

Pancreatic Cancer 3D Cell Line Organoids (CLOs) Maintain the Phenotypic Characteristics of Organoids and Accurately Reflect the Cellular Architecture and Heterogeneity In Vivo
 , , , ,
, , , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Establishment of Isogenic Matched 2D Organoid-Derived Cell Line
2.2. Cell Line Organoid (CLO) Recapitulation to Organoid
2.3. Cell Viability Assays
2.4. Quantitative Reverse Transcription PCR (RT-qPCR)
2.5. Slide Preparation for Immunofluorescence Staining
2.5.1. Cell Lines
2.5.2. Organoids and CLOs
2.6. Immunofluorescence Staining
2.7. In Vivo Xenograft Study
2.8. Immunohistochemistry
2.9. Statistical Methods
3. Results
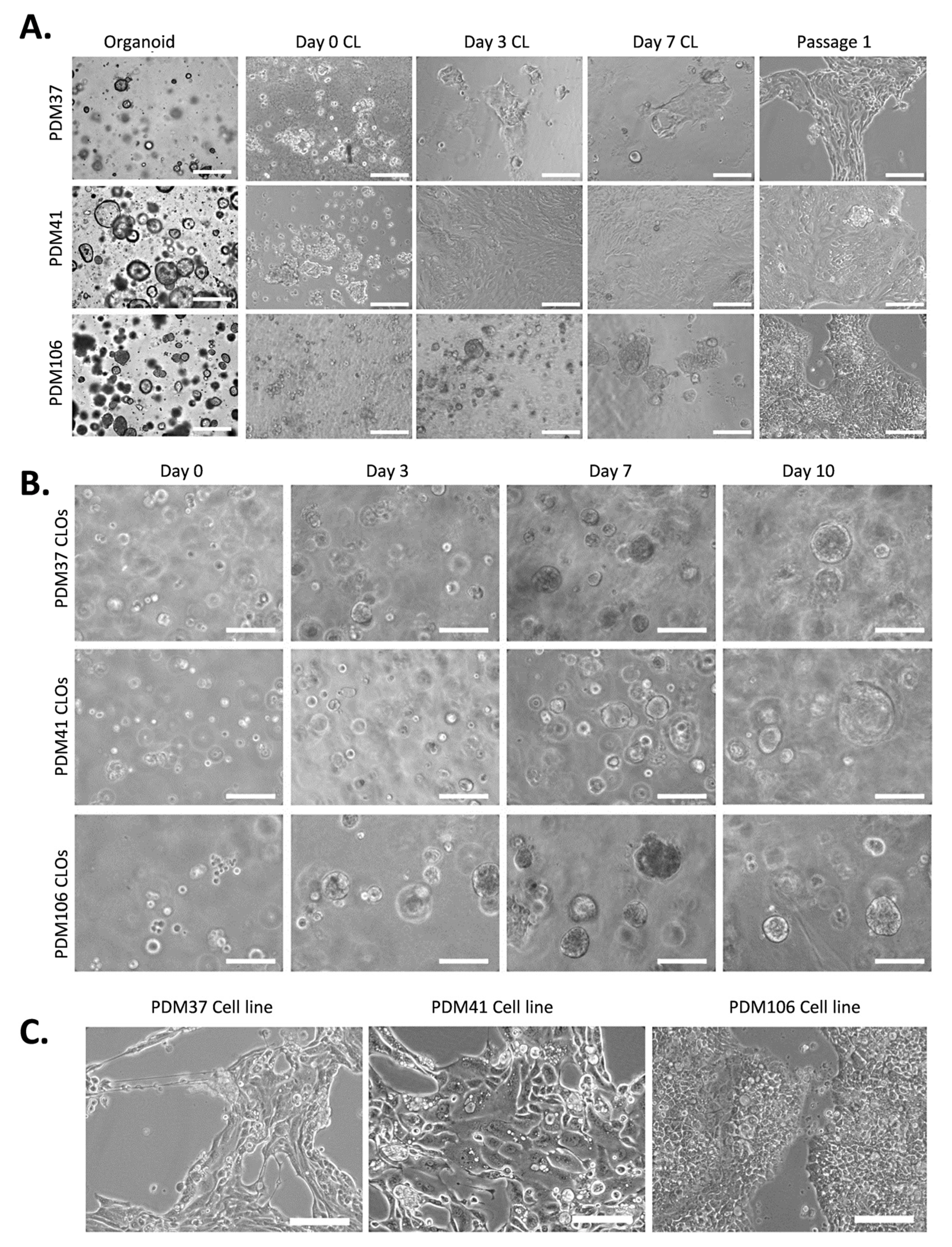
3.1. Generation of 2.5D Organoid-Derived Cell Lines and 3D Cell Line Organoids (CLOs) from PDAC Organoids
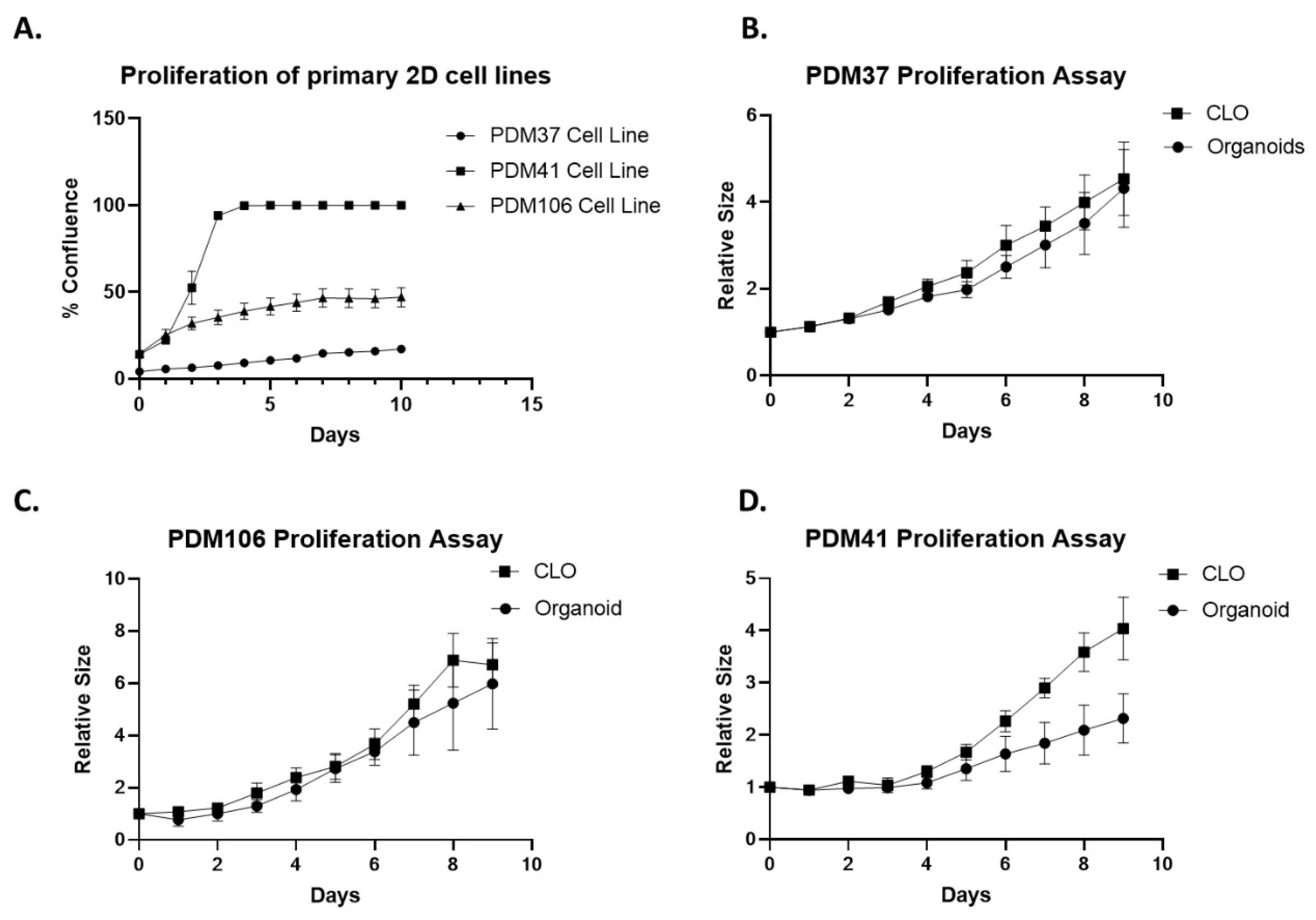
3.2. Relative Proliferation of Newly Established Primary 2D Cell Lines, Organoids, and CLOs
3.3. Therapeutic Response between 2D, CLOs, and Organoids
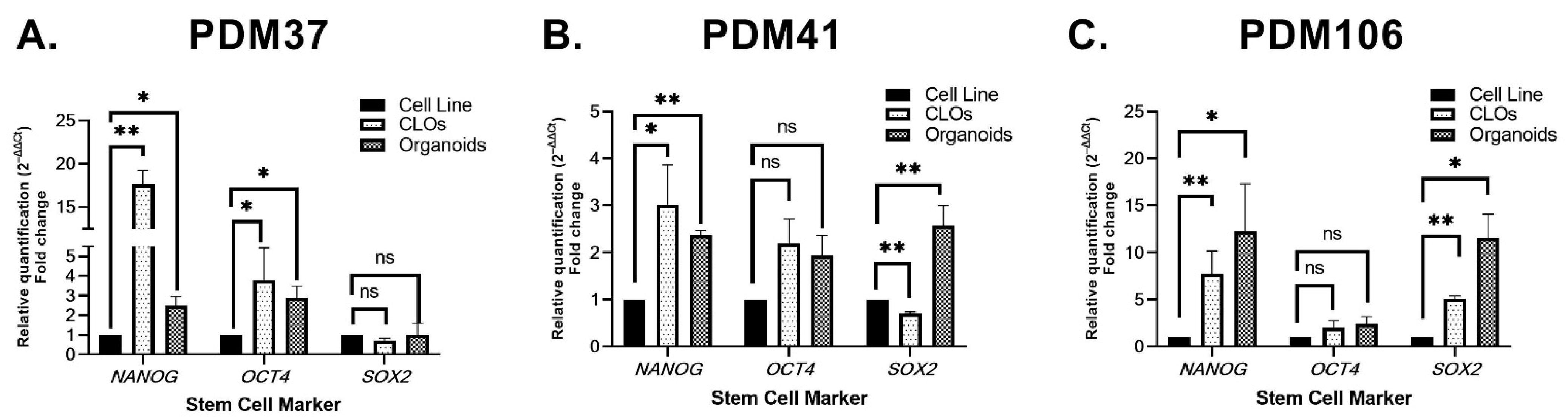
3.4. Stem Cell Marker Expression between Isogenic 2D Primary Cell Lines, Organoids, and CLOs
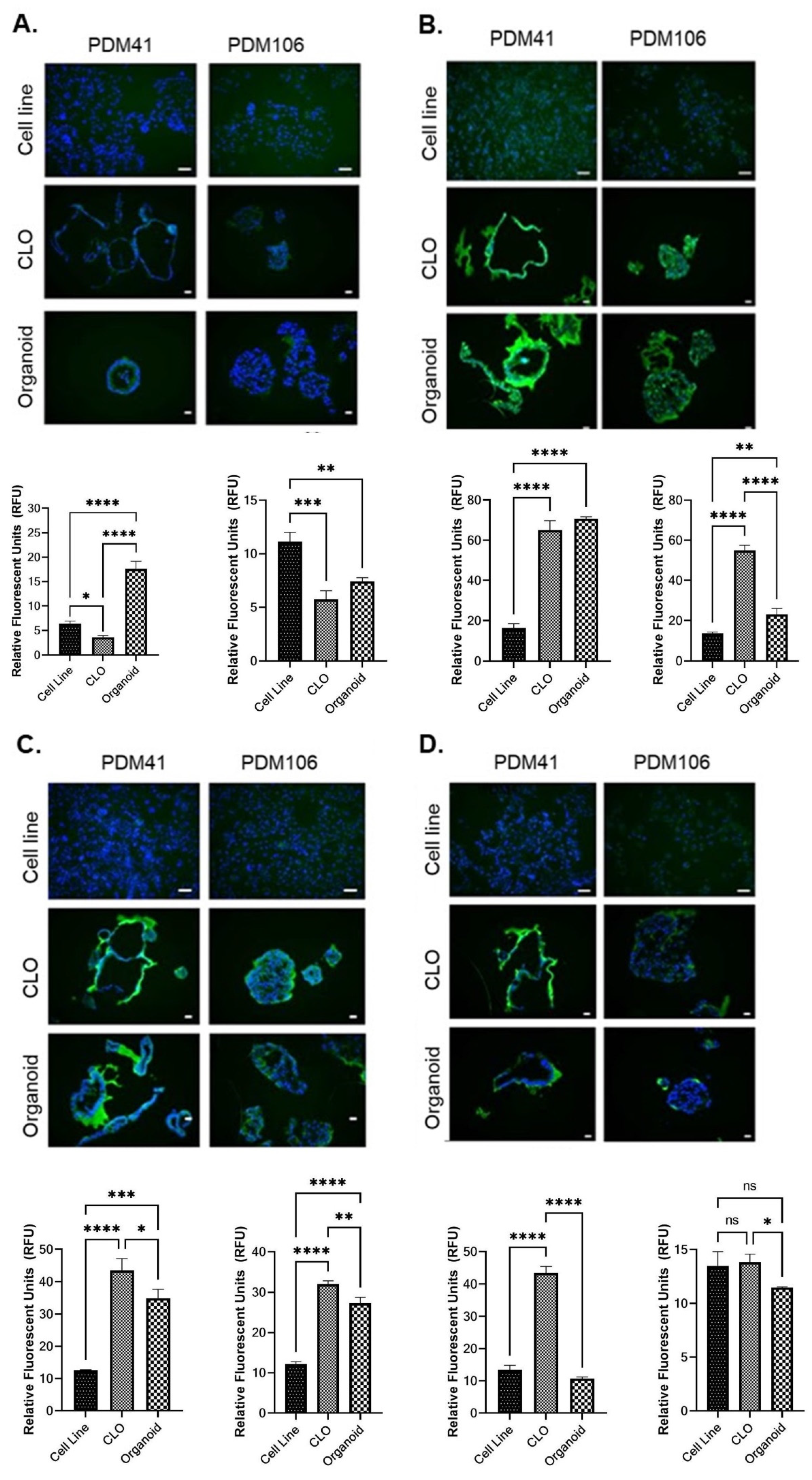
3.5. Immunofluorescence Analysis of Cancer Stem Cell Markers
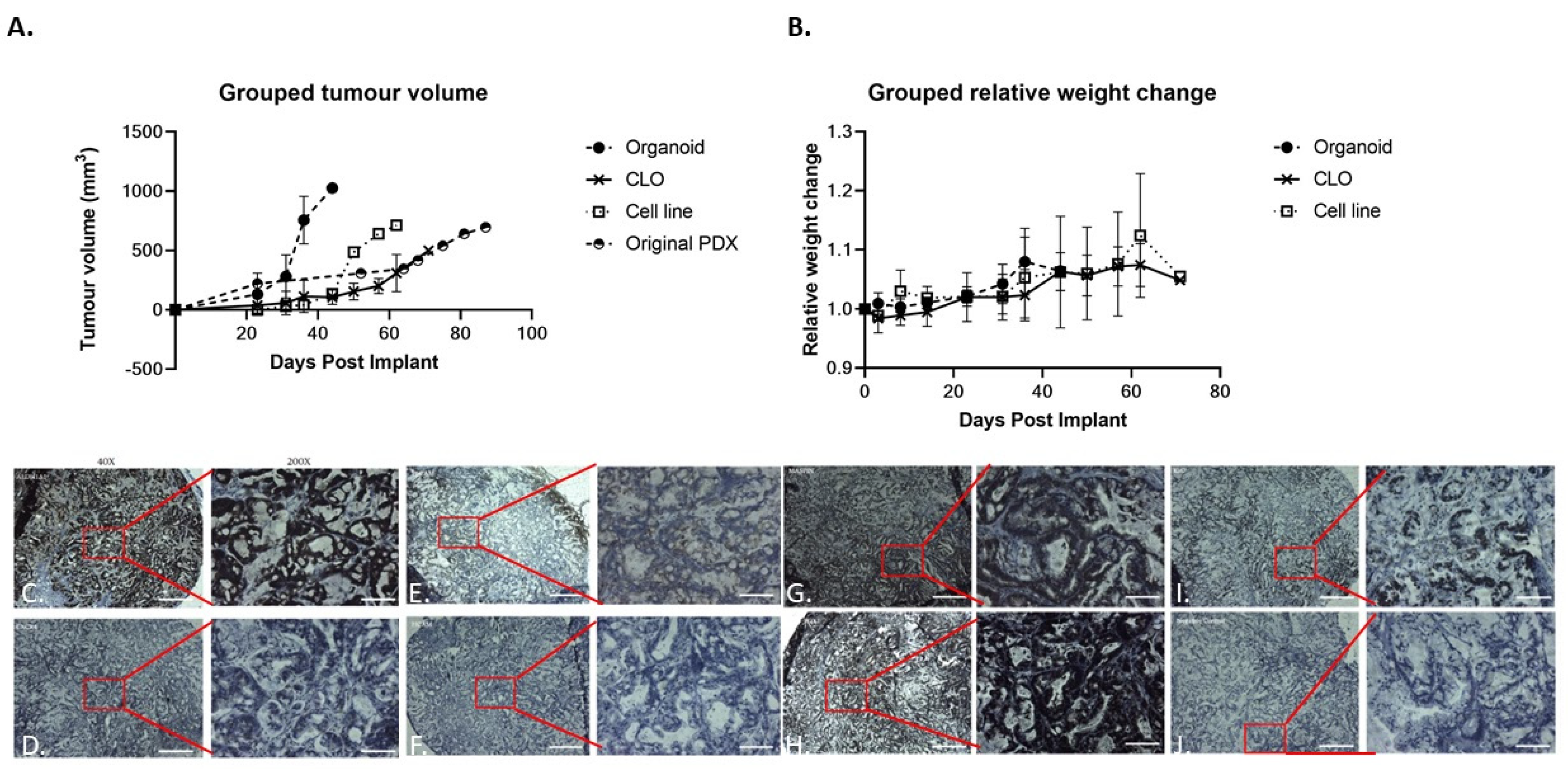
3.6. In Vivo Tumorigenesis Comparison in Novel 2D Cell Line, CLO, and Organoid
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Rahib, L.; Smith, B.D.; Aizenberg, R.; Rosenzweig, A.B.; Fleshman, J.M.; Matrisian, L.M. Projecting cancer incidence and deaths to 2030: The unexpected burden of thyroid, liver, and pancreas cancers in the United States. Cancer Res. 2014, 74, 2913–2921. [Google Scholar] [CrossRef] [Green Version]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2016. CA Cancer J. Clin. 2016, 66, 7–30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ushio, J.; Kanno, A.; Ikeda, E.; Ando, K.; Nagai, H.; Miwata, T.; Kawasaki, Y.; Tada, Y.; Yokoyama, K.; Numao, N.; et al. Pancreatic Ductal Adenocarcinoma: Epidemiology and Risk Factors. Diagnostics 2021, 11, 562. [Google Scholar] [CrossRef]
- Hackeng, W.M.; Hruban, R.H.; Offerhaus, G.J.A.; Brosens, L.A.A. Surgical and molecular pathology of pancreatic neoplasms. Diagn. Pathol. 2016, 11, 47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raphael, B.J.; Hruban, R.H.; Aguirre, A.J.; Moffitt, R.A.; Yeh, J.J.; Stewart, C.; Robertson, A.G.; Cherniack, A.D.; Gupta, M.; Getz, G.; et al. Integrated Genomic Characterization of Pancreatic Ductal Adenocarcinoma. Cancer Cell 2017, 32, 185–203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jones, S.; Zhang, X.; Parsons, D.W.; Lin, J.C.-H.; Leary, R.J.; Angenendt, P.; Mankoo, P.; Carter, H.; Kamiyama, H.; Jimeno, A.; et al. Core Signaling Pathways in Human Pancreatic Cancers Revealed by Global Genomic Analyses. Science 2008, 321, 1801–1806. [Google Scholar] [CrossRef] [Green Version]
- Gillen, S.; Schuster, T.; Büschenfelde, C.M.Z.; Friess, H.; Kleeff, J. Preoperative/Neoadjuvant Therapy in Pancreatic Cancer: A Systematic Review and Meta-analysis of Response and Resection Percentages. PLoS Med. 2010, 7, e1000267. [Google Scholar] [CrossRef] [Green Version]
- La Torre, M.; Nigri, G.; Cavallini, M.; Mercantini, P.; Ziparo, V.; Ramacciato, G. The Glasgow Prognostic Score as a Predictor of Survival in Patients with Potentially Resectable Pancreatic Adenocarcinoma. Ann. Surg. Oncol. 2012, 19, 2917–2923. [Google Scholar] [CrossRef]
- Nishio, K.; Kimura, K.; Amano, R.; Yamazoe, S.; Ohrira, G.; Nakata, B.; Hirakawa, K.; Ohira, M. Preoperative predictors for early recurrence of resectable pancreatic cancer. World J. Surg. Oncol. 2017, 15, 16. [Google Scholar] [CrossRef] [Green Version]
- Adamska, A.; Domenichini, A.; Falasca, M. Pancreatic Ductal Adenocarcinoma: Current and Evolving Therapies. Int. J. Mol. Sci. 2017, 18, 1338. [Google Scholar] [CrossRef]
- Christenson, E.S.; Jaffee, E.; Azad, N.S. Current and emerging therapies for patients with advanced pancreatic ductal adenocarcinoma: A bright future. Lancet Oncol. 2020, 21, e135–e145. [Google Scholar] [CrossRef] [PubMed]
- Nevala-Plagemann, C.; Hidalgo, M.; Garrido-Laguna, I. From state-of-the-art treatments to novel therapies for advanced-stage pancreatic cancer. Nat. Rev. Clin. Oncol. 2020, 17, 108–123. [Google Scholar] [CrossRef] [PubMed]
- Liang, C.; Shi, S.; Meng, Q.; Liang, D.; Ji, S.; Zhang, B.; Qin, Y.; Xu, J.; Ni, Q.; Yu, X. Complex roles of the stroma in the intrinsic resistance to gemcitabine in pancreatic cancer: Where we are and where we are going. Exp. Mol. Med. 2017, 49, e406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ishiwata, T.; Matsuda, Y.; Yoshimura, H.; Sasaki, N.; Ishiwata, S.; Ishikawa, N.; Takubo, K.; Arai, T.; Aida, J. Pancreatic cancer stem cells: Features and detection methods. Pathol. Oncol. Res. 2018, 24, 797–805. [Google Scholar] [CrossRef] [PubMed]
- Quiñonero, F.; Mesas, C.; Doello, K.; Cabeza, L.; Perazzoli, G.; Jimenez-Luna, C.; Rama, A.R.; Melguizo, C.; Prados, J. The challenge of drug resistance in pancreatic ductal adenocarcinoma: A current overview. Cancer Biol. Med. 2019, 16, 688–699. [Google Scholar] [CrossRef]
- Stopa, K.B.; Kusiak, A.A.; Szopa, M.D.; Ferdek, P.E.; Jakubowska, M.A. Pancreatic Cancer and Its Microenvironment—Recent Advances and Current Controversies. Int. J. Mol. Sci. 2020, 21, 3218. [Google Scholar] [CrossRef]
- Knudsen, E.S.; Balaji, U.; Mannakee, B.; Vail, P.; Eslinger, C.; Moxom, C.; Mansour, J.; Witkiewicz, A.K. Pancreatic cancer cell lines as patient-derived avatars: Genetic characterisation and functional utility. Gut 2018, 67, 508–520. [Google Scholar] [CrossRef] [Green Version]
- Hosein, A.N.; Brekken, R.A.; Maitra, A. Pancreatic cancer stroma: An update on therapeutic targeting strategies. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 487–505. [Google Scholar] [CrossRef]
- Wang, S.; Gao, D.; Chen, Y. The potential of organoids in urological cancer research. Nat. Rev. Urol. 2017, 14, 401–414. [Google Scholar] [CrossRef]
- Watters, K.M.; Bajwa, P.; Kenny, H.A. Organotypic 3D Models of the Ovarian Cancer Tumor Microenvironment. Cancers 2018, 10, 265. [Google Scholar] [CrossRef]
- Kim, M.; Mun, H.; Sung, C.O.; Cho, E.J.; Jeon, H.-J.; Chun, S.-M.; Jung, D.J.; Shin, T.H.; Jeong, G.S.; Kim, D.K.; et al. Patient-derived lung cancer organoids as in vitro cancer models for therapeutic screening. Nat. Commun. 2019, 10, 3991. [Google Scholar] [CrossRef] [Green Version]
- Bs, A.M.G.; Ci, X.; Lin, D.; Wang, Y. A synopsis of prostate organoid methodologies, applications, and limitations. Prostate 2020, 80, 518–526. [Google Scholar] [CrossRef]
- Seidlitz, T.; Koo, B.-K.; Stange, D.E. Gastric organoids—An in vitro model system for the study of gastric development and road to personalized medicine. Cell Death Differ. 2021, 28, 68–83. [Google Scholar] [CrossRef]
- Linkous, A.; Balamatsias, D.; Snuderl, M.; Edwards, L.; Miyaguchi, K.; Milner, T.; Reich, B.; Cohen-Gould, L.; Storaska, A.; Nakayama, Y.; et al. Modeling Patient-Derived Glioblastoma with Cerebral Organoids. Cell Rep. 2019, 26, 3203–3211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reidy, E.; Leonard, N.; Treacy, O.; Ryan, A. A 3D View of Colorectal Cancer Models in Predicting Therapeutic Responses and Resistance. Cancers 2021, 13, 227. [Google Scholar] [CrossRef] [PubMed]
- Dekkers, J.F.; van Vliet, E.J.; Sachs, N.; Rosenbluth, J.M.; Kopper, O.; Rebel, H.G.; Wehrens, E.J.; Piani, C.; Visvader, J.E.; Verissimo, C.S.; et al. Long-term culture, genetic manipulation and xenotransplantation of human normal and breast cancer organoids. Nat. Protoc. 2021, 16, 1936–1965. [Google Scholar] [CrossRef] [PubMed]
- Brooks, A.; Liang, X.; Zhang, Y.; Zhao, C.-X.; Roberts, M.S.; Wang, H.; Zhang, L.; Crawford, D.H. Liver organoid as a 3D in vitro model for drug validation and toxicity assessment. Pharmacol. Res. 2021, 169, 105608. [Google Scholar] [CrossRef]
- Clevers, H. Modeling Development and Disease with Organoids. Cell 2016, 165, 1586–1597. [Google Scholar] [CrossRef] [Green Version]
- Nelson, S.R.; Zhang, C.; Roche, S.; O’Neill, F.; Swan, N.; Luo, Y.; Larkin, A.; Crown, J.; Walsh, N. Modelling of pancreatic cancer biology: Transcriptomic signature for 3D PDX-derived organoids and primary cell line organoid development. Sci. Rep. 2020, 10, 2778. [Google Scholar] [CrossRef] [Green Version]
- Herreros-Villanueva, M.; Bujanda, L.; Billadeau, D.D.; Zhang, J.-S. Embryonic stem cell factors and pancreatic cancer. World J. Gastroenterol. 2014, 20, 2247–2254. [Google Scholar] [CrossRef]
- Gzil, A.; Zarębska, I.; Bursiewicz, W.; Antosik, P.; Grzanka, D.; Szylberg, Ł. Markers of pancreatic cancer stem cells and their clinical and therapeutic implications. Mol. Biol. Rep. 2019, 46, 6629–6645. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roche, S.; O’Neill, F.; Murphy, J.; Swan, N.; Meiller, J.; Conlon, N.T.; Geoghegan, J.; Conlon, K.; McDermott, R.; Rahman, R.; et al. Establishment and Characterisation by Expression Microarray of Patient-Derived Xenograft Panel of Human Pancreatic Adenocarcinoma Patients. Int. J. Mol. Sci. 2020, 21, 962. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jensen, C.; Teng, Y. Is It Time to Start Transitioning From 2D to 3D Cell Culture? Front. Mol. Biosci. 2020, 7, 33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kapałczyńska, M.; Kolenda, T.; Przybyła, W.; Zajączkowska, M.; Teresiak, A.; Filas, V.; Ibbs, M.; Bliźniak, R.; Łuczewski, L.; Lamperska, K. 2D and 3D cell cultures—A comparison of different types of cancer cell cultures. Arch. Med. Sci. 2018, 14, 910–919. [Google Scholar] [CrossRef]
- Edmondson, R.; Broglie, J.J.; Adcock, A.F.; Yang, L. Three-Dimensional Cell Culture Systems and Their Applications in Drug Discovery and Cell-Based Biosensors. ASSAY Drug Dev. Technol. 2014, 12, 207–218. [Google Scholar] [CrossRef] [Green Version]
- Duval, K.; Grover, H.; Han, L.-H.; Mou, Y.; Pegoraro, A.F.; Fredberg, J.; Chen, Z. Modeling Physiological Events in 2D vs. 3D Cell Culture. Physiology 2017, 32, 266–277. [Google Scholar] [CrossRef] [Green Version]
- Adcock, A.F.; Trivedi, G.; Edmondson, R.; Yang, C.S.A.L. Three-Dimensional (3D) Cell Cultures in Cell-based Assays for in-vitro Evaluation of Anticancer Drugs. J. Anal. Bioanal. Tech. 2015, 6, 1. [Google Scholar] [CrossRef]
- Durand, R.E.; Olive, P.L. Resistance of tumor cells to chemo- and radiotherapy modulated by the three-dimensional architecture of solid tumors and spheroids. Methods Cell Biol. 2001, 64, 211–233. [Google Scholar] [CrossRef]
- Hirt, C.K.; Booij, T.H.; Grob, L.; Simmler, P.; Toussaint, N.C.; Keller, D.; Taube, D.; Ludwig, V.; Goryachkin, A.; Pauli, C.; et al. Drug screening and genome editing in human pancreatic cancer organoids identifies drug-gene interactions and candidates for off-label therapy. Cell Genom. 2022, 2, 100095. [Google Scholar] [CrossRef]
- Patil, K.; Khan, F.B.; Akhtar, S.; Ahmad, A.; Uddin, S. The plasticity of pancreatic cancer stem cells: Implications in therapeutic resistance. Cancer Metastasis Rev. 2021, 40, 691–720. [Google Scholar] [CrossRef]
- Li, C.; Heidt, D.G.; Dalerba, P.; Burant, C.F.; Zhang, L.; Adsay, V.; Wicha, M.; Clarke, M.F.; Simeone, D.M. Identification of Pancreatic Cancer Stem Cells. Cancer Res. 2007, 67, 1030–1037. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hermann, P.C.; Huber, S.L.; Herrler, T.; Aicher, A.; Ellwart, J.W.; Guba, M.; Bruns, C.J.; Heeschen, C. Distinct Populations of Cancer Stem Cells Determine Tumor Growth and Metastatic Activity in Human Pancreatic Cancer. Cell Stem Cell 2007, 1, 313–323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morimoto, M.; Matsuo, Y.; Koide, S.; Tsuboi, K.; Shamoto, T.; Sato, T.; Saito, K.; Takahashi, H.; Takeyama, H. Enhancement of the CXCL12/CXCR4 axis due to acquisition of gemcitabine resistance in pancreatic cancer: Effect of CXCR4 antagonists. BMC Cancer 2016, 16, 305. [Google Scholar] [CrossRef] [Green Version]
- RL Sleightholm, BK Neilsen, J Li, MM Steele Emerging roles of the CXCL12/CXCR4 axis in pancreatic cancer progression and therapy. Pharmacol. Ther. 2017, 179, 158–170. [CrossRef] [PubMed]
- Sun, J.-S.; Zhang, X.-L.; Yang, Y.-J.; Nie, Z.-G.; Zhang, Y. Hypoxia promotes C-X-C chemokine receptor type 4 expression through microRNA-150 in pancreatic cancer cells. Oncol. Lett. 2015, 10, 835–840. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zagzag, D.; Lukyanov, Y.; Lan, L.; Ali, M.A.; Esencay, M.; Mendez, O.; Yee, H.; Voura, E.B.; Newcomb, E.W. Hypoxia-inducible factor 1 and VEGF upregulate CXCR4 in glioblastoma: Implications for angiogenesis and glioma cell invasion. Lab. Investig. 2006, 86, 1221–1232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Erkan, M.; Kurtoglu, M.; Kleeff, J. The role of hypoxia in pancreatic cancer: A potential therapeutic target? Expert Rev. Gastroenterol. Hepatol. 2016, 10, 301–316. [Google Scholar] [CrossRef]
- Hubert, C.G.; Rivera, M.; Spangler, L.C.; Wu, Q.; Mack, S.C.; Prager, B.C.; Couce, M.; McLendon, R.E.; Sloan, A.E.; Rich, J.N. A Three-Dimensional Organoid Culture System Derived from Human Glioblastomas Recapitulates the Hypoxic Gradients and Cancer Stem Cell Heterogeneity of Tumors Found In Vivo. Cancer Res. 2016, 76, 2465–2477. [Google Scholar] [CrossRef] [Green Version]
- Patriarca, C.; Macchi, R.M.; Marschner, A.K.; Mellstedt, H. Epithelial cell adhesion molecule expression (CD326) in cancer: A short review. Cancer Treat. Rev. 2012, 38, 68–75. [Google Scholar] [CrossRef]
- Wang, M.-H.; Sun, R.; Zhou, X.-M.; Zhang, M.-Y.; Lu, J.-B.; Yang, Y.; Zeng, L.-S.; Yang, X.-Z.; Shi, L.; Xiao, R.-W.; et al. Epithelial cell adhesion molecule overexpression regulates epithelial-mesenchymal transition, stemness and metastasis of nasopharyngeal carcinoma cells via the PTEN/AKT/mTOR pathway. Cell Death Dis. 2018, 9, 2. [Google Scholar] [CrossRef]
- Fong, D.; Steurer, M.; Obrist, P.; Barbieri, V.; Margreiter, R.; Amberger, A.; Laimer, K.; Gastl, G.; Tzankov, A.; Spizzo, G. Ep-CAM expression in pancreatic and ampullary carcinomas: Frequency and prognostic relevance. J. Clin. Pathol. 2008, 61, 31–35. [Google Scholar] [CrossRef] [PubMed]
- Skoda, J.; Hermanova, M.; Loja, T.; Nemec, P.; Neradil, J.; Karasek, P.; Veselska, R. Co-Expression of Cancer Stem Cell Markers Corresponds to a Pro-Tumorigenic Expression Profile in Pancreatic Adenocarcinoma. PLoS ONE 2016, 11, e0159255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martowicz, A.; Spizzo, G.; Gastl, G.; Untergasser, G. Phenotype-dependent effects of EpCAM expression on growth and invasion of human breast cancer cell lines. BMC Cancer 2012, 12, 501. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Litvinov, S.V.; Velders, M.P.; A Bakker, H.; Fleuren, G.J.; O Warnaar, S. Ep-CAM: A human epithelial antigen is a homophilic cell-cell adhesion molecule. J. Cell Biol. 1994, 125, 437–446. [Google Scholar] [CrossRef]
- Wu, K.; Xu, H.; Tian, Y.; Yuan, X.; Wu, H.; Liu, Q.; Pestell, R. The role of CD44 in epithelial–mesenchymal transition and cancer development. OncoTargets Ther. 2015, 8, 3783–3792. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fontana, F.; Raimondi, M.; Marzagalli, M.; Sommariva, M.; Limonta, P.; Gagliano, N. Epithelial-To-Mesenchymal Transition Markers and CD44 Isoforms Are Differently Expressed in 2D and 3D Cell Cultures of Prostate Cancer Cells. Cells 2019, 8, 143. [Google Scholar] [CrossRef] [Green Version]
- Rasheed, Z.A.; Yang, J.; Wang, Q.; Kowalski, J.; Freed, I.; Murter, C.; Hong, S.-M.; Koorstra, J.-B.; Rajeshkumar, N.V.; He, X.; et al. Prognostic Significance of Tumorigenic Cells With Mesenchymal Features in Pancreatic Adenocarcinoma. Gynecol. Oncol. 2010, 102, 340–351. [Google Scholar] [CrossRef]
- Tomita, H.; Tanaka, K.; Tanaka, T.; Hara, A. Aldehyde dehydrogenase 1A1 in stem cells and cancer. Oncotarget 2016, 7, 11018–11032. [Google Scholar] [CrossRef] [Green Version]
- Boj, S.F.; Hwang, C.-I.; Baker, L.A.; Chio, I.I.C.; Engle, D.D.; Corbo, V.; Jager, M.; Ponz-Sarvise, M.; Tiriac, H.; Spector, M.S.; et al. Organoid Models of Human and Mouse Ductal Pancreatic Cancer. Cell 2015, 160, 324–338. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 5-FU (µM) | Cisplatin (µM) | Nab-Paclitaxel (ng/mL) | |
|---|---|---|---|
| PDM41 | |||
| Organoids | 51.76 | 9.51 | 58.24 |
| CLOs | 95.06 | 14.35 | 190.2 |
| Cell line | 324.9 | Not responsive | Not responsive |
| PDM106 | |||
| Organoids | 164.7 | Not responsive | 380.3 |
| CLOs | 144.7 | 17.43 | 223.2 |
| Cell line | 207.4 | 11.26 | Not responsive |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Noorani, S.; Nelson, S.R.; Conlon, N.T.; Meiller, J.; Shcheglova, E.; Usai, A.; Stoof, J.; Palanga, L.; O’Neill, F.; Roche, S.; et al. Pancreatic Cancer 3D Cell Line Organoids (CLOs) Maintain the Phenotypic Characteristics of Organoids and Accurately Reflect the Cellular Architecture and Heterogeneity In Vivo. Organoids 2022, 1, 168-183. https://doi.org/10.3390/organoids1020013
Noorani S, Nelson SR, Conlon NT, Meiller J, Shcheglova E, Usai A, Stoof J, Palanga L, O’Neill F, Roche S, et al. Pancreatic Cancer 3D Cell Line Organoids (CLOs) Maintain the Phenotypic Characteristics of Organoids and Accurately Reflect the Cellular Architecture and Heterogeneity In Vivo. Organoids. 2022; 1(2):168-183. https://doi.org/10.3390/organoids1020013
Chicago/Turabian StyleNoorani, Sara, Shannon R. Nelson, Neil T. Conlon, Justine Meiller, Ekaterina Shcheglova, Alice Usai, Jojanneke Stoof, Letizia Palanga, Fiona O’Neill, Sandra Roche, and et al. 2022. "Pancreatic Cancer 3D Cell Line Organoids (CLOs) Maintain the Phenotypic Characteristics of Organoids and Accurately Reflect the Cellular Architecture and Heterogeneity In Vivo" Organoids 1, no. 2: 168-183. https://doi.org/10.3390/organoids1020013