
Applications for Colon Organoid Models in Cancer Research

1
Gillies McIndoe Research Institute, Newtown, Wellington 6242, New Zealand
2
Wellington Regional Plastic, Maxillofacial & Burns Unit, Hutt Hospital, Lower Hutt 5010, New Zealand
3
Department of Surgery, The Royal Melbourne Hospital, The University of Melbourne, Melbourne 3010, Australia
*
Author to whom correspondence should be addressed.
Organoids 2023, 2(1), 37-49; https://doi.org/10.3390/organoids2010003
Submission received: 11 October 2022
/
Revised: 13 November 2022
/
Accepted: 4 January 2023
/
Published: 12 January 2023
(This article belongs to the Special Issue Advances in Organoid Technology—Selected Papers from "Organoids Are Us 2022")
Abstract
:Organoids are 3D organ-like structures grown from stem cells in vitro that mimic the organ or disease from which they are derived. Due to their stem cell origin, organoids contain a heterogeneous population of cells reflecting the diversity of cell types seen in vivo. Similarly, tumour organoids reflect intratumoural heterogeneity in a way that traditional 2D cell culture and cell lines do not, and, therefore, they show greater promise as a more relevant model for effective disease modelling and drug testing. Tumour organoids arise from cancer stem cells, which contribute to many of the greatest challenges to cancer treatment, including therapy resistance, tumour recurrence, and metastasis. In this review, we outline methods for generating colon organoids from patient-derived normal and tumour tissues. Furthermore, we discuss organoid biobanking, applications of organoids in disease modelling, and a range of platforms applicable to high-throughput drug testing, including apical-out/reverse-polarity colon organoids.
1. Introduction
Cancer stem cells (CSCs) are thought to arise through mutation of tissue-resident adult stem cells, dedifferentiation of transformed cancer cells, and/or therapy-induced senescence-associated reprogramming [1,2,3,4,5]. CSCs contribute to many of the greatest challenges to cancer treatment, including therapy resistance, loco-regional recurrence, and metastasis [1,2]. They divide asymmetrically to produce identical daughter CSCs as well as progenitor cells that go on to form the tumour bulk [6,7]. CSCs resist conventional cancer therapies which are directed at proliferating cells because they alternate between two different states—a proliferative state that produces progenitor cells, and a quiescent state that enables therapy resistance and subsequent recurrence [8,9]. Furthermore, they upregulate drug efflux proteins and DNA damage-induced death mechanisms [4]. CSCs have a migratory capacity that promotes the spread of cancer to distant organs. Circulating tumour cells (CTCs) have been detected in the blood of cancer patients and exhibit hallmarks of CSCs, including a hybrid epithelial-mesenchymal signature [10]. Specific biomarkers represent a powerful method of identifying and isolating CSCs. However, the heterogeneity of marker expression between different cancers and within cancer types and the crossover with normal stem cells [6] suggests that a more reliable method of CSC enrichment, and, indeed, of targeting CSCs, is through functional means [7,9].
Organoids are 3D organ-like structures grown in culture from human tissue-derived stem cells. Organoids contain diverse cell types reflecting those seen in vivo, produced by the stem cells. For example, colon organoids contain goblet cells (mucin-2 [MUC2]), enterocytes (carbonic anhydrase 2 [CA2]), endocrine cells (chromogranin A [CHGA]), Paneth cells (lysozyme [LYZ]), and transit amplifying cells (ephrin B2 [EPHB2]), as well as stem cells (LGR5) [11,12]. They self-assemble into complex structures to partially recapitulate the physiology seen in vivo, making them a promising tool for translating preclinical research into the clinic [13]. In line with this, organoids have become a valuable resource for in vitro drug testing. There are great hopes that organoids will facilitate progress in understanding diseases that have traditionally been difficult or impossible to faithfully model [14].
2. Protocols for Colon Organoid Culture
2.1. Establishing Normal and Tumour Organoids from Colon Tissues
In 2011, Sato et al. adapted a protocol for growing murine intestinal organoids to successfully culture human normal colon, Barrett’s epithelium, and colon adenoma and adenocarcinoma organoids [15]. Normal colon organoids require a wide range of media supplements to mimic the influence of signalling and niche factors, including Wnt3A, R-spondin-1/3 and Noggin [15]. However, colon tumour organoids possess mutations that render certain factors unnecessary. For example, the adenomatous polyposis coli (APC) gene is the most mutated gene in colon cancer and leads to constitutive Wnt signalling; this removes the dependence of the tumour on Wnt agonists (Wnt3A and R-spondins).
The L-WRN cell line (ATCC CRL-3276) was created by stably transfecting the Wnt3A-producing L-Wnt3A cell line (ATCC CRL-2647) with a vector expressing R-spondin-3 and Noggin [16]. Conditioned media harvested from this cell line contains these three factors with greater stability and at lower cost than if they were reconstituted from a lyophilised powder [16]. Batches of conditioned medium from L-WRN cells are highly reproducible [17].
Our protocol for generating organoids from patient-derived colon tumour tissue and patient-matched normal colonic mucosa will be outlined. This is based on methods developed and optimised previously by laboratories worldwide [15,18,19,20,21,22].
Tissue specimens are received from local hospitals on ice on the day of surgery, and a sample is taken for histological analysis. Colon tumour tissue is washed with PBS, minced into a paste using dissecting scissors and incubated in 10 mL of digestion buffer (1 mg/mL collagenase VI and 0.2 mg/mL DNase I) at 37 °C for 45–60 min. The digest is mechanically disrupted every 15 min using a 10 mL serological pipette by pipetting up and down ~10 times, expelling the contents with the tip almost flat on the bottom of the dish, until cells have been sufficiently released. The reaction is stopped by adding 1 mL of foetal calf serum and 10 mL of PBS, the digest is filtered through a 100 µM filter to remove large chunks, connective tissue, and mucus, and a red blood cell lysis is performed using a buffer of ammonium chloride (155 mM), sodium bicarbonate (12 mM), and EDTA (0.1 mM). Cells are pelleted by centrifugation at 400 g and 4 °C for 5 min, counted, and embedded in 40 µL domes of Cultrex® 3D RGF BME Culture Matrix (R&D Systems, #3445-005-01), a basement membrane gel composed primarily of laminin and collagen IV and optimised for the development of 3D organoid structures, at a density of ~1000 cells per µL (~40,000 cells per dome). In each well of a standard 24-well culture plate, 1–2 domes are seeded.
When processing samples of normal colon, the mucosa is isolated from the investing muscle layer using dissecting scissors, cut into smaller pieces (~2–5 mm) and washed with PBS by pipetting up and down ~10 times. Tissue pieces are incubated in EDTA buffer (7.5 mM EDTA in PBS with antibiotic-antimycotic [penicillin, streptomycin, amphotericin B]) at 4°C on a rocker for 60 min. Wash steps are then performed using 10 mL of PBS and a 10 mL serological pipette, by drawing the tissue pieces up and down 10 times to release crypts, retaining the 10 mL of PBS wash buffer in a 50 mL tube lined with 1% BSA, and then repeating this wash procedure a further 5 times to give a total of 50 mL of PBS containing isolated crypts. The crypts are pelleted by centrifugation at 200 g and 4 °C for 3 min, counted, and seeded in Cultrex domes at a density of 50–200 crypts per dome.
Cultrex domes are left to set at 37°C for at least 30 min before adding 0.5 mL culture medium to each well. Media recipes can be found in Table 1.
2.2. Organoid Characterisation and Phenotype
Organoids generated from the stem cells of normal colon tissue form cystic structures consisting of a hollow lumen surrounded by columnar epithelium and can exhibit a budding pattern (Figure 1) [15]. Noggin, a BMP inhibitor, is required for the maintenance of LGR5+ colon stem cells within the organoids [15]. Nicotinamide is essential for extending organoid growth beyond 1 week [15]. Furthermore, A83-01 (TGF-β receptor [Alk4/5/7] inhibitor) and either SB202190 (p38 inhibitor) or IGF1 + FGF2 allow the organoids to persist for at least 6 months [15]. Removing Wnt3A, R-spondin, Noggin, and Nicotinamide from the culture medium, and adding the γ-secretase inhibitor DAPT, causes organoids to differentiate [15,22].
Colon cancer organoids tend to develop as compact masses of cells without the lumens observed in normal colon organoids (Figure 2) [15]. Most colon tumour organoids do not require Noggin in the culture medium [15]. Approximately 80% of colon cancers have mutations to APC, AXIN2, CTNNB1, or TCF7L2 causing constitutive Wnt signalling, thus removing the requirement for Wnt3A or R-spondin; the remaining 20% have wild-type APC genes and are, therefore, dependent on the addition of Wnt3A in the culture medium [23]. Furthermore, some colon tumour organoids require both exogenous EGF and SB202190, and others require neither [23]. Fujii et al. suggest trialling each new organoid culture on an array of different media formulations to determine the optimal conditions for each individual sample [23].
Characterisation is a vital aspect of organoid development and validation. To validate stem cell-derived organoids and ensure that they contain a heterogeneous population of cells, markers of the various colon cell types can be visualised using fluorescence confocal microscopy [11,12]. Colon stem cells give rise to the other colonic cell types and express LGR5 on their surface [24]. They are supported by Paneth cells that express LYZ and give rise to transit amplifying cells that express EPHB2. The transit amplifying cells differentiate into the other colonic cell types as they migrate up from the crypt base towards the lumenal epithelial surface. Goblet cells are secretory cells, identified by their expression of MUC2. The absorptive cells, enterocytes, can be visualised by their expression of Villin (VIL1) or CA2. CHGA is used to identify colonic endocrine cells. The epithelial barrier function of organoids can be assayed using an uptake assay using FITC-labelled dextran [20].
2.3. Reversed-Polarity Organoids
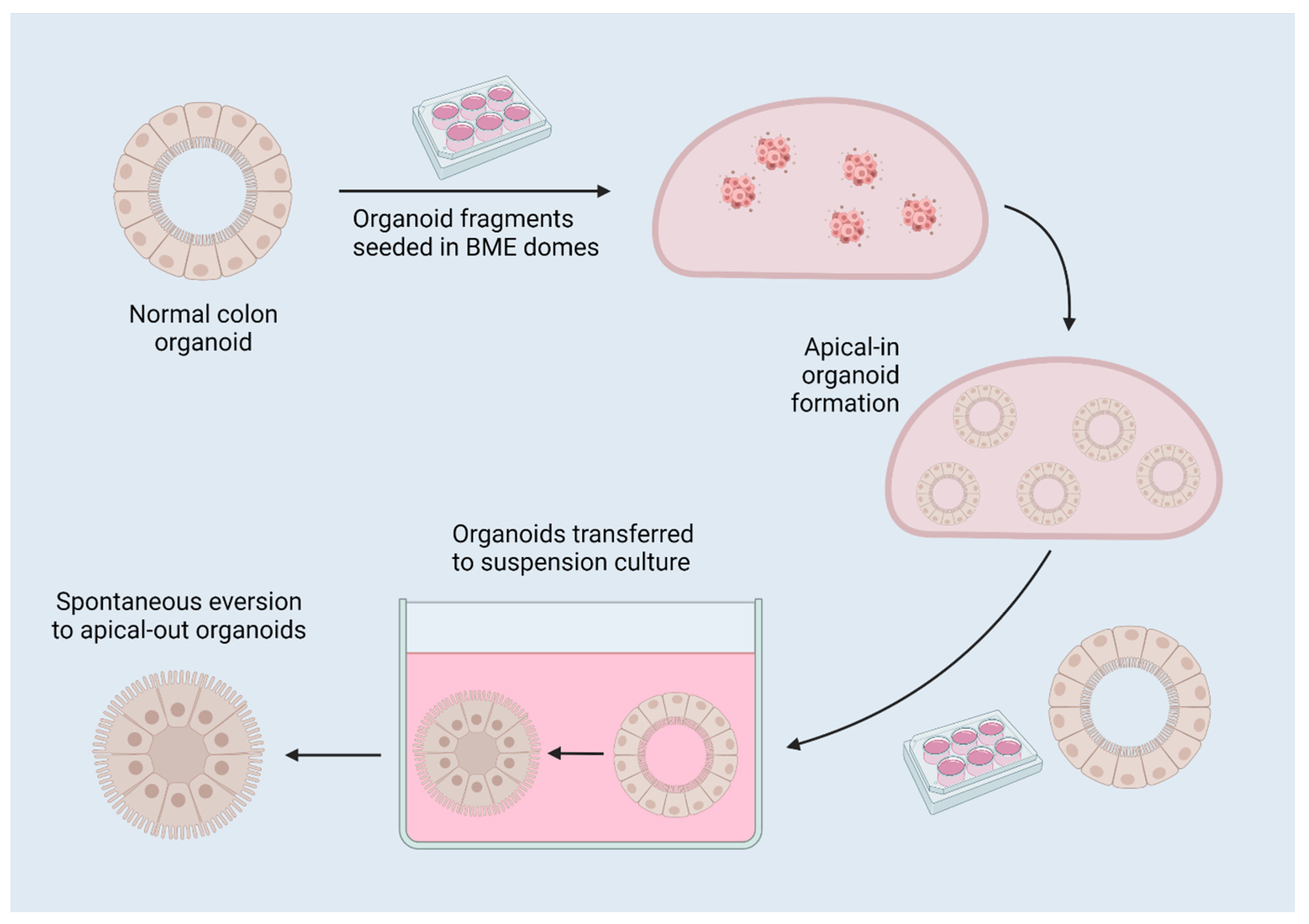
The apical surface of the epithelium that faces the lumen of the gut is on the interior surface of the organoid. This limits the validity of organoid assays, such as drug exposure and bacterial co-culture, because the pertinent interface is not accessible if factors are simply added to the media. The apical surface of epithelial cells facilitates absorption, secretion, and binding/detection, whereas the basolateral surface is responsible for anchorage, nutrient delivery to the bloodstream, and intercellular communication [20]. This issue has been circumvented by microinjection of drugs or bacteria into the organoid lumen, or alternatively by breaking up the organoids, adding the drug or bacteria to the culture medium, and allowing the organoids to reform [25,26]. However, microinjection requires a high degree of technical expertise, and breaking and reforming organoids can lead to inconsistent amounts of drug or bacteria in the lumen of each organoid. One elegant solution is the creation of reverse-polarity organoids [20,21]. When removed from their extracellular matrix (ECM) dome (Matrigel, Cultrex, etc.) and placed in suspension culture, the typical basal-out organoids spontaneously evert to become apical-out, reverse-polarity organoids (Figure 3), as described by Co et al. [20,21]. Apical-out organoids have been proven to retain their barrier integrity and allow increased bacterial invasion compared to basal-out organoids, representing a more relevant model for colonic function, and facilitating the targeting of receptors on the apical/lumenal epithelial surface by pathogens and for drug screening [20].
2.4. Cryopreservation and Recovery
Whole organoids or organoid fragments are preferred over an organoid-derived single-cell digest for cryopreservation due to their enhanced viability when thawed [27]. Similarly, it has been proposed that adding ROCKi to the cryopreservation media increases the viability of thawed organoids [27]. A common cryopreservation media formula includes 10% DMSO and 20% foetal calf serum in basal medium, or, alternatively, a commercial solution such as CryoStor (StemCell Technologies, Vancouver, BC, Canada) may be used.
3. Applications of Colon Organoids
3.1. Disease Modelling
Organoids are an in vitro version of the tissue from which they are established. As such, they are a useful system for disease modelling. Each tumour-derived organoid is a model of the tumour from which it is produced and can be used for drug testing in the personalised medicine context. They incorporate a range of cell types seen in the original organ, thereby constituting a more robust and physiologically relevant model than more homogenous 2D cell lines, which tend to contain a single dominant cell type that outcompetes the others. The complexity and physiological relevance of such models can be enhanced by co-culture of organoids with other cell types including immune cells, endothelium, fibroblasts, and microbes, often utilising an organ-on-a-chip model, as will be discussed later.
A model system for researching cystic fibrosis in intestinal organoids with and without a mutated CFTR gene has been described, whereby wild-type organoids swell when exposed to forskolin, but those with a CFTR mutation as seen in cystic fibrosis patients do not respond to forskolin [28]. This model allows drug assays to be performed in order to find compounds that reverse the effects of defective cystic fibrosis transmembrane conductance regulator protein (CFTR) and identify candidates for treating cystic fibrosis [28].
Colon organoids have also been used to model SARS-CoV-2 infection. COVID-19 infections primarily occur in the respiratory and digestive tracts, and various colon cell types express the ACE2 protein that SARS-CoV-2 utilises to invade cells. Colon organoids are susceptible to SARS-CoV-2 infection, and this can be prevented through treatment with various compounds to identify “entry inhibitors” [11].
Matano et al. [29] utilised organoids derived from normal human intestinal tissue to model the initiation of colorectal cancer (CRC). The influence of pathways known to play a role in CRC were elucidated by introducing mutations via CRISPR-Cas9 site-directed mutagenesis. Sequential mutation of APC, SMAD4, TP53, KRAS, and PIK3CA produced organoids with no dependence on Wnt, R-spondin, EGF, Noggin, or A83-01 in the culture media, and that could form tumours when transplanted into immunodeficient mice [29]. This highlights the potential of organoids as a model for delineating the mechanisms of early tumourigenesis.
The colon air–liquid interface (ALI) model was extensively characterised by Wang et al. [30], who grew colon spheroids, dissociated them into single cells, and seeded them on Transwell inserts. After being submerged for 7 days in L-WRN conditioned medium, the cells were cultured as an ALI, with the bottom surface of the insert in contact with media and the top surface exposed to air, for 28 days. During this time, the cells differentiated into a heterogenous 2D monolayer consisting of MUC2+ goblet cells (by day 4), CHGA+ enteroendocrine cells (day 7), and SLC26A3+ enterocytes/colonocytes (day 14–21) [30]. Day 21 ALI cultures could be reformed following passage and recover from cryopreservation, and performed mucosal repair following damage [30]. These experiments present spheroid- or organoid-derived ALI cultures as a robust in vitro model for colon function, including an intact epithelial barrier, for applications such as co-culture with bacteria [31] or studies of colonic metabolism [32].
Colon tumour patient-derived organoids (PDOs) have been co-cultured with stromal cells to simulate the in vivo influence of the tumour microenvironment (TME) in drug response. Dijkstra et al. [33] developed organoids from mismatch repair-deficient colon tumours, which have higher mutational burden, and isolated peripheral blood from the same patients to establish a co-culture system. Following two weeks of co-culture, levels of tumour-reactive T-cells with specific killing activity increased from undetectable levels in some cases to around 1–3% of the total pool of CD8+ T cells across samples [33]. Neal et al. [34] developed an ALI model utilising tumour tissue samples that had been minced but not digested enzymatically, thereby retaining tumour architecture in terms of associations between tumour cells and stromal cells. The stroma contained fibroblasts, which persisted for up to 4 weeks after ALI culture establishment, and immune cells including tumour-infiltrating lymphocytes and macrophages, which steadily decreased in number over time but could still be detected for 30–60 days and retained the immune cell diversity observed in the parental tissue [34]. These results lay the groundwork for future organoid investigations that will allow for increased translation of medicines from the lab to the clinic.
3.2. Drug Screening
The use of organoids for drug screening has recently been reviewed by Rae et al. [35]. For testing an array of compounds on colon tumour organoids in a high-throughput screening (HTS) setting, Boehnke et al. [18] successfully established organoids in 384-well plates. Tumour organoids were first established in 12-well plates as described above, then digested to a single-cell suspension and seeded in Matrigel domes at a density of 5000 cells per dome. To measure the response of tumour organoids to a range of compounds, the CellTitre-Glo viability assay (Promega) was used to report on ATP consumption as a measure of the metabolic activity of the cells, as a proxy for cell viability [18]. Toshimitsu et al. reported a similar protocol utilising 384-well plates, but rather than embedding colon tumour cells in solid Matrigel domes, cells were grown in suspension culture in media supplemented with 2% Matrigel [36]. Organoids formed from this single-cell suspension, and the plates were incubated on a rotating platform. This system greatly enhances the scalability and establishment speed of HTS assays. To ensure that an equal number of cells were seeded per well, they were labelled with GFP and dispensed by a modified FACS system. The GFP-labelled cells allowed organoid growth to be tracked before, during and after drug treatment, and this method of drug effect measurement was comparable to ATP-based luminescence assays [36]. The results of drug testing performed on the organoids established in a suspension culture based on 2% Matrigel in culture medium were very similar to the results seen in vivo [36] and may be a useful alternative to the reverse-polarity method described above.
Vlachogiannis et al. [37] carried out a comprehensive comparison of PDOs with their parental tissues, and utilised these organoids to test the feasibility of genomically-guided personalised treatment. PDOs closely phenocopied the morphology and expression of CDX2 and CK7 seen in the parental tissues [37]. Furthermore, 96% of mutations detected in the PDOs and tissues were shared [37]. PDOs were treated with a library of 55 drugs either under investigation in clinical trials or already used clinically. Response to these compounds was directly correlated with genetic alterations to the drug target; for instance, the strongest response to the ERBB2/EGFR inhibitor lapatinib was seen in a PDO with an ERBB2 amplification, a PDO with an AKT1 amplification and E17K mutation displayed the greatest response to two AKT inhibitors (MK-2206 and GSK690693), and those PDOs with RB1 amplifications responded to CDK4/CDK6 inhibitor palbociclib [37]. In vitro PDO models and PDO-xenograft mouse models both replicated the responses to multi-kinase inhibitor regorafenib and ATM/ATR inhibitor VX-970 seen in clinical trials in humans [37]. Finally, this study shows that organoids reliably replicate both the histopathology of tumours and their response to drugs as seen in patients, suggesting that organoids may be valuable for screening drugs for personalised treatment or to be used in a co-clinical trial context. Similarly, Schnalzger et al. [38] utilised PDOs and live-cell fluorescence imaging to trial chimeric antigen receptor (CAR) lymphocytes engineered to be tumour-specific, revealing efficient killing of tumour organoids but with accompanying off-target effects on normal colon organoids, highlighting the utility of organoids for testing the safety of new treatments before administration to humans.
Drug response can also be followed by tracking organoid phenotypes over time using microscopy, including organoid size and architecture (solid vs. cystic) [39]; however, to mitigate the inherent variability between organoids derived from different patients, assays tracking cell death, senescence and growth rate from a baseline value established before treatment would improve the accuracy, success and utility of HTS assays.
4. Experimental and Statistical Considerations
Statistical and experimental design is a major factor of cell culture and organoid research. Many experiments are well thought out, but are undermined by a poor design, and, likewise, a well-designed experiment may be made less robust by choice of statistical model. Important factors such as randomisation, blinding, appropriate ‘n’ number, robustness of data, and potential sex effects are readily used in human and animal studies, but routinely overlooked in in vitro cell culture and organoid studies.
The number of replicates or experimental units (‘n’ number) must be considered when planning PDO experiments. Where an appropriate number of biological replicates has been collected, an appropriate number of technical replicates for each individual biological replicate is also an important consideration. The ‘n’ number becomes a larger issue when experiments using cell lines or organoids are designed. When using organoids, replicates from the same lines or primary tissue that have been frozen and reconstituted into different aliquots are the same sample, and considered technical replicates, as all cells are produced from the same parent tissue are not different from one another. This problem can be balanced by generating cell lines from different individual human or animal tissue samples. Technical replicates are more appropriate when applied to a biological sample and a mean value calculated to ensure more robust representation of each individual PDO. Individually, technical replicates cannot be considered as an experimental unit. Schurch et al. stated that in cell line experiments, a minimum of three technical replicates of each biological replicate/sample should be used, greatly reducing the chance of a Type I statistical error [40]. Whilst the perfect experiment does not exist, in our opinion, PDO experiments should be designed with both adequate numbers of biological samples and technical replicates. Therefore, each biological replicate/sample should have a minimum of three technical replicates, whereby the mean of the technical replicates is a measure of the specific outcome within that biological sample. Additionally, a minimum of six biological replicates/samples per group has been suggested to provide a robust data set for analysis.
Further considerations of statistical design should be carefully evaluated too. Separating experimental groups by sex and treatment and adding in factors of analysis, such as age, ethnicity, environmental factors, and time, can all be built into statistical models. Therefore, it is important to consider more advanced statistical methods such as repeated-measures, factorial/mixed effects, and nested analysis when performing cell culture and organoid experiments. These tools are often underutilised but are a necessary part of study design to ensure the robustness of experimental and statistical interpretation of data. Implementing sound statistical considerations will ensure that PDO-based assays have the greatest chance of producing valid, reproducible results and achieving successful translation to the clinic.
5. Organoid Libraries and Biobanks
When establishing a biobank, samples must be collected, processed, and stored in a standardised and highly controlled manner to ensure that all samples are equally viable [41,42,43,44]. Furthermore, samples stored in the biobank must be characterised. It is typical to record patient demographics including the age, gender, ethnicity, and family history, as well as medical history, comorbidities, medications, and previous treatments, and to investigate and report on information such as clinical phenotype, histological and pathological reports, and molecular profiling (genetic, epigenetic, proteomic, etc.) [23,45,46,47,48,49]. Access to a biobank reduces the time and resources required for researchers to populate a study. Organoid biobanks specifically enable future research projects to include functional studies [42,45]. Considerations when establishing a biobank include: forming and following guidelines and standard operating procedures, including strict process standardisation to reduce variability and increase viability of biological samples; appropriate management, storage, and security of samples and data; an internal governance system and committee to define and enforce the scope, context, ethical considerations, and guiding principles of the biobank; and staff with applicable qualifications, training, and understanding [42,44].
Organoid biobanks are a useful source of material for performing drug screening, particularly when deriving patient-matched normal colon and colon tumour organoids. Colon organoid biobanks or libraries have previously been established, described, characterised, and reviewed [23,41,45,46,47,48]. In 2015, van der Wetering et al. reported on the establishment of an organoid library consisting of 22 tumour organoids and 19 normal colon organoids from 20 individual patients, with the samples displaying variation in their growth success ranging from a yield of 10–20 organoids to thousands of organoids [47]. All organoids were subjected to whole exome sequencing to identify tumour-specific somatic mutations. Furthermore, they established a screening platform utilising BME-coated 384-well plates and a culture medium containing 2% BME, allowing the organoids to adhere to the bottom of the plate. Fujii et al. established a similar library consisting of 55 organoids derived from 52 colorectal tumours, and 41 normal colon organoids, from 43 individual patients, on which 8 different culture conditions were tested [23]. The best-suited media formulation for each organoid depended on the mutations detected in each tumour; KRAS/BRAF mutants did not require SB202190 or EGF, and organoids with Wnt pathway alterations (APC, CTNNB1, TCF7L2) did not require Wnt3A or R-spondin. This study highlights the importance of obtaining clinical information and performing genetic analysis on each sample in the biobank. Schutte et al. developed a biobank of 116 tumour tissues, consisting of 89 primary CRC and 27 CRC metastases, from which 35 organoids and 59 xenografts were established [48]. When examined using whole genome, exome, and transcriptome sequencing, the majority of the organoids and xenografts were shown to accurately reflect the genetic characteristics of the original tissue [48]. The few clonal and sub-clonal differences that developed in organoid cultures were attributed to intratumoural heterogeneity and subsequent sampling from the tissue when establishing each model [48]. Similarly, our organoid biobank holds approximately 38 patient-derived organoids from a range of tissues and tumours, including glioblastoma, meningioma, colon, lung, and liver. Overall, organoid biobanks represent a valuable resource for research requiring functional assays such as drug screening, but the development of standardised protocols for tissue collecting, processing, and storage, and subsequent organoid culture is necessary.
6. Future Perspectives
Organoids have emerged as a vital tool in biomedical research due to their recapitulation of in vivo biology, including cell type heterogeneity and mutational signature. Importantly, tumour organoids reflect intratumoural heterogeneity, as a relevant model for cancer research, including drug screening, by retaining rare clones. Methods for generating normal tissue and tumour organoids can be tailored to each patient-derived tissue, and patient-derived organoids can be stored in biobanks for future use. They are already being utilised for next-generation applications such as organ-on-a-chip.
The next iteration of physiologically relevant in vitro models is the organ-on-a-chip, which in the context of the colon is called a gut- or intestine-on-a-chip. This topic has recently been reviewed elsewhere [35]. These systems involve growing tissue-derived cells within hollow channels on microfluidic chips. The chips can be manufactured by using a mould to cast two channels made from clear, flexible polydimethylsiloxane and joining these with a porous membrane between them. To simulate the mechanical strain that occurs within the gut, vacuum chambers can be attached adjacent to the channels to cyclically distort the flexible walls to an extent similar to that seen in vivo (~10% mean cell strain at 0.15 Hz, equivalent to ~6 s per cycle) [50].
Different cell types can be grown in each of the adjacent channels with the permeable membrane between them to study the interplay between an organ or tumour and other surrounding tissue types. This system has been established and described in detail by Kim et al. [50,51]. A media flow is established through the channels, in conjunction with the vacuum-induced mechanical strain, to simulate the peristaltic movement of liquid through the lumen of the gut with resultant shear stress on the epithelial cells [50]. Applications have included colonic epithelial cells (from normal colon or colon tumour tissues or organoids) grown in one channel with endothelial cells or fibroblasts grown in the other. CaCo-2 cells seeded within a channel form a columnal layer with barrier integrity, and spontaneously polarise and develop villus-like folds [50]. Furthermore, the apical/lumenal surface of the villus structures secrete mucus as seen in vivo [50]. This differentiated columnar morphology develops much more quickly in the presence of media flow than in a static Transwell setting [50]. The complexity of this model can be increased by introducing microbes to the epithelial channel and immune cells to the endothelial channel. Microbes can be stably co-cultured with epithelial cells in microfluidic devices for several weeks without overgrowth, allowing studies of chronic exposure to specific microorganisms [51]. The perfusion of immune cells into the endothelial-lined channel simulates inflammation, and the effect of inflammation on the epithelial cells in the neighbouring channel may be examined [51].
This model offers advantages over organoids, such as enabling interactions with other cell types, mimicking vascularisation, modelling peristalsis to ensure normal differentiation and structure, controlling exposure to specific gradients or combinations of factors in the media, and allowing sustainable co-culture with microbes.
A recent application of this system involved seeding fragmented organoids inside one channel and human intestinal microvascular endothelial cells in the other [52]. Organoids were produced from normal duodenal tissue and once seeded inside the channel, they formed a confluent layer with villi within 12 days. A transcriptome-wide analysis found that the intestine chip was more like the parent tissue than the organoids were, suggesting that the chip is even more representative of the gut than organoids [52]. Organoid fragments reached confluency within the channels quicker than single cells did. Similarly, colon tumour organoids grew more quickly and had greater viability within the chip than they did when grown in a matrix dome [53], highlighting the continued importance and relevance of organoids in these new applications.
Collectively, these studies demonstrate that microfluidic chips can better reproduce the in vivo conditions within the gut than organoids, while organoids serve as an effective starting point for seeding the chips, probably because the organoid system preserves biological and cellular diversity in a manner that 2D cell culture may not.
Author Contributions
Conceptualization, M.J.M. and C.G.; data curation, M.J.M.; writing—original draft preparation, M.J.M.; writing—review and editing, M.J.M., S.T.T. and C.G. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Our colon cancer programme was approved by the Central Health and Disability Ethics Committee (ethics approval code 15/CEN/96).
Informed Consent Statement
Informed consent was obtained for all subjects recruited to the GMRI Tissue Bank and used for this study.
Data Availability Statement
The data presented in this review are representative images from our laboratory that are as yet unpublished, and are available on request from the corresponding author.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Zheng, S.; Xin, L.; Liang, A.; Fu, Y. Cancer stem cell hypothesis: A brief summary and two proposals. Cytotechnology 2013, 65, 505–512. [Google Scholar] [CrossRef] [Green Version]
- Kreso, A.; Dick, J.E. Evolution of the cancer stem cell model. Cell Stem Cell 2014, 14, 275–291. [Google Scholar] [CrossRef] [Green Version]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [Green Version]
- Walcher, L.; Kistenmacher, A.K.; Suo, H.; Kitte, R.; Dluczek, S.; Strauss, A.; Blaudszun, A.R.; Yevsa, T.; Fricke, S.; Kossatz-Boehlert, U. Cancer Stem Cells-Origins and Biomarkers: Perspectives for Targeted Personalized Therapies. Front. Immunol. 2020, 11, 1280. [Google Scholar] [CrossRef]
- Milanovic, M.; Fan, D.N.Y.; Belenki, D.; Dabritz, J.H.M.; Zhao, Z.; Yu, Y.; Dorr, J.R.; Dimitrova, L.; Lenze, D.; Monteiro Barbosa, I.A.; et al. Senescence-associated reprogramming promotes cancer stemness. Nature 2018, 553, 96–100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, D.G. Understanding cancer stem cell heterogeneity and plasticity. Cell Res. 2012, 22, 457–472. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seaberg, R.M.; van der Kooy, D. Stem and progenitor cells: The premature desertion of rigorous definitions. Trends Neurosci. 2003, 26, 125–131. [Google Scholar] [CrossRef]
- Hayat, H.; Hayat, H.; Dwan, B.F.; Gudi, M.; Bishop, J.O.; Wang, P. A Concise Review: The Role of Stem Cells in Cancer Progression and Therapy. OncoTargets Ther. 2021, 14, 2761–2772. [Google Scholar] [CrossRef] [PubMed]
- Pastrana, E.; Silva-Vargas, V.; Doetsch, F. Eyes wide open: A critical review of sphere-formation as an assay for stem cells. Cell Stem Cell 2011, 8, 486–498. [Google Scholar] [CrossRef] [Green Version]
- De Angelis, M.L.; Francescangeli, F.; Nicolazzo, C.; Signore, M.; Giuliani, A.; Colace, L.; Boe, A.; Magri, V.; Baiocchi, M.; Ciardi, A.; et al. An organoid model of colorectal circulating tumor cells with stem cell features, hybrid EMT state and distinctive therapy response profile. J. Exp. Clin. Cancer Res. 2022, 41, 86. [Google Scholar] [CrossRef]
- Han, Y.; Duan, X.; Yang, L.; Nilsson-Payant, B.E.; Wang, P.; Duan, F.; Tang, X.; Yaron, T.M.; Zhang, T.; Uhl, S.; et al. Identification of SARS-CoV-2 inhibitors using lung and colonic organoids. Nature 2021, 589, 270–275. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, S.; Miwa, H.; Kawachi, T.; Kume, S.; Takahashi, K. Generation of intestinal organoids derived from human pluripotent stem cells for drug testing. Sci. Rep. 2020, 10, 5989. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fujii, M.; Sato, T. Somatic cell-derived organoids as prototypes of human epithelial tissues and diseases. Nat. Mater. 2021, 20, 156–169. [Google Scholar] [CrossRef] [PubMed]
- Barbachano, A.; Fernandez-Barral, A.; Bustamante-Madrid, P.; Prieto, I.; Rodriguez-Salas, N.; Larriba, M.J.; Munoz, A. Organoids and Colorectal Cancer. Cancers 2021, 13, 2657. [Google Scholar] [CrossRef]
- Sato, T.; Stange, D.E.; Ferrante, M.; Vries, R.G.; Van Es, J.H.; Van den Brink, S.; Van Houdt, W.J.; Pronk, A.; Van Gorp, J.; Siersema, P.D.; et al. Long-term expansion of epithelial organoids from human colon, adenoma, adenocarcinoma, and Barrett′s epithelium. Gastroenterology 2011, 141, 1762–1772. [Google Scholar] [CrossRef]
- Miyoshi, H.; Stappenbeck, T.S. In vitro expansion and genetic modification of gastrointestinal stem cells in spheroid culture. Nat. Protoc. 2013, 8, 2471–2482. [Google Scholar] [CrossRef]
- VanDussen, K.L.; Sonnek, N.M.; Stappenbeck, T.S. L-WRN conditioned medium for gastrointestinal epithelial stem cell culture shows replicable batch-to-batch activity levels across multiple research teams. Stem Cell Res. 2019, 37, 101430. [Google Scholar] [CrossRef]
- Boehnke, K.; Iversen, P.W.; Schumacher, D.; Lallena, M.J.; Haro, R.; Amat, J.; Haybaeck, J.; Liebs, S.; Lange, M.; Schafer, R.; et al. Assay Establishment and Validation of a High-Throughput Screening Platform for Three-Dimensional Patient-Derived Colon Cancer Organoid Cultures. J. Biomol. Screen 2016, 21, 931–941. [Google Scholar] [CrossRef] [Green Version]
- Sato, T.; van Es, J.H.; Snippert, H.J.; Stange, D.E.; Vries, R.G.; van den Born, M.; Barker, N.; Shroyer, N.F.; van de Wetering, M.; Clevers, H. Paneth cells constitute the niche for Lgr5 stem cells in intestinal crypts. Nature 2011, 469, 415–418. [Google Scholar] [CrossRef] [Green Version]
- Co, J.Y.; Margalef-Catala, M.; Li, X.; Mah, A.T.; Kuo, C.J.; Monack, D.M.; Amieva, M.R. Controlling Epithelial Polarity: A Human Enteroid Model for Host-Pathogen Interactions. Cell Rep. 2019, 26, 2509–2520.e4. [Google Scholar] [CrossRef]
- Co, J.Y.; Margalef-Catala, M.; Monack, D.M.; Amieva, M.R. Controlling the polarity of human gastrointestinal organoids to investigate epithelial biology and infectious diseases. Nat. Protoc. 2021, 16, 5171–5192. [Google Scholar] [CrossRef]
- Wilson, S.S.; Mayo, M.; Melim, T.; Knight, H.; Patnaude, L.; Wu, X.; Phillips, L.; Westmoreland, S.; Dunstan, R.; Fiebiger, E.; et al. Optimized Culture Conditions for Improved Growth and Functional Differentiation of Mouse and Human Colon Organoids. Front. Immunol. 2020, 11, 547102. [Google Scholar] [CrossRef]
- Fujii, M.; Shimokawa, M.; Date, S.; Takano, A.; Matano, M.; Nanki, K.; Ohta, Y.; Toshimitsu, K.; Nakazato, Y.; Kawasaki, K.; et al. A Colorectal Tumor Organoid Library Demonstrates Progressive Loss of Niche Factor Requirements during Tumorigenesis. Cell Stem Cell 2016, 18, 827–838. [Google Scholar] [CrossRef] [Green Version]
- Barker, N.; van Es, J.H.; Kuipers, J.; Kujala, P.; van den Born, M.; Cozijnsen, M.; Haegebarth, A.; Korving, J.; Begthel, H.; Peters, P.J.; et al. Identification of stem cells in small intestine and colon by marker gene Lgr5. Nature 2007, 449, 1003–1007. [Google Scholar] [CrossRef] [PubMed]
- Puschhof, J.; Pleguezuelos-Manzano, C.; Martinez-Silgado, A.; Akkerman, N.; Saftien, A.; Boot, C.; de Waal, A.; Beumer, J.; Dutta, D.; Heo, I.; et al. Intestinal organoid cocultures with microbes. Nat. Protoc. 2021, 16, 4633–4649. [Google Scholar] [CrossRef]
- Bartfeld, S.; Clevers, H. Organoids as Model for Infectious Diseases: Culture of Human and Murine Stomach Organoids and Microinjection of Helicobacter Pylori. J. Vis. Exp. 2015, 105, e53359. [Google Scholar] [CrossRef] [Green Version]
- Han, S.H.; Shim, S.; Kim, M.J.; Shin, H.Y.; Jang, W.S.; Lee, S.J.; Jin, Y.W.; Lee, S.S.; Lee, S.B.; Park, S. Long-term culture-induced phenotypic difference and efficient cryopreservation of small intestinal organoids by treatment timing of Rho kinase inhibitor. World J. Gastroenterol. 2017, 23, 964–975. [Google Scholar] [CrossRef] [PubMed]
- Dekkers, J.F.; Wiegerinck, C.L.; de Jonge, H.R.; Bronsveld, I.; Janssens, H.M.; de Winter-de Groot, K.M.; Brandsma, A.M.; de Jong, N.W.; Bijvelds, M.J.; Scholte, B.J.; et al. A functional CFTR assay using primary cystic fibrosis intestinal organoids. Nat. Med. 2013, 19, 939–945. [Google Scholar] [CrossRef] [PubMed]
- Matano, M.; Date, S.; Shimokawa, M.; Takano, A.; Fujii, M.; Ohta, Y.; Watanabe, T.; Kanai, T.; Sato, T. Modeling colorectal cancer using CRISPR-Cas9-mediated engineering of human intestinal organoids. Nat. Med. 2015, 21, 256–262. [Google Scholar] [CrossRef]
- Wang, Y.; Chiang, I.L.; Ohara, T.E.; Fujii, S.; Cheng, J.; Muegge, B.D.; Ver Heul, A.; Han, N.D.; Lu, Q.; Xiong, S.; et al. Long-Term Culture Captures Injury-Repair Cycles of Colonic Stem Cells. Cell 2019, 179, 1144–1159.e15. [Google Scholar] [CrossRef]
- In, J.G.; Foulke-Abel, J.; Clarke, E.; Kovbasnjuk, O. Human Colonoid Monolayers to Study Interactions Between Pathogens, Commensals, and Host Intestinal Epithelium. J. Vis. Exp. 2019, 146, e59357. [Google Scholar] [CrossRef] [PubMed]
- Richiardone, E.; Van den Bossche, V.; Corbet, C. Metabolic Studies in Organoids: Current Applications, Opportunities and Challenges. Organoids 2022, 1, 8. [Google Scholar] [CrossRef]
- Dijkstra, K.K.; Cattaneo, C.M.; Weeber, F.; Chalabi, M.; van de Haar, J.; Fanchi, L.F.; Slagter, M.; van der Velden, D.L.; Kaing, S.; Kelderman, S.; et al. Generation of Tumor-Reactive T Cells by Co-culture of Peripheral Blood Lymphocytes and Tumor Organoids. Cell 2018, 174, 1586–1598.e12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neal, J.T.; Li, X.; Zhu, J.; Giangarra, V.; Grzeskowiak, C.L.; Ju, J.; Liu, I.H.; Chiou, S.H.; Salahudeen, A.A.; Smith, A.R.; et al. Organoid Modeling of the Tumor Immune Microenvironment. Cell 2018, 175, 1972–1988.e16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rae, C.; Amato, F.; Braconi, C. Patient-Derived Organoids as a Model for Cancer Drug Discovery. Int. J. Mol. Sci. 2021, 22, 3483. [Google Scholar] [CrossRef] [PubMed]
- Toshimitsu, K.; Takano, A.; Fujii, M.; Togasaki, K.; Matano, M.; Takahashi, S.; Kanai, T.; Sato, T. Organoid screening reveals epigenetic vulnerabilities in human colorectal cancer. Nat. Chem. Biol. 2022, 18, 605–614. [Google Scholar] [CrossRef]
- Vlachogiannis, G.; Hedayat, S.; Vatsiou, A.; Jamin, Y.; Fernandez-Mateos, J.; Khan, K.; Lampis, A.; Eason, K.; Huntingford, I.; Burke, R.; et al. Patient-derived organoids model treatment response of metastatic gastrointestinal cancers. Science 2018, 359, 920–926. [Google Scholar] [CrossRef] [Green Version]
- Schnalzger, T.E.; de Groot, M.H.; Zhang, C.; Mosa, M.H.; Michels, B.E.; Roder, J.; Darvishi, T.; Wels, W.S.; Farin, H.F. 3D model for CAR-mediated cytotoxicity using patient-derived colorectal cancer organoids. EMBO J. 2019, 38, e100928. [Google Scholar] [CrossRef]
- Betge, J.; Rindtorff, N.; Sauer, J.; Rauscher, B.; Dingert, C.; Gaitantzi, H.; Herweck, F.; Srour-Mhanna, K.; Miersch, T.; Valentini, E.; et al. The drug-induced phenotypic landscape of colorectal cancer organoids. Nat. Commun. 2022, 13, 3135. [Google Scholar] [CrossRef]
- Schurch, N.J.; Schofield, P.; Gierlinski, M.; Cole, C.; Sherstnev, A.; Singh, V.; Wrobel, N.; Gharbi, K.; Simpson, G.G.; Owen-Hughes, T.; et al. How many biological replicates are needed in an RNA-seq experiment and which differential expression tool should you use? RNA 2016, 22, 839–851. [Google Scholar] [CrossRef]
- Botti, G.; Di Bonito, M.; Cantile, M. Organoid biobanks as a new tool for pre-clinical validation of candidate drug efficacy and safety. Int. J. Physiol. Pathophysiol. Pharmacol. 2021, 13, 17–21. [Google Scholar] [PubMed]
- Mendy, M.; Caboux, E.; Lawlor, R.T.; Wright, J.; Wild, C.P. Common Minimum Technical Standards and Protocols for Biobanks Dedicated to Cancer Research; International Agency for Research on Cancer: Lyon, France, 2017. [Google Scholar]
- Hallmans, G.; Vaught, J.B. Best practices for establishing a biobank. Methods Mol. Biol. 2011, 675, 241–260. [Google Scholar] [CrossRef] [PubMed]
- Shabihkhani, M.; Lucey, G.M.; Wei, B.; Mareninov, S.; Lou, J.J.; Vinters, H.V.; Singer, E.J.; Cloughesy, T.F.; Yong, W.H. The procurement, storage, and quality assurance of frozen blood and tissue biospecimens in pathology, biorepository, and biobank settings. Clin. Biochem. 2014, 47, 258–266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perrone, F.; Zilbauer, M. Biobanking of human gut organoids for translational research. Exp. Mol. Med. 2021, 53, 1451–1458. [Google Scholar] [CrossRef] [PubMed]
- Engel, R.M.; Jarde, T.; Oliva, K.; Kerr, G.; Chan, W.H.; Hlavca, S.; Nickless, D.; Archer, S.K.; Yap, R.; Ranchod, P.; et al. Modeling colorectal cancer: A bio-resource of 50 patient-derived organoid lines. J. Gastroenterol. Hepatol. 2022, 37, 898–907. [Google Scholar] [CrossRef] [PubMed]
- van de Wetering, M.; Francies, H.E.; Francis, J.M.; Bounova, G.; Iorio, F.; Pronk, A.; van Houdt, W.; van Gorp, J.; Taylor-Weiner, A.; Kester, L.; et al. Prospective derivation of a living organoid biobank of colorectal cancer patients. Cell 2015, 161, 933–945. [Google Scholar] [CrossRef] [Green Version]
- Schutte, M.; Risch, T.; Abdavi-Azar, N.; Boehnke, K.; Schumacher, D.; Keil, M.; Yildiriman, R.; Jandrasits, C.; Borodina, T.; Amstislavskiy, V.; et al. Molecular dissection of colorectal cancer in pre-clinical models identifies biomarkers predicting sensitivity to EGFR inhibitors. Nat. Commun. 2017, 8, 14262. [Google Scholar] [CrossRef] [Green Version]
- Sidorenkov, G.; Nagel, J.; Meijer, C.; Duker, J.J.; Groen, H.J.M.; Halmos, G.B.; Oonk, M.H.M.; Oostergo, R.J.; van der Vegt, B.; Witjes, M.J.H.; et al. The OncoLifeS data-biobank for oncology: A comprehensive repository of clinical data, biological samples, and the patient′s perspective. J. Transl. Med. 2019, 17, 374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, H.J.; Huh, D.; Hamilton, G.; Ingber, D.E. Human gut-on-a-chip inhabited by microbial flora that experiences intestinal peristalsis-like motions and flow. Lab Chip 2012, 12, 2165–2174. [Google Scholar] [CrossRef]
- Kim, H.J.; Li, H.; Collins, J.J.; Ingber, D.E. Contributions of microbiome and mechanical deformation to intestinal bacterial overgrowth and inflammation in a human gut-on-a-chip. Proc. Natl. Acad. Sci. USA 2016, 113, E7–E15. [Google Scholar] [CrossRef]
- Kasendra, M.; Tovaglieri, A.; Sontheimer-Phelps, A.; Jalili-Firoozinezhad, S.; Bein, A.; Chalkiadaki, A.; Scholl, W.; Zhang, C.; Rickner, H.; Richmond, C.A.; et al. Development of a primary human Small Intestine-on-a-Chip using biopsy-derived organoids. Sci. Rep. 2018, 8, 2871. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pinho, D.; Santos, D.; Vila, A.; Carvalho, S. Establishment of Colorectal Cancer Organoids in Microfluidic-Based System. Micromachines 2021, 12, 497. [Google Scholar] [CrossRef] [PubMed]
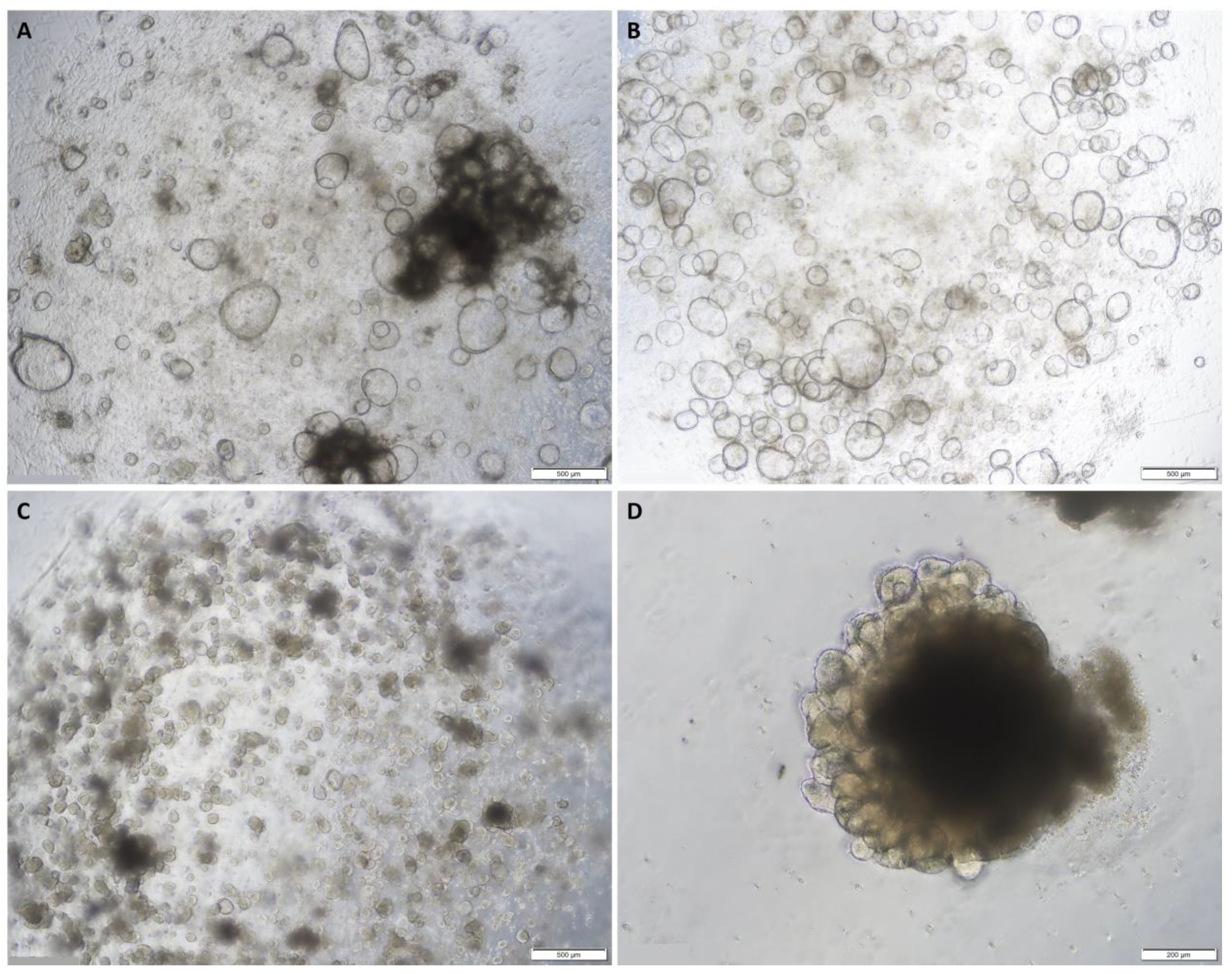
Figure 1.
Representative images of normal colon organoids: (A) on day 7 of passage 1 showing cystic organoids with large hollow lumens; (B) on day 8 of passage 1 showing cystic organoids with large hollow lumens; (C) on day 7 of passage 2 showing more compact organoids; (D) on day 5 of passage 6 showing epithelial budding. Original magnification: 4× (A–C) and 10× (D).
Figure 1.
Representative images of normal colon organoids: (A) on day 7 of passage 1 showing cystic organoids with large hollow lumens; (B) on day 8 of passage 1 showing cystic organoids with large hollow lumens; (C) on day 7 of passage 2 showing more compact organoids; (D) on day 5 of passage 6 showing epithelial budding. Original magnification: 4× (A–C) and 10× (D).

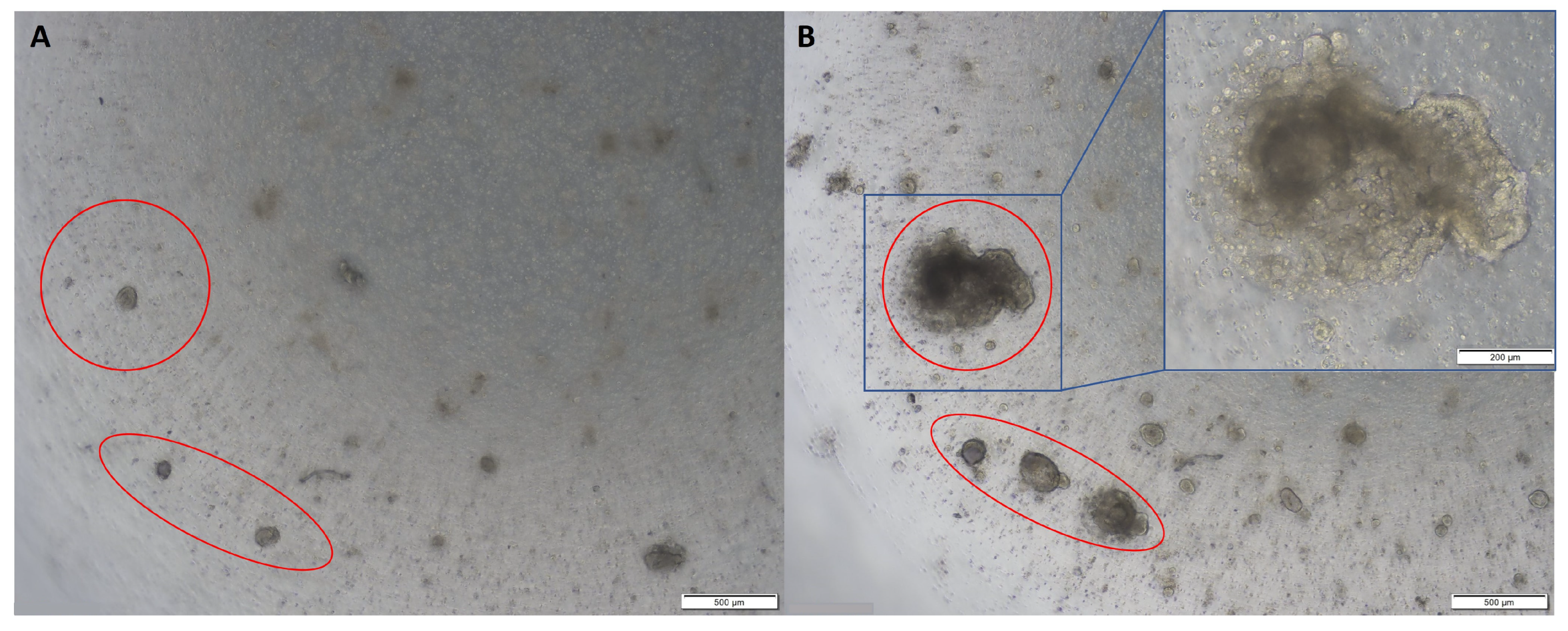
Figure 2.
Representative images of colon tumour organoids: (A) on day 2 of passage 2; (B) on day 10 of passage 2. Original magnification: 4× and 10× (inset).
Figure 2.
Representative images of colon tumour organoids: (A) on day 2 of passage 2; (B) on day 10 of passage 2. Original magnification: 4× and 10× (inset).

Figure 3.
A diagram of reverse-polarity colon organoids. Organoids are established in 3D matrix domes, then released for culture in suspension, which induces spontaneous eversion to apical-out organoids due to the removal of interactions with surrounding matrix. Created with BioRender.com 17 July 2022.
Figure 3.
A diagram of reverse-polarity colon organoids. Organoids are established in 3D matrix domes, then released for culture in suspension, which induces spontaneous eversion to apical-out organoids due to the removal of interactions with surrounding matrix. Created with BioRender.com 17 July 2022.


{kind=link}
{kind=link}
{kind=link}
Table 1.
Media formulations for culture of normal colon organoids and colon tumour organoids.
| Normal Colon Organoid Media Formulations | Colon Tumour Organoid Media Formulations | ||||
|---|---|---|---|---|---|
| Advanced DMEM/F12 | 50% | 1× | 1× | 1× | 1× |
| L-WRN Conditioned Medium | 50% | - | - | - | - |
| Human recombinant Wnt3a | - | 100 ng/mL | - | - | - |
| Human recombinant R-spondin-1 | - | 1 ug/mL | - | - | - |
| Human recombinant Noggin | - | 100 ng/mL | - | 100 ng/mL | - |
| HEPES | 10 mM | 10 mM | 10 mM | 10 mM | 10 mM |
| Anti-Anti | 2× | 2× | 1× | 1× | 1× |
| GlutaMAX | 1× | 1× | 1× | 1× | 1× |
| B27 | 1× | 1× | - | 1× | 1× |
| N2 | - | - | - | - | 1× |
| EGF | 50 ng/mL | 50 ng/mL | 50 ng/mL | 50 ng/mL | 20 ng/mL |
| Gastrin | 10 nM | 10 nM | 10 nM | 10 nM | - |
| N-acetyl-L-cysteine | 1.25 mM | 1 mM | 1 mM | 1 mM | - |
| Nicotinamide | 10 mM | 10 mM | 10 mM | 10 mM | - |
| A83-01 (Alk4,5,7 inhibitor) | 500 nM | 500 nM | 500 nM | 500 nM | - |
| SB202190 (p38 inhibitor) | - | 10 uM | - | - | - |
| IGF-1 | 100 ng/mL | - | - | - | - |
| FGF-2 | 50 ng/mL | - | - | - | 10 ng/mL |
| Heparin | - | - | - | - | 4 ng/mL |
| Y-27632 (ROCKi)—first 48 h | 10 mM | 10 mM | 10 mM | 10 mM | - |
L-WRN = Wnt3a-producing mouse L-cell line, genetically modified to express R-spondin and Noggin; Anti-Anti = antibiotic-antimycotic (penicillin, streptomycin, amphotericin B); EGF = epidermal growth factor; IGF = insulin-like growth factor; FGF = fibroblast growth factor; ROCKi = Rho kinase inhibitor.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Munro, M.J.; Tan, S.T.; Gray, C. Applications for Colon Organoid Models in Cancer Research. Organoids 2023, 2, 37-49. https://doi.org/10.3390/organoids2010003
AMA Style
Munro MJ, Tan ST, Gray C. Applications for Colon Organoid Models in Cancer Research. Organoids. 2023; 2(1):37-49. https://doi.org/10.3390/organoids2010003
Chicago/Turabian StyleMunro, Matthew J., Swee T. Tan, and Clint Gray. 2023. "Applications for Colon Organoid Models in Cancer Research" Organoids 2, no. 1: 37-49. https://doi.org/10.3390/organoids2010003