way ANOVA in SPSS version 21. Significance established at p < 0.05. Post hoc Bonferroni test showed no significant difference between preserved sGAG content among the treatment groups (p = 1.000). (C) DNA content as a measure of cellular clearance with 4 decellularization protocols (DP1–4) compared to untreated kidney. Data presented as Mean +/− Standard error. Variance analyzed using one-way ANOVA in SPSS version 21. Significance established at p < 0.05. Post hoc Bonferroni analysis revealed statistically significant differences (p < 0.0001) between all decellularized cohort combinations, except for between DP2 and DP3 where statistical significance was p = 0.01. Statistically significant differences were observed between all the decellularized cohorts with the native (untreated) controls. Abbreviations: ECM = Extracellular Matrix; hiPSC = Human Induced Pluripotent Stem Cells; sGAG = Sulphated Glycosaminoglycan; ANOVA = Analysis of Variance; SPSS = Statistical Package for Social Sciences; DP = Decellularization Protocol; DP1 = 0.5% w/v SDS (36 h) + 1% v/v Triton-X (24 h); DP2 = 0.5% w/v SDS (48 h); DP3 = 1% v/v Triton-X (48 h) + 0.5% w/v SDS (24 h); DP4 = 1% v/v Triton-X (96 h).

Figure 3.
3D renal cell organoids generated from hiPSC-derived embryoid bodies (EBs) cultured in renal extracellular matrix (rECM) gel express putative markers of renal progenitor cells and mesenchyme to epithelial transition (MET). (A) hiPSC-derived EBs prior to renal differentiation protocol. (B) Image of rECM gel plated in a well of a 48-well tissue culture plate, overlaid with cell culture medium. (C) hiPSC-derived EBs were subjected to a renal differentiation protocol to drive them to develop into metanephric mesenchyme (MM-EBs). MM-EBs were then embedded in a murine renal extracellular matrix (rECM) gel comprising 10% w/v gelatin and homogenized decellularized murine kidneys. (D) Imaging of a stage 5 hiPSC-derived metanephric mesenchyme embryoid body following 12 days of culture in rECM gel. MM-EBs embedded within rECM gel for 12 days were immunostained for renal markers and imaged using an EVOS® Cell imaging system at 10× magnification to examine expression of renal cell markers indicative of MET. (a) Transmitted light; (b) DAPI nuclear counterstain; (c) WT1 (488 nm, Green fluorescent protein channel); (d) K-cadherin (tubular epithelium, 594 nm, Texas Red Channel); (e) Nephrin (podocyte marker, 637 nm, Cy5 channel); (f) Overlay of DAPI, WT1, K-cadherin and Nephrin expression; (g) mouse kidney stained for WT1, K-cadherin, Nephrin and DAPI. rECM = renal extracellular matrix gel. (Magnification 10×. Scale Bar 400 μm).
Figure 3.
3D renal cell organoids generated from hiPSC-derived embryoid bodies (EBs) cultured in renal extracellular matrix (rECM) gel express putative markers of renal progenitor cells and mesenchyme to epithelial transition (MET). (A) hiPSC-derived EBs prior to renal differentiation protocol. (B) Image of rECM gel plated in a well of a 48-well tissue culture plate, overlaid with cell culture medium. (C) hiPSC-derived EBs were subjected to a renal differentiation protocol to drive them to develop into metanephric mesenchyme (MM-EBs). MM-EBs were then embedded in a murine renal extracellular matrix (rECM) gel comprising 10% w/v gelatin and homogenized decellularized murine kidneys. (D) Imaging of a stage 5 hiPSC-derived metanephric mesenchyme embryoid body following 12 days of culture in rECM gel. MM-EBs embedded within rECM gel for 12 days were immunostained for renal markers and imaged using an EVOS® Cell imaging system at 10× magnification to examine expression of renal cell markers indicative of MET. (a) Transmitted light; (b) DAPI nuclear counterstain; (c) WT1 (488 nm, Green fluorescent protein channel); (d) K-cadherin (tubular epithelium, 594 nm, Texas Red Channel); (e) Nephrin (podocyte marker, 637 nm, Cy5 channel); (f) Overlay of DAPI, WT1, K-cadherin and Nephrin expression; (g) mouse kidney stained for WT1, K-cadherin, Nephrin and DAPI. rECM = renal extracellular matrix gel. (Magnification 10×. Scale Bar 400 μm).

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Nag, S.; Boyd, A.S. Decellularization of Mouse Kidneys to Generate an Extracellular Matrix Gel for Human Induced Pluripotent Stem Cell Derived Renal Organoids. Organoids 2023, 2, 66-78. https://doi.org/10.3390/organoids2010005
AMA Style
Nag S, Boyd AS. Decellularization of Mouse Kidneys to Generate an Extracellular Matrix Gel for Human Induced Pluripotent Stem Cell Derived Renal Organoids. Organoids. 2023; 2(1):66-78. https://doi.org/10.3390/organoids2010005
Chicago/Turabian StyleNag, Sparshita, and Ashleigh S. Boyd. 2023. "Decellularization of Mouse Kidneys to Generate an Extracellular Matrix Gel for Human Induced Pluripotent Stem Cell Derived Renal Organoids" Organoids 2, no. 1: 66-78. https://doi.org/10.3390/organoids2010005