
Adenoma-Derived Organoids for Precision Therapy

Abstract
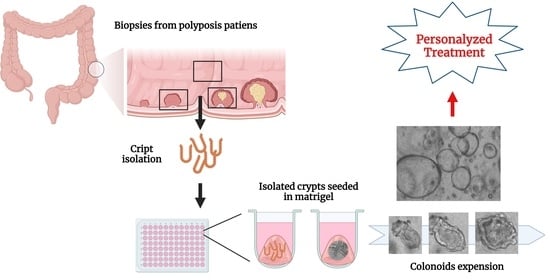
:
1. Introduction
2. Materials and Methods
2.1. Human Tissue Samples
2.2. Isolation of Intestinal Crypts from Human Colorectal Biopsies
2.3. LWRN Complete Medium
2.4. Cryopreservation and Thawing of Colon Organoids
2.5. Immunofluorescence (IF)
2.6. Microscopy and Imaging
2.7. RNA Isolation and RT-qPCR Analysis
2.8. qPCR Primers
2.9. Western Blot Analysis (WB)
2.10. Statistical Methods
3. Results
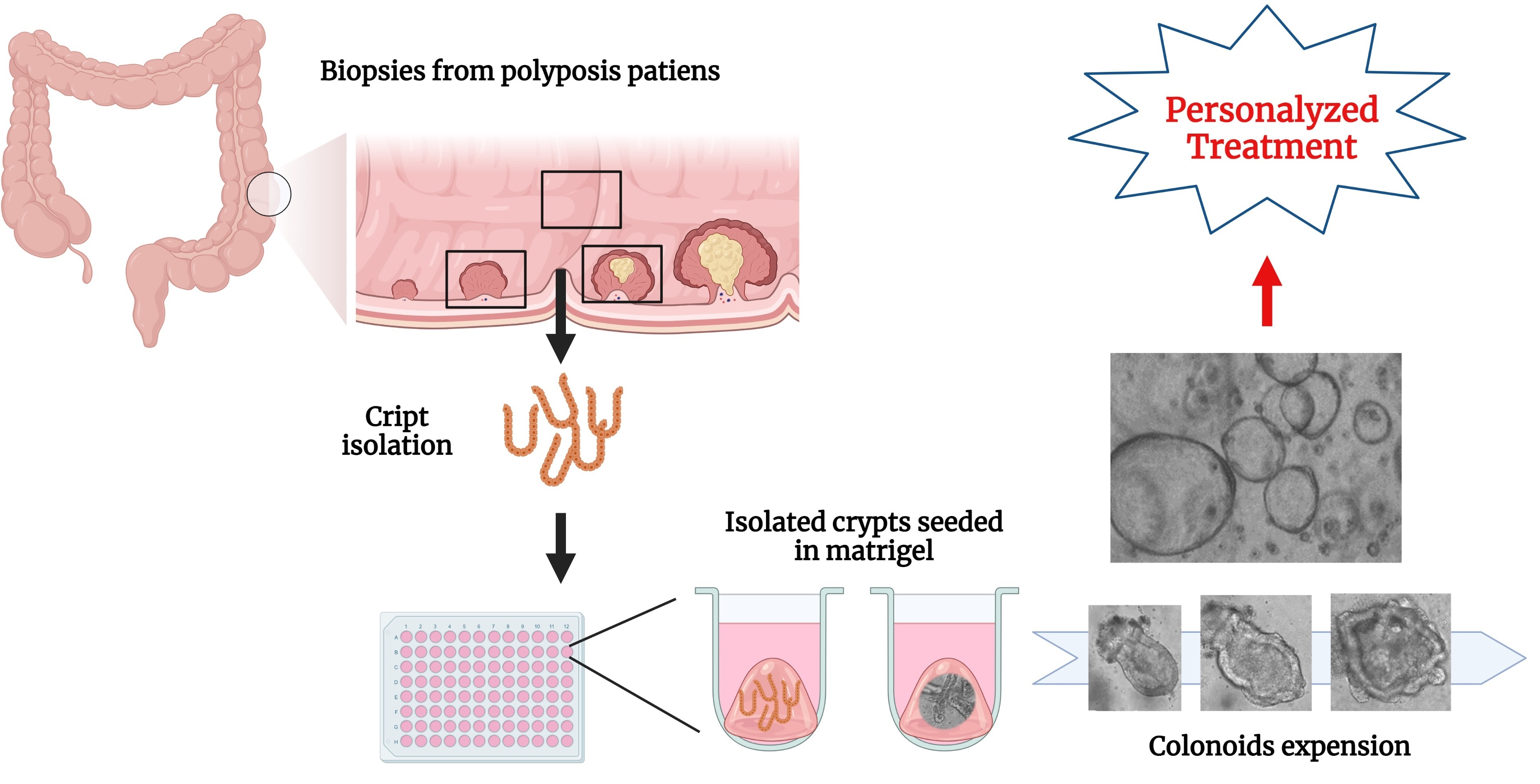
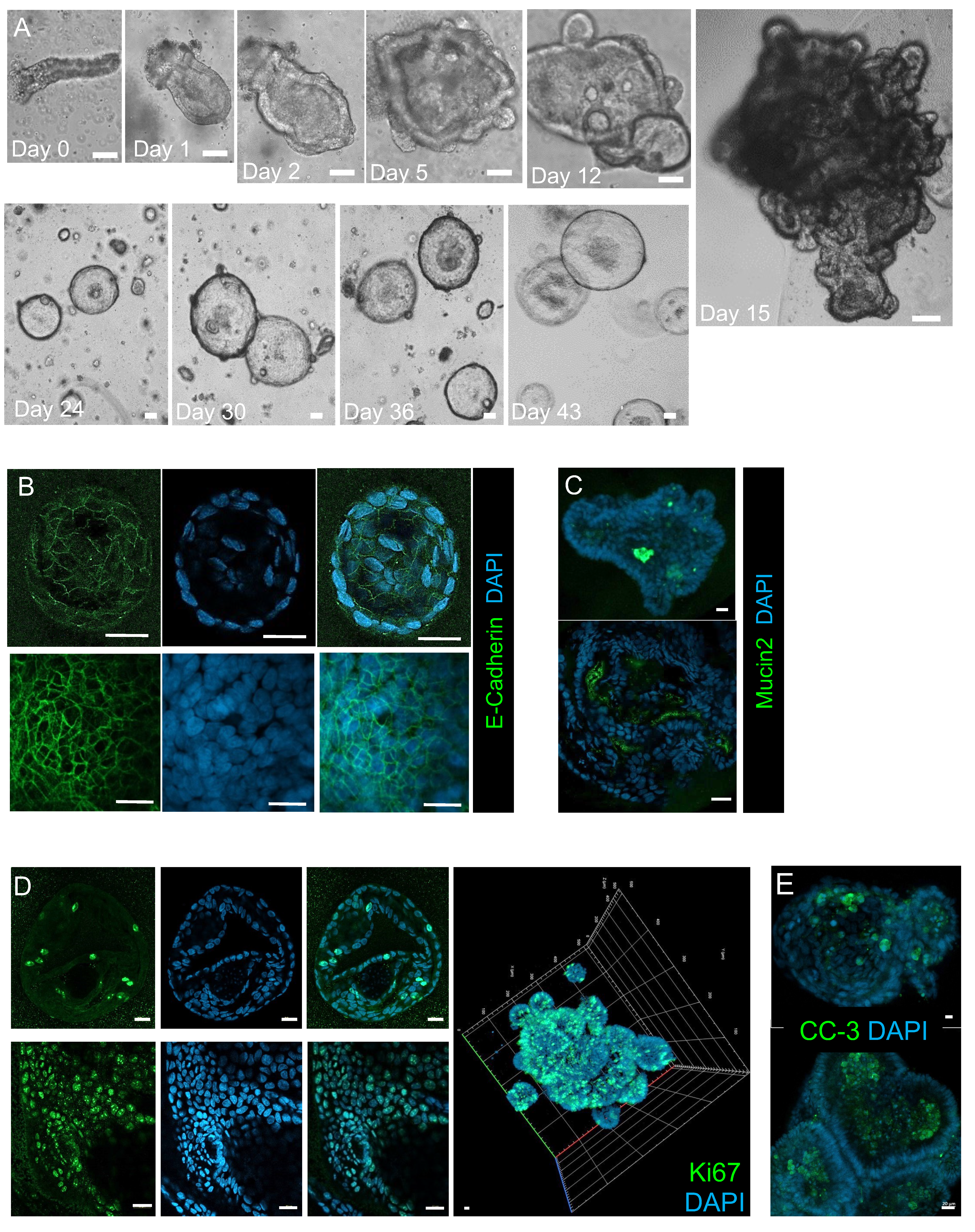
3.1. Establishing and Characterizing Adenoma-Derived Colonoids
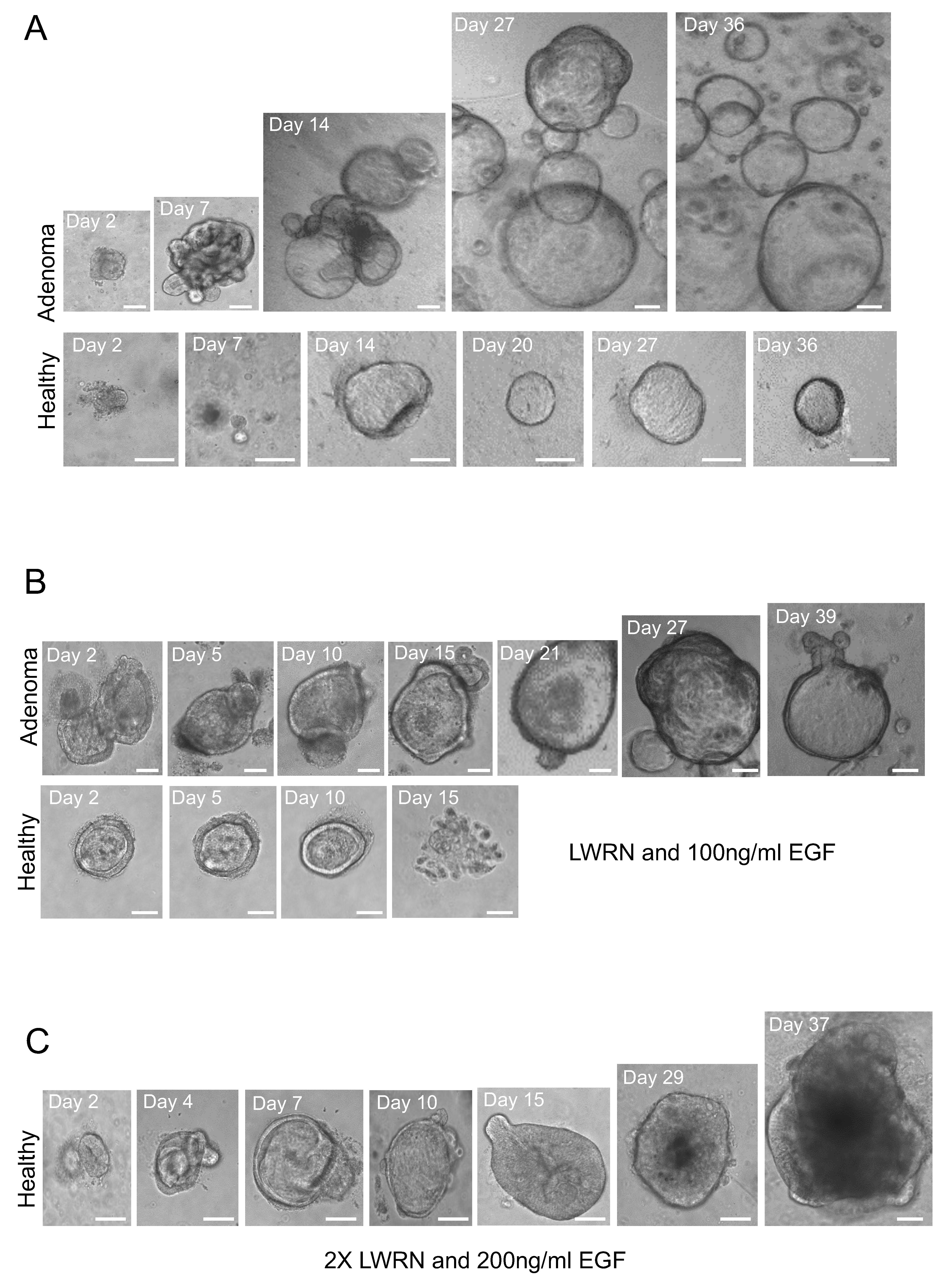
3.2. Enhancing the Growth of Healthy Tissue-Derived Organoids
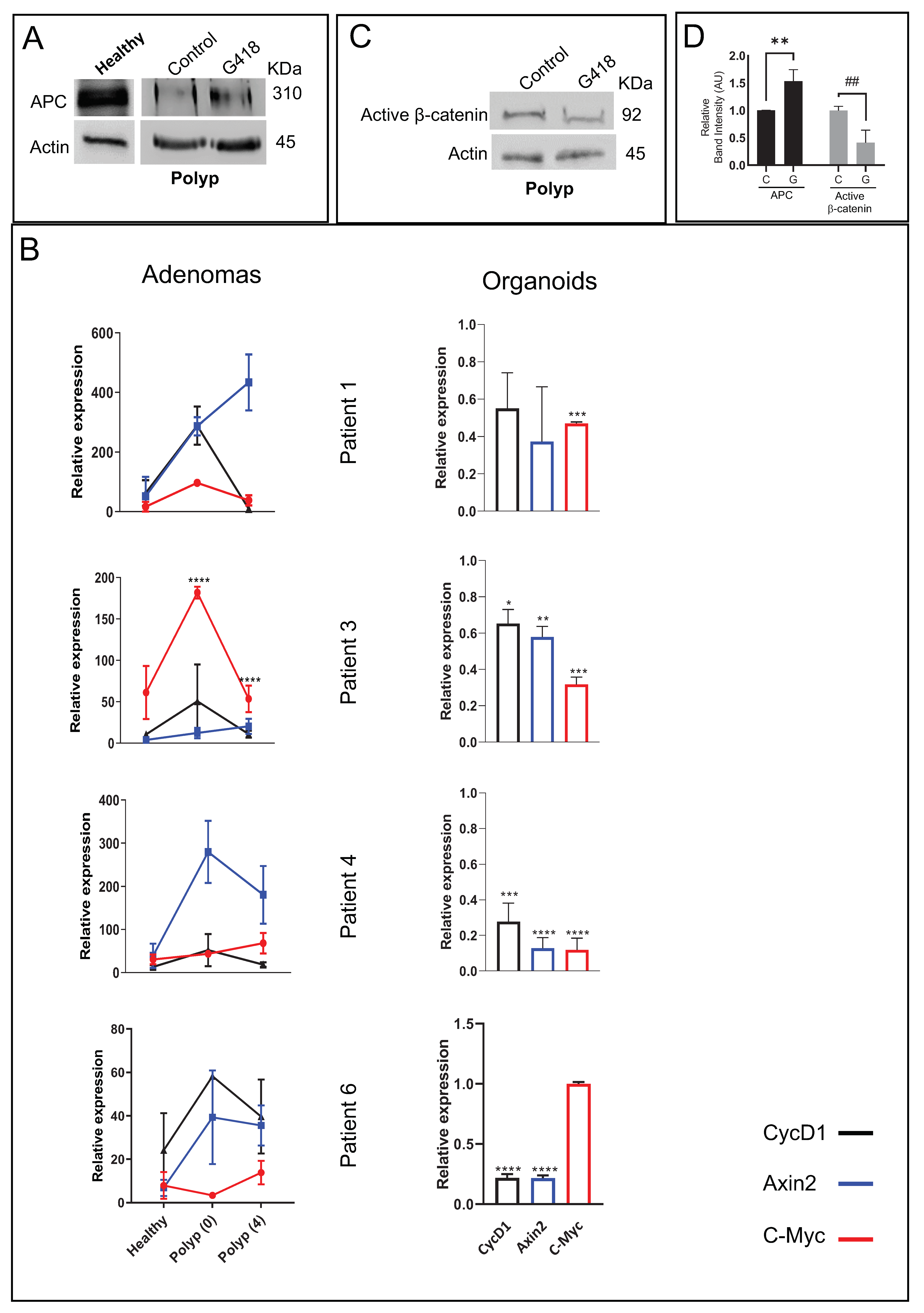
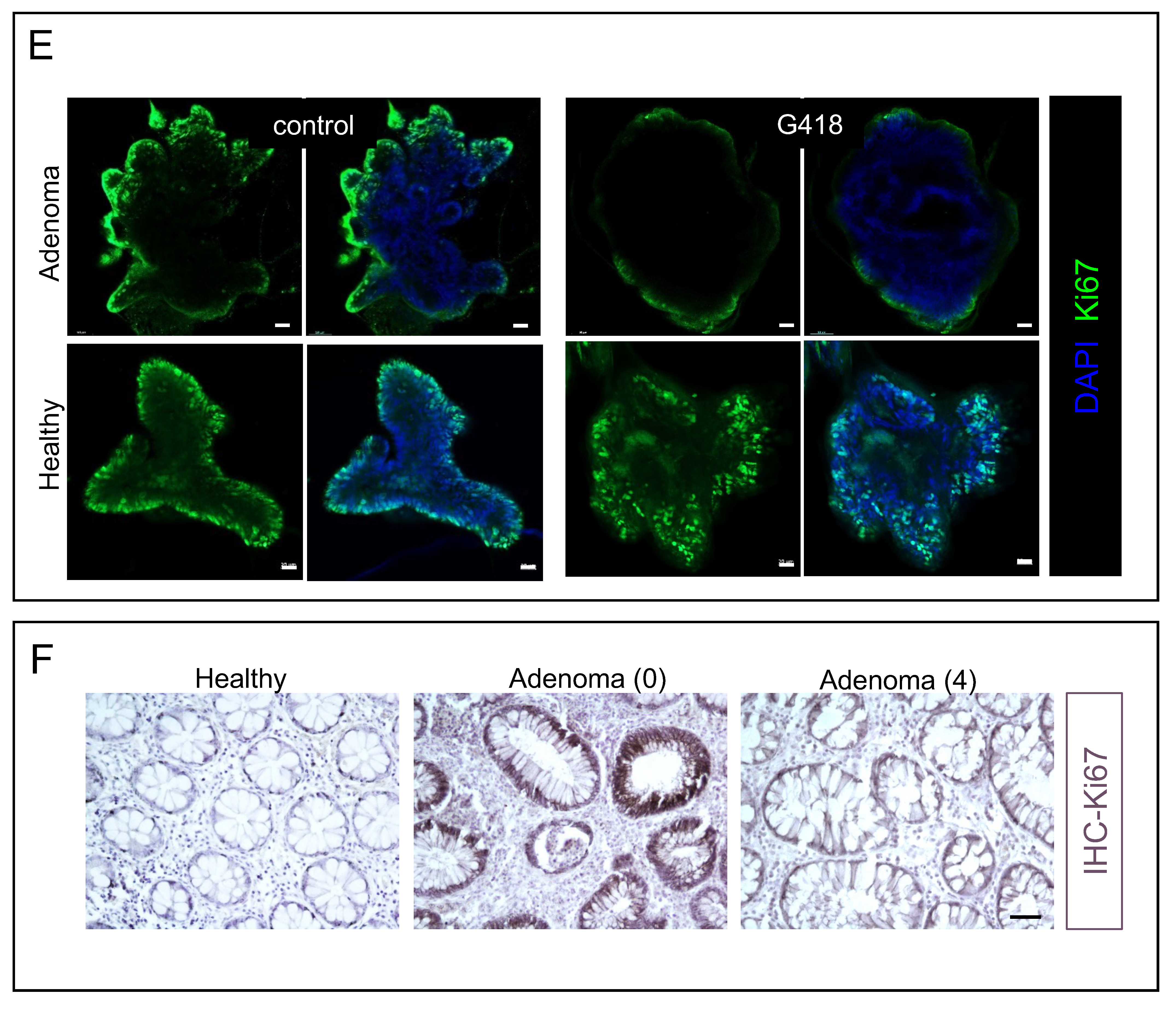
3.3. G418 Induces APC Restoration Decreasing Canonical Wnt Signaling and Cell Proliferation in Treated Colonoids
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer Statistics, 2021. CA Cancer J. Clin. 2021, 71, 7–33. [Google Scholar] [CrossRef] [PubMed]
- Mao, R.; Krautscheid, P.; Graham, R.P.; Ganguly, A.; Shankar, S.; Ferber, M.; Hegde, M.; Committee, A.L.Q.A. Genetic testing for inherited colorectal cancer and polyposis, 2021 revision: A technical standard of the American College of Medical Genetics and Genomics (ACMG). Genet. Med. 2021, 23, 1807–1817. [Google Scholar] [CrossRef] [PubMed]
- Pearlman, R.; Frankel, W.L.; Swanson, B.; Zhao, W.; Yilmaz, A.; Miller, K.; Bacher, J.; Bigley, C.; Nelsen, L.; Goodfellow, P.J.; et al. Prevalence and Spectrum of Germline Cancer Susceptibility Gene Mutations Among Patients with Early-Onset Colorectal Cancer. JAMA Oncol. 2017, 3, 464–471. [Google Scholar] [CrossRef] [PubMed]
- Seifert, B.A.; McGlaughon, J.L.; Jackson, S.A.; Ritter, D.I.; Roberts, M.E.; Schmidt, R.J.; Thompson, B.A.; Jimenez, S.; Trapp, M.; Lee, K.; et al. Determining the clinical validity of hereditary colorectal cancer and polyposis susceptibility genes using the Clinical Genome Resource Clinical Validity Framework. Genet. Med. 2019, 21, 1507–1516. [Google Scholar] [CrossRef] [PubMed]
- Lynch, H.T.; de la Chapelle, A. Hereditary colorectal cancer. N. Engl. J. Med. 2003, 348, 919–932. [Google Scholar] [CrossRef]
- Leoz, M.L.; Carballal, S.; Moreira, L.; Ocana, T.; Balaguer, F. The genetic basis of familial adenomatous polyposis and its implications for clinical practice and risk management. Appl. Clin. Genet. 2015, 8, 95–107. [Google Scholar]
- Kinzler, K.W.; Vogelstein, B. Lessons from hereditary colorectal cancer. Cell 1996, 87, 159–170. [Google Scholar] [CrossRef] [Green Version]
- Lamlum, H.; Ilyas, M.; Rowan, A.; Clark, S.; Johnson, V.; Bell, J.; Frayling, I.; Efstathiou, J.; Pack, K.; Payne, S.; et al. The type of somatic mutation at APC in familial adenomatous polyposis is determined by the site of the germline mutation: A new facet to Kxnudson’s ‘two-hit’ hypothesis. Nat. Med. 1999, 5, 1071–1075. [Google Scholar] [CrossRef]
- Burt, R.W.; DiSario, J.A.; Cannon-Albright, L. Genetics of colon cancer: Impact of inheritance on colon cancer risk. Annu. Rev. Med. 1995, 46, 371–379. [Google Scholar] [CrossRef]
- Christie, M.; Jorissen, R.N.; Mouradov, D.; Sakthianandeswaren, A.; Li, S.; Day, F.; Tsui, C.; Lipton, L.; Desai, J.; Jones, I.T.; et al. Different APC genotypes in proximal and distal sporadic colorectal cancers suggest distinct WNT/beta-catenin signalling thresholds for tumourigenesis. Oncogene 2013, 32, 4675–4682. [Google Scholar] [CrossRef]
- Caspi, M.; Wittenstein, A.; Kazelnik, M.; Shor-Nareznoy, Y.; Rosin-Arbesfeld, R. Therapeutic targeting of the oncogenic Wnt signaling pathway for treating colorectal cancer and other colonic disorders. Adv. Drug Deliv. Rev. 2021, 169, 118–136. [Google Scholar] [CrossRef] [PubMed]
- Nathke, I.S. The adenomatous polyposis coli protein: The Achilles heel of the gut epithelium. Annu. Rev. Cell Dev. Biol. 2004, 20, 337–366. [Google Scholar] [CrossRef] [PubMed]
- Nusse, R. Wnt signaling in disease and in development. Cell Res. 2005, 15, 28–32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van der Vaart, J.; Clevers, H. Airway organoids as models of human disease. J. Intern. Med. 2021, 289, 604–613. [Google Scholar] [CrossRef]
- Sato, T.; Clevers, H. Growing self-organizing mini-guts from a single intestinal stem cell: Mechanism and applications. Science 2013, 340, 1190–1194. [Google Scholar] [CrossRef] [Green Version]
- Fujii, M.; Sato, T. Somatic cell-derived organoids as prototypes of human epithelial tissues and diseases. Nat. Mater. 2021, 20, 156–169. [Google Scholar] [CrossRef]
- Sato, T.; Stange, D.E.; Ferrante, M.; Vries, R.G.; Van Es, J.H.; Van den Brink, S.; Van Houdt, W.J.; Pronk, A.; Van Gorp, J.; Siersema, P.D.; et al. Long-term expansion of epithelial organoids from human colon, adenoma, adenocarcinoma, and Barrett’s epithelium. Gastroenterology 2011, 141, 1762–1772. [Google Scholar] [CrossRef]
- De Lau, W.; Barker, N.; Low, T.Y.; Koo, B.K.; Li, V.S.; Teunissen, H.; Kujala, P.; Haegebarth, A.; Peters, P.J.; van de Wetering, M.; et al. Lgr5 homologues associate with Wnt receptors and mediate R-spondin signalling. Nature 2011, 476, 293–297. [Google Scholar] [CrossRef]
- Haramis, A.P.; Begthel, H.; van den Born, M.; van Es, J.; Jonkheer, S.; Offerhaus, G.J.; Clevers, H. De novo crypt formation and juvenile polyposis on BMP inhibition in mouse intestine. Science 2004, 303, 1684–1686. [Google Scholar] [CrossRef] [Green Version]
- Sato, T.; Vries, R.G.; Snippert, H.J.; van de Wetering, M.; Barker, N.; Stange, D.E.; van Es, J.H.; Abo, A.; Kujala, P.; Peters, P.J.; et al. Single Lgr5 stem cells build crypt-villus structures in vitro without a mesenchymal niche. Nature 2009, 459, 262–265. [Google Scholar] [CrossRef]
- Fearnhead, N.S.; Britton, M.P.; Bodmer, W.F. The ABC of APC. Hum. Mol. Genet. 2001, 10, 721–733. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kariv, R.; Caspi, M.; Fliss-Isakov, N.; Shorer, Y.; Shor, Y.; Rosner, G.; Brazowski, E.; Beer, G.; Cohen, S.; Rosin-Arbesfeld, R. Resorting the function of the colorectal cancer gatekeeper adenomatous polyposis coli. Int. J. Cancer 2020, 146, 1064–1074. [Google Scholar] [CrossRef] [PubMed]
- Zilberberg, A.; Lahav, L.; Rosin-Arbesfeld, R. Restoration of APC gene function in colorectal cancer cells by aminoglycoside- and macrolide-induced read-through of premature termination codons. Gut 2010, 59, 496–507. [Google Scholar] [CrossRef] [PubMed]
- Dame, M.K.; Jiang, Y.; Appelman, H.D.; Copley, K.D.; McClintock, S.D.; Aslam, M.N.; Attili, D.; Elmunzer, B.J.; Brenner, D.E.; Varani, J.; et al. Human colonic crypts in culture: Segregation of immunochemical markers in normal versus adenoma-derived. Lab. Investig. 2014, 94, 222–234. [Google Scholar] [CrossRef] [Green Version]
- Jung, P.; Sato, T.; Merlos-Suarez, A.; Barriga, F.M.; Iglesias, M.; Rossell, D.; Auer, H.; Gallardo, M.; Blasco, M.A.; Sancho, E.; et al. Isolation and in vitro expansion of human colonic stem cells. Nat. Med. 2011, 17, 1225–1227. [Google Scholar] [CrossRef]
- Tsai, Y.H.; Czerwinski, M.; Wu, A.; Dame, M.K.; Attili, D.; Hill, E.; Colacino, J.A.; Nowacki, L.M.; Shroyer, N.F.; Higgins, P.D.R.; et al. A Method for Cryogenic Preservation of Human Biopsy Specimens and Subsequent Organoid Culture. Cell Mol. Gastroenterol. Hepatol. 2018, 6, 218–222.e217. [Google Scholar] [CrossRef] [Green Version]
- Kashfi, S.M.H.; Almozyan, S.; Jinks, N.; Koo, B.K.; Nateri, A.S. Morphological alterations of cultured human colorectal matched tumour and healthy organoids. Oncotarget 2018, 9, 10572–10584. [Google Scholar] [CrossRef] [Green Version]
- Knapp, D.; Kannan, N.; Pellacani, D.; Eaves, C.J. Mass Cytometric Analysis Reveals Viable Activated Caspase-3(+) Luminal Progenitors in the Normal Adult Human Mammary Gland. Cell Rep. 2017, 21, 1116–1126. [Google Scholar] [CrossRef] [Green Version]
- Skovdahl, H.K.; Gopalakrishnan, S.; Svendsen, T.D.; Granlund, A.V.B.; Bakke, I.; Ginbot, Z.G.; Thorsvik, S.; Flatberg, A.; Sporsheim, B.; Ostrop, J.; et al. Patient Derived Colonoids as Drug Testing Platforms-Critical Importance of Oxygen Concentration. Front. Pharmacol. 2021, 12, 679741. [Google Scholar] [CrossRef]
- Wilson, S.S.; Mayo, M.; Melim, T.; Knight, H.; Patnaude, L.; Wu, X.; Phillips, L.; Westmoreland, S.; Dunstan, R.; Fiebiger, E.; et al. Optimized Culture Conditions for Improved Growth and Functional Differentiation of Mouse and Human Colon Organoids. Front. Immunol. 2020, 11, 547102. [Google Scholar] [CrossRef]
- Zachos, N.C.; Kovbasnjuk, O.; Foulke-Abel, J.; In, J.; Blutt, S.E.; de Jonge, H.R.; Estes, M.K.; Donowitz, M. Human Enteroids/Colonoids and Intestinal Organoids Functionally Recapitulate Normal Intestinal Physiology and Pathophysiology. J. Biol. Chem. 2016, 291, 3759–3766. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.; Koo, B.K.; Knoblich, J.A. Human organoids: Model systems for human biology and medicine. Nat. Rev. Mol. Cell Biol. 2020, 21, 571–584. [Google Scholar] [CrossRef] [PubMed]
- Schutgens, F.; Clevers, H. Human Organoids: Tools for Understanding Biology and Treating Diseases. Annu. Rev. Pathol. 2020, 15, 211–234. [Google Scholar] [CrossRef] [Green Version]
- Clevers, H. Modeling Development and Disease with Organoids. Cell 2016, 165, 1586–1597. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rookmaaker, M.B.; Schutgens, F.; Verhaar, M.C.; Clevers, H. Development and application of human adult stem or progenitor cell organoids. Nat. Rev. Nephrol. 2015, 11, 546–554. [Google Scholar] [CrossRef] [PubMed]
- Fujii, M.; Matano, M.; Nanki, K.; Sato, T. Efficient genetic engineering of human intestinal organoids using electroporation. Nat. Protoc. 2015, 10, 1474–1485. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.Y.; Huang, J.J.; Liu, Y.; Zhao, Y.; Wu, X.W.; Ren, J.A. Current knowledge on the multiform reconstitution of intestinal stem cell niche. World J. Stem Cells 2021, 13, 1564–1579. [Google Scholar] [CrossRef]
- Wallach, T.E.; Bayrer, J.R. Intestinal Organoids: New Frontiers in the Study of Intestinal Disease and Physiology. J. Pediatr. Gastroenterol. Nutr. 2017, 64, 180–185. [Google Scholar] [CrossRef] [Green Version]
- In, J.G.; Foulke-Abel, J.; Estes, M.K.; Zachos, N.C.; Kovbasnjuk, O.; Donowitz, M. Human mini-guts: New insights into intestinal physiology and host-pathogen interactions. Nat. Rev. Gastroenterol. Hepatol. 2016, 13, 633–642. [Google Scholar] [CrossRef] [Green Version]
- Nusse, R.; Clevers, H. Wnt/beta-Catenin Signaling, Disease, and Emerging Therapeutic Modalities. Cell 2017, 169, 985–999. [Google Scholar] [CrossRef]
- Laurent-Puig, P.; Beroud, C.; Soussi, T. APC gene: Database of germline and somatic mutations in human tumors and cell lines. Nucleic Acids Res. 1998, 26, 269–270. [Google Scholar] [CrossRef] [PubMed]
- Burke, J.F.; Mogg, A.E. Suppression of a nonsense mutation in mammalian cells in vivo by the aminoglycoside antibiotics G-418 and paromomycin. Nucleic Acids Res. 1985, 13, 6265–6272. [Google Scholar] [CrossRef] [PubMed]
- Crespo, M.; Vilar, E.; Tsai, S.Y.; Chang, K.; Amin, S.; Srinivasan, T.; Zhang, T.; Pipalia, N.H.; Chen, H.J.; Witherspoon, M.; et al. Colonic organoids derived from human induced pluripotent stem cells for modeling colorectal cancer and drug testing. Nat. Med. 2017, 23, 878–884. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Patient/Isolation | Gender | Genetic Background | Growth (days) | Structure (days) | ||
|---|---|---|---|---|---|---|
| H | P | H | P | |||
| 1-1 | F | APC ns S874X | 3 | 21 | U | B (1–15); C (15–21) |
| 1-2 | 43 | 43 | B (1–43) | B (1–15); C (15–43) | ||
| 2 | F | APC ns L77X | X * | 15 | X | U (1–15) |
| 3 | M | APC ns L77X | − | 20 | − | B&C (1–20) |
| 4-1 | M | APC ns Q341X | 7 | − | C (1–7) | − |
| 4-2 | 3 | 39 | X | B&C (1–39) | ||
| 5-1 | M | APC ns Q341X | 3 * | 14 | X | C (1–14) |
| 5-2 | 27 | − | C (1–27) | − | ||
| 6-1 | M | APC ns R302X | 6 * | 6 * | C (1–6) | X |
| 6-2 | 7 | 19 | B (1–7) | B (1–19) | ||
| 6-3 | 8 | 22 | B (1–8) | B&C (1–22) | ||
| 7 | F | APC fs Z1379R | 21 | 29 | C (1–21) | B&C (1–29) |
| 8 | F | APC fs Q264 | 5 | 19 | C (1–5) | B (1–19) |
| 9-1 | APC fs R332 | 4 * | 7 * | U | B (1–7) | |
| 9-2 | 8 | 8 | B (1–8) | B (1–8) | ||
| 10 | F | APC fs 2688delC in exon 15 | 10 | − | B (1–10) | − |
| 11 | M | MYH G382D −/− | X | 39 | X | B (1–11); C (11–39) |
| 12 | MYH 1145G>A +/− RAD50 326_329delCAGA +/− | 7 | 21 | B&C (1–7) | B&C (1–21) | |
| 13 | M | POLD1 V759I +/− | − | 9 | − | 2D (7<) |
| 14 | PTEN 697C>T +/− | − | 25 | − | 2D (23<) | |
| 15 | M | Ulcerative Colitis | 7 | − | U (1–7) | − |
| 16 | M | Unknown | 3 | 36 | U | B (1–11); C (11-36) |
| 17 | Unknown | X | 36 | X | B (1–18); C (18–36) | |
| 18 | F | Unknown | 7 | 7 | B (1–7) | B (1–7) |
| Gene Symbol | Fw Primer | Rv Primer |
|---|---|---|
| GAPDH | GCACCGTCAAGGCTGAGAAC | ATGGTGGTGAAGACGCCAGT |
| MYC | CTCGGTGGTCTTCCCCTACCCT | TGTCCAACTTGACCCTCTTGGC |
| AXIN2 | GCAGCTCAGCAAAAAGGGAAAT | TACATCGGGAGCACCGTCTCAT |
| CCND1 | CATCTACACCGACAACTCCATC | TCTGGCATTTTGGAGAGGAAG |
| SOX9 | ACTTGCACAACGCCGAG | CTGGTACTTGTAATCCGGGTG |
| LEF1 | AGACAAGCACAAACCTCTCAG | TCATTATGTACCCGGAATAACTC |
| ACTIN | CCTGGCACCCAGCACAAT | GGGCCGGACTCGTCATACT |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Evron-Levy, T.; Caspi, M.; Wittenstein, A.; Shorer-Arbel, Y.; Shomron, O.; Hirschberg, K.; Kariv, R.; Rosin-Arbesfeld, R. Adenoma-Derived Organoids for Precision Therapy. Organoids 2022, 1, 54-68. https://doi.org/10.3390/organoids1010006
Evron-Levy T, Caspi M, Wittenstein A, Shorer-Arbel Y, Shomron O, Hirschberg K, Kariv R, Rosin-Arbesfeld R. Adenoma-Derived Organoids for Precision Therapy. Organoids. 2022; 1(1):54-68. https://doi.org/10.3390/organoids1010006
Chicago/Turabian StyleEvron-Levy, Tamar, Michal Caspi, Amnon Wittenstein, Yamit Shorer-Arbel, Olga Shomron, Koret Hirschberg, Revital Kariv, and Rina Rosin-Arbesfeld. 2022. "Adenoma-Derived Organoids for Precision Therapy" Organoids 1, no. 1: 54-68. https://doi.org/10.3390/organoids1010006