
Programmable Proteins: Target Specificity, Programmability and Future Directions


Department of Molecular and Cellular Biology, Harvard University, Cambridge, MA 02138, USA
SynBio 2023, 1(1), 65-76; https://doi.org/10.3390/synbio1010005
Submission received: 17 October 2022
/
Revised: 25 October 2022
/
Accepted: 27 October 2022
/
Published: 28 October 2022
(This article belongs to the Special Issue Programmable Proteins in Synthetic Biology)
{kind=link}
Abstract
:Programmable proteins to detect, visualize, modulate, or eliminate proteins of selection in vitro and in vivo are essential to study the targets recognized and the biology that follows. The specificity of programmable proteins can be easily altered by designing their sequences and structures. The flexibility and modularity of these proteins are currently pivotal for synthetic biology and various medical applications. There exist numerous reviews of the concept and application of individual programmable proteins, such as programmable nucleases, single-domain antibodies, and other protein scaffolds. This review proposes an expanded conceptual framework of such programmable proteins based on their programmable principle and target specificity to biomolecules (nucleic acids, proteins, and glycans) and overviews their advantages, limitations, and future directions.
Keywords:
programmable protein; nuclease; CRISPR; TALEN; Cas9; Argonaute; nanobody; DARPin; glycan-binding protein; lectin1. Introduction: What Are Programmable Proteins?
The term “programmable” is generally used to describe a computer or machine that can accept a set of instructions or rules to perform a range of tasks as intended. The instructions or rules are usually written in a particular programming language. In biological organisms, “programmable” refers to the artificial modification of biomolecules and intermolecular and intercellular circuits, which leads to the conversion of specificity and functions. The function of nucleic acids and proteins is programmable by changing their sequences. With the superior programmability of DNA molecules, it is possible to confer unique chemical properties on nucleic acids [1]. Aptamers are designable oligonucleotide sequences capable of recognizing various molecules with specificity and affinity that rival those of antibodies [2,3,4]. Riboswitches, ribozymes, and deoxyribozymes are functional nucleic acids potentially designable by changing their nucleotide sequences [5,6]. However, functions such as fluorogenicity, ligand-binding, and catalytic activity are still not entirely predictable without the aid of the systematic evolution of ligands by exponential enrichment (SELEX) [7].
Proteins are important biomolecules that have diverse chemical characteristics and biological roles. Protein-biomolecule interactions play pivotal roles in biology, and approaches to designing proteins that inhibit or change these interactions would have great utility. The recent deep learning approach, including AlphaFold2, indicated that the structure of proteins can be computationally predictable [8,9]. In some cases, new binding proteins can be designed using only the knowledge of the structure of the target, without requiring prior knowledge of binding [10,11,12]. This raises the possibility that diverse proteins will become artificially programmable as functional proteins in the near future. However, in the meantime, the templates used for programmable proteins are limited. As summarized in Figure 1, current programmable proteins can be categorized into two major types: nucleic-acid-guided and protein-guided. They can also be classified based on the targets they interact with: nucleic acid, protein, and other molecules, including glycans.
Such programmable proteins are widely used in biology, synthetic biology, and medicine. Numerous applications of the CRISPR/Cas9 (clustered regularly interspaced short palindromic repeats, CRISPR-associated protein 9) system are good examples [13,14,15,16]. Accordingly, there already exist copious reviews of individual programmable proteins such as programmable nucleases and single-domain antibodies. Moreover, the field is rapidly advancing. Thus, any review articles on this topic will be quickly outdated. Instead, this review proposes to extend the idea of programmable proteins to other unique binding molecules and overviews their advantages, shortcomings, and future directions.
2. Programmable Nucleases, Modifiers, and Nucleic Acid-Binding Proteins
Several types of programmable nucleases of natural and synthetic origin have been reported [17,18,19,20]. Two groups of such enzymes are used for genome engineering: protein-guided nucleases that recognize the specific sequence using protein module–DNA interactions (e.g., TALEN: Section 2.1) and nucleic-acid-guided nucleases that recognize the specific sequence via an attached short complementary DNA or RNA (e.g., Cas9: Section 2.2, Section 2.3, Section 2.4, Section 2.5 and Section 2.6).
2.1. ZFNs and TALENs
The first proof-of-concept programmable nuclease was an artificial hybrid deoxyribonuclease produced by connecting the DNA-binding homeodomain (Drosophila Ultrabithorax) to a non-specific DNA cleavage domain of Fok1 [21]. Fok1, discovered in Flavobacterium okeanokoites, is a type IIS restriction endonuclease consisting of an N-terminal DNA-binding domain and a DNA cleavage domain (~200 amino acids) at the C-terminus. However, earlier approaches to create programmable nucleases such as meganucleases did not gain popularity because of their technical limitations (e.g., modification of homing enzymes and FEN1 (Flap structure-specific endonuclease-1)) [19,20,22]. Subsequently, engineered zinc-finger nucleases (ZFNs) and transcription activator-like effector nucleases (TALENs) were invented as two types of protein-guided programmable nucleases.
ZFNs use a programmable DNA binding protein recognizing ~3 bp DNA. ZFNs form dimers from monomers composed of an endonuclease FokI domain fused to a zinc finger array programmed to recognize a specific target DNA sequence [23]. The DNA-binding domain of a ZFN is usually composed of 3–4 zinc finger arrays. Developing the methods used to create new ZFNs has addressed many of the technical challenges; however, it remains a limitation of ZFNs.
TALENs are constructed similarly to ZFNs, composed of a DNA-binding domain and a FokI DNA cleavage domain [24]. They are derived from naturally occurring plant bacterial pathogens of the genus Xanthomonas and contain DNA-binding proteins called TALEs [25]. Each TALE is ~34 amino acids long and recognizes a single base pair of DNA, as opposed to a triplet for ZFNs, giving TALENs higher flexibility over ZFNs. TALEs are tandemly connected to form a chain capable of targeting a specific DNA sequence. However, constructing a TALEN array requires the assembly of multiple, nearly identical repeat sequences, which is technically demanding. This issue has led to the development of several elegant laboratory methods. Although nucleic-acid-guided CRISPRs are widely used programmable nucleases, protein-guided TALENs show fewer off-targets and target the mitochondrial DNA, where guide RNA of CRISPR is difficult to import [26].
2.2. Cas9 (Type II CRISPR-Cas)
ZFNs and TALENs use a strategy of linking endonuclease domains to building block-guided DNA-binding modules for inducing targeted DNA double-stranded breaks (DSBs). In contrast, Cas9 is a nucleic-acid-guided nuclease through base-pairing with target DNA, offering a system that makes it easier to design specific, efficient, and well-suited for high-throughput and multiplexed gene editing for diverse cell types and organisms. The CRISPR-Cas9 strategy originated from a naturally occurring genome editing system that bacteria and archaeon use for adaptive immunity to viruses and plasmids [27,28].
The CRISPR/Cas9 system uses a Cas9 monomeric nuclease from various bacterial species (e.g., Streptococcus pyogenes Cas9 (SpCas9), Staphylococcus aureus Cas9 (SaCas9), Streptococcus thermophilus Cas9 (St1Cas9 and St3Cas9), Campylobacter jejuni Cas9 (CjCas9), Neisseria meningitides (NmCas9)), a specificity-determining CRISPR RNA (crRNA) and an auxiliary trans-activating RNA (tracrRNA). crRNA and tracrRNA are used as a dual RNA or single-guide RNA (sgRNA), which are synthesized in vitro or in vivo [13,14,15,16]. Although SpCas9 is the most widely used, Cas9, SaCas9, CjCas9, and NmCas9 are smaller than SpCas9, allowing packaging into adeno-associated viral vectors. Cas9 contains an HNH nuclease domain that cuts the DNA strand complementary to the guide RNA (target strand), and a RuvC nuclease domain required for cutting the noncomplementary strand (non-target strand), resulting in DSBs. The most widely characterized SpCas9-RNA complex recognizes the strands, including a protospacer adjacent motif (PAM), NGG. Among the targeted 20 bps in the upstream of the PAM, 8 to 12 bps are critical for recognition. The blunt-ended cleavage site is at the third bps upstream of the PAM. DSBs were subsequently repaired by non-homologous end joining (NHEJ) or homology-directed repair (HDR) [29]. By electroporating crRNA-Cas9 protein complexes, GFP reporters can be somatically integrated into specific genes in the chick genome by HDR, showing that this approach is possible in a variety of species [30].
2.3. Cas12 (Type V CRISPR-Cas)
CRISPR-Cas12a (Cpf1) is another CRISPR/Cas system that has diversified the genome editing toolbox [34,35]. Compared with CRISPR/Cas9, the CRISPR/Cas12a system has a smaller size, requires only crRNA and no tracrRNA, uses a T-rich PAM, and creates sticky ends at the cut site. It functions as both a deoxyribonuclease and a ribonuclease that can process multiple functional crRNAs from a single transcript. Thus, CRISPR/Cas9 uses RNA guides to recognize and cleave DNA (R-D nuclease), but CRISPR/Cas12a uses RNA guides to recognize and cleave both DNA and RNA (R-D/R nuclease). The most commonly used Cas12a originates from Francisella novicida (FnCas12a), Acidaminococcus sp. (AsCas12a), and Lachnospiraceae bacterium (LbCas12a).
Cas12b (also known as C2c1) proteins are smaller than Cas9 and Cas12a. Similar to Cas9, Cas12b requires both crRNA and tracrRNA, which can be combined as sgRNA, for DNA targeting. The recent development of mesophilic Cas12b from Alicyclobacillus acidiphilus (AaCas12b) and Bacillus hisashii (BhCas12b) can be adapted for mammalian genome editing [36,37].
2.4. Cas13 (Type VI CRISPR-Cas)
Cas13 (also known as C2c2) is an RNA-guided ribonuclease (R-R nuclease) that uses a crRNA to identify its target, single-stranded RNA (cis-recognition and cleavage), and exhibits trans-cleavage ribonuclease activity (trans-cleavage/collateral effect), holding promise for RNA gene silencing comparable to RNAi or CRISPRi without changing the genome sequence [38,39,40]. There are four subtypes identified in the Cas13 family, including Cas13a, Cas13b, Cas13c, and Cas13d. All Cas13 family members are smaller than Cas9 and require a crRNA to ensure target specificity. Like Cas12a, this nucleic-acid-guided nuclease has often been used for sequence-specific detection of RNA or DNA targets for diagnostics [41].
2.5. OMEGA (TnpB, IscB)
IscB and TnpB are recently characterized RNA-guided deoxyribonucleases (R-D nuclease), likely to be the ancestral forms of Cas9 and Cas12, respectively. IscB was found in a distinct family of prokaryotic IS200/IS605 transposons [42]. TnpB was from the same transposon in an extremophilic bacterium Deinococcus [42,43]. IscB and TnpB proteins IscB and TnpB are guided by non-coding short ωRNA and right end RNA (reRNA) encoded by the transposon. Both transposon-encoded RNA-guided deoxyribonucleases cleave dsDNA in human cells and expand the genome-editing toolbox by providing a new group of small and programmable non-Cas nucleases. Feng Zhang’s group proposed calling these widespread nucleases OMEGA (obligate mobile element-guided activity) [43].
2.6. Argonaute
Ago (Argonaute) proteins are the second type of nucleic-acid-guided programmable proteins [20,44]. Similar to bacterial CRISPR, Ago plays a pivotal role in genetic immune systems that protect host cells from invading nucleic acids in eukaryotes and prokaryotes. Eukaryotic Argonaute proteins (eAgos) play a role in RNA interference (RNAi) and use guide RNAs for the recognition of RNA targets (R-R nuclease). In contrast, prokaryotic Ago (pAgo) nucleases have a natural specificity for DNA guides and DNA targets (D-D nuclease), and a small group of CRISPR-associated pAgos are programmable with DNA guides or RNA guides to cleave DNA targets (D/R-D nuclease).
However, no success has been reported using Ago for programmable genome editing in mammalian cells. This is because pAgos initially characterized from thermophilic prokaryotes are most effective at high temperatures (>65 °C) but not at 37 °C (e.g., pAgos from Thermus thermophilus (TtAgo), Methanocaldococcus jannaschii (MjAgo) and Pyrococcus furiosus (PfAgo)). Nonetheless, the diverse structures and functions of pAgo proteins in various prokaryotes suggest that they will soon provide the next-generation tools for genome editing along with Cas nucleases. Some pAgo proteins are indeed demonstrated to cut DNA sequences at 37 °C in a DNA guide-dependent manner (e.g., pAgos from Clostridium perfringens (CpAgo) and Intestinibacter bartlettii (IbAgo)) [45]. If the attempt is successful, it could expand the range of CRISPR-Cas9 tools, whose application is often limited owing to the tolerance of guide-target mismatches, possible RNA secondary structures, and the PAM requirement.
3. Programmable Protein-Binding Proteins
In addition to aiming at nucleic acids, diverse classes of programmable proteins can detect, disrupt, or modulate protein interactions that have essential roles in biology [46,47,48,49]. Antibodies are old immunological tools with various applications. In particular, with the advent of hybridoma and recombinant DNA technologies, successful applications of monoclonal antibodies and recombinant antibodies including single-chain variable fragments (scFvs) have inspired the development of diverse types of immunological reagents and therapeutic drugs [50]. The invention of in vitro selection methods such as phage display, yeast display, mRNA display, ribosome display, directed evolution, and affinity maturation have not only enabled further antibody engineering but also facilitated the development of novel binding proteins [51,52,53,54,55]. However, they principally differ from nucleic-acid-guided programmable proteins in that their precise specificity is not readily predictable. To date, the efficient programmability depends on various display methods and directed evolution in vitro. Nonetheless, emerging computational approaches raise the possibility that they will become programmable [10,11,12,56].
The first class of such programmable binding proteins is a single-chain fragment from an unusual antibody called VHH or nanobody (Section 3.1). The second class is based on a protein scaffold that offers two structural features that were viewed as the hallmark of immunoglobulins: a variable segment that provides the structural adaptability to design novel binding sites and a constant region that offers folding stability. Such bipartite building blocks are achievable when starting from a domain architecture that already exhibits variable loop motifs. Thus, repetitive domains such as ankyrin repeats and fibronectin type III (FN3) repeats appeared captivating as a robust scaffold for a general binding protein. Accordingly, a series of protein-guided programmable proteins such as DARPins and monobodies were created (Section 3.2 and Section 3.3). The third class is based on ligand-binding unrepeated proteins with high thermal and proteolytic stability. Programmable proteins dubbed affibodies and anticalins were derived from Staphylococcus aureus protein A and a family of transport proteins, lipocalins, respectively (Section 3.4 and Section 3.5). This third class is typically used as a single protein, although the first and second classes are frequently applied as a recombinant fusion protein as well as a solitary protein.
There are wide-ranging examples of constructing programmable proteins (e.g., αRep proteins containing HEAT-like repeats [57], repebodies based on leucine-rich repeats [58], Fynomers based on SH3 motifs in Fyn [59], affimer/adhirons based on cystatins [60], affilins based on ubiquitin/gamma-B crystallin [61], avimers based on various membrane receptors [62], and Kunitz domain peptides [47], among others). These scaffolds expanded the repertoire of programmable protein scaffolds and have provided significant added value regarding their diverse chemical properties, pharmacokinetics, and penetrance to unique tissues [46,47,48,49]. Here, I briefly focus on five protein-guided programmable proteins as exemplars because they have been demonstrated as practically useful.
3.1. Single-Domain Antibody, Nanobody, VHH
Single-domain antibodies, also called nanobodies, are small antigen-binding polypeptides having a molecular weight of ~15 kDa and ~2–4 nm in size. They comprise the variable domain of a heavy chain-only antibody (VHH), which was first described in the serum of camelids (camels and llamas) in the 1990s [63]. Similar single-chain antibodies are found in cartilaginous fishes (VNAR, from sharks) [64,65].
Thus, nanobodies are the smallest intact antigen-binding protein fragments derived from an active immunoglobulin [66,67]. Nanobodies are advantageous alternatives to conventional antibodies owing to their tiny size, high solubility, and high stability across a variety of applications. Furthermore, phage display, ribosome display, and mRNA display methods can be used for the efficient generation and optimization of binding molecules in vitro. The nanobodies can be genetically encoded, tagged, and expressed as recombinant intrabodies in cells or reporter fusion bodies for in vivo localization and functional studies of target proteins [68,69,70]. There are currently several nanobodies undergoing clinical trials, and one was approved by the FDA for acquired thrombotic thrombocytopenic purpura in 2019 [71].
3.2. DARPin
Designed ankyrin repeat proteins (DARPins) are genetically engineered antibody mimetic proteins typically exhibiting highly specific and high-affinity target protein binding [72,73]. Most natural ankyrin repeat (AR) proteins contain 4–6 ARs stacked onto each other. DARPins contain 2–3 internal ARs sandwiched between the N- and C-terminal capping modules. Each internal AR module consists of 27 defined framework residues and 6 potential protein-binding residues that form a β-turn followed by two antiparallel helices and a loop connecting to the β-turn of the next AR [74]. DARPins are small in size (14–18 kDa, depending on the number of internal ARs), thermostable, resistant to proteases and chemical denaturants, and can be expressed in bacteria. DARPins have a concave binding interface, adding value by expanding the synthetic ligand landscape because their binding interface differs from the nanobodies’ convex interface [75].
3.3. Monobody, Adnectin®, FingR
Monobodies were initially designed based on the tenth FN3 domain, which has an immunoglobulin β-sandwich fold with seven strands connected by six loops but no disulfide bonds [76,77]. The original tenth FN3 consists of 94 amino acids, has a molecular mass of ~10 kDa, and contains the adhesive RGD sequence that binds integrins. Three of the six flexible loops on one side of the FN3 are surface-exposed and have been shown to be a synthetic interface for binding ligands of interest. Subsequently, their commercial equivalence, Adnectin®, and several monobody-like scaffolds have been developed [77,78], reiterating the robustness of the FN3 approach for creating programmable proteins. Various display techniques have selected ligand-binding proteins with binding affinities in the nanomolar to picomolar range. Like nanobody-based intrabodies, monobody-derived proteins can also be used intracellularly (e.g., FingR (=Monobody) fused to GFP that colocalized to endogenous PSD95 at neuronal synapses [79]).
Note: Adnectin® is a registered trademark of Adnexus, a Bristol-Myers Squibb R&D Company.
3.4. Affibody
Affibodies are designed based on the Z-domain of the immunoglobulin-binding region of Staphylococcus aureus protein A [80,81], which adopts a three-helix scaffold and contains no disulfide bonds. The programmable ligand-binding surface is composed of 13 amino acid residues scattered between two of the helices. Their compactness (58 amino acids, ~7 kDa in size) allows them to be simply expressed in bacteria or produced by chemical synthesis.
3.5. Anticalin®
Anticalin® was designed based on lipocalins, a large group of secreted proteins that typically transport or store small compounds, including vitamins, steroids, odorants, and various metabolites [82,83]. The anticalin scaffold adopts an eight-stranded antiparallel β-barrel, which is open to the solvent at one end and contains 160–180 amino acids (~20 kDa in size). Anticalins are not glycosylated and possess no disulfide bonds. An anticalin library for screening ligand-binding proteins contains 16–24 randomized amino acids in each loop [82]. Ligand-specific anticalins have been engineered via phage and bacterial surface displays and are expressed in either bacteria or yeasts [82].
Note: Anticalin® is a registered trademark of Pieris Pharmaceuticals.
4. Glycan-Binding Proteins (GBPs)
Glycans—a general term describing carbohydrates, including oligosaccharides and polysaccharides—are the third class of essential biological macromolecules, following nucleic acids and proteins [84]. They exist as free sugars but are more commonly found as complex glycoconjugates, including glycoproteins, proteoglycans, and glycolipids. The broad importance of glycans has driven the interest in developing new glycan-binding molecules [85,86,87].
Glycan-binding proteins (GBPs) include antibodies, lectins, pseudoenzymes, and carbohydrate-binding modules. Antibodies against glycans are also seen in nature, and many monoclonal antibodies are remarkably useful reagents for recognizing the specific structure of glycans [88]. However, glycans are poorly immunogenic in general. On the other hand, lectins are non-immunoglobulin proteins containing at least one non-catalytic domain that often displays a specific glycan binding. For example, numerous plant lectins that recognize distinct glycans have been used as tools for detecting glycans [88,89,90]. GBPs also include carbohydrate-binding modules that are similar to lectins but are small binding domains typically found in carbohydrate-active enzymes such as glycosidases and glycosyltransferases [91]. Some carbohydrate-active enzymes have evolved to pseudoenzymes that have lost their enzymatic activity but retain their glycan-binding feature, providing a potential GBP scaffold [92].
Numerous pioneering works in lectin engineering have changed the binding specificity of eukaryotic and prokaryotic lectins. For example, using random mutagenesis and ribosome display techniques, a novel sialic acid-binding protein was created from a galactose-binding R-type lectin (galectin) from earthworms [93]. A sugar-binding spectrum of two different mushroom (Agrocybe cylindracea and Aleuria aurantia) lectins was also changed by mutagenesis [94,95]. These earlier studies demonstrated that new GBPs could be generated [96,97], as in the case of naturally occurring legume lectins [98], supporting the idea that lectins are potentially programmable. Moreover, other common protein scaffolds, including the protein-guided programmable proteins described in Section 3, can be employed to acquire a novel glycan-binding specificity. Lastly, a plethora of new computational and data science approaches can be used to expand the toolboxes to distinguish further complex glycans [90,92,99], along with the state-of-the-art progress in glycoconjugates research [84].
5. Perspectives
Programmable proteins to bind, label, inhibit, or remove biomolecules of choice in vitro and in vivo are critical for understanding the fundamental roles of the biomolecules recognized. The specificity of programmable proteins can be flexibly designed by changing their sequences in vitro. In particular, with the superb synthesizability of DNA, the programmability of nucleic-acid-guided proteins for recognizing specific nucleic acid sequences is practically finalized, although many significant issues such as off-target and inefficiency remain unsolved. In contrast, the programmability of protein-guided programmable proteins is still in development, except for building-block-guided modular nucleases. Nonetheless, the emerging deep learning approach that includes AlphaFold2 demonstrates that many protein structures can be accurately predictable in silico [8]. Thus, in limited cases, binding proteins were designed using only information on the structure of the target [10,11,12], raising the possibility that diverse proteins will be designable as useful to recognizing specific sequences and structures in proteins or glycans. Developing such computational algorithms and massive, open-source databases will be the key to creating a cutting-edge armamentarium in synthetic biology.
This review focused on programmable proteins that recognize three important biopolymers: nucleic acid, protein, and glycan. However, there are different types of programmablity. For example, a unique group of programmable proteins are enzymes involved in the biosynthesis of bioactive natural products, such as polyketides, non-ribosomal peptides (NRPs), and ribosomally synthesized and post-translationally modified peptides (RiPPs), widespread throughout bacteria, fungi, and plants [100,101,102]. These biosynthetic enzymes produce diverse natural products using a modular synthetic scheme that resembles an assembly pipeline. The modularity of these megaenzymes is useful from a synthetic biology viewpoint. The substitution or rearrangement of modules can lead to new natural products, suggesting that the biosynthesis of bioactive small molecules is programmable [101,102,103]. Taken together, the flexibility and modularity of programmable proteins are crucial for industrial and medical applications such as diagnostic reagents and therapeutic drugs.
Funding
This research received no external funding.
Conflicts of Interest
The author has no conflict of interest directly relevant to the content of this article.
References
- Micura, R.; Höbartner, C. Fundamental studies of functional nucleic acids: Aptamers, riboswitches, ribozymes and DNAzymes. Chem. Soc. Rev. 2020, 49, 7331–7353. [Google Scholar] [CrossRef] [PubMed]
- Sun, W.; Du, L.; Li, M. Aptamer-based carbohydrate recognition. Curr. Pharm. Des. 2010, 16, 2269–2278. [Google Scholar] [CrossRef]
- Nimjee, S.M.; White, R.R.; Becker, R.C.; Sullenger, B.A. Aptamers as Therapeutics. Annu. Rev. Pharmacol. Toxicol. 2017, 57, 61–79. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Rossi, J. Aptamers as targeted therapeutics: Current potential and challenges. Nat. Rev. Drug Discov. 2017, 16, 181–202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, L.; Liu, J. Catalytic Nucleic Acids: Biochemistry, Chemical Biology, Biosensors, and Nanotechnology. iScience 2020, 23, 100815. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Breaker, R.R. The Biochemical Landscape of Riboswitch Ligands. Biochemistry 2022, 61, 137–149. [Google Scholar] [CrossRef]
- Lee, G.; Jang, G.H.; Kang, H.Y.; Song, G. Predicting aptamer sequences that interact with target proteins using an aptamer-protein interaction classifier and a Monte Carlo tree search approach. PLoS ONE 2021, 16, e0253760. [Google Scholar] [CrossRef]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Židek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with Alpha Fold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef]
- Baek, M.; DiMaio, F.; Anishchenko, I.; Dauparas, J.; Ovchinnikov, S.; Lee, G.R.; Wang, J.; Cong, Q.; Kinch, L.N.; Schaeffer, R.D.; et al. Accurate prediction of protein structures and interactions using a three-track neural network. Science 2021, 373, 871–876. [Google Scholar] [CrossRef]
- Pan, X.; Kortemme, T. Recent advances in de novo protein design: Principles, methods, and applications. J. Biol. Chem. 2021, 296, 100558. [Google Scholar] [CrossRef]
- Cao, L.; Coventry, B.; Goreshnik, I.; Huang, B.; Sheffler, W.; Park, J.S.; Jude, K.M.; Marković, I.; Kadam, R.U.; Verschueren, K.H.G.; et al. Design of protein-binding proteins from the target structure alone. Nature 2022, 605, 551–560. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Lisanza, S.; Juergens, D.; Tischer, D.; Watson, J.L.; Castro, K.M.; Ragotte, R.; Saragovi, A.; Milles, L.F.; Baek, M.; et al. Scaffolding protein functional sites using deep learning. Science 2022, 377, 387–394. [Google Scholar] [CrossRef] [PubMed]
- Doudna, J.A.; Charpentier, E. Genome editing. The new frontier of genome engineering with CRISPR-Cas9. Science 2014, 346, 1258096. [Google Scholar] [CrossRef] [PubMed]
- Hsu, P.D.; Lander, E.S.; Zhang, F. Development and applications of CRISPR-Cas9 for genome engineering. Cell 2014, 157, 1262–1278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adli, M. The CRISPR tool kit for genome editing and beyond. Nat. Commun. 2018, 9, 1911. [Google Scholar] [CrossRef] [Green Version]
- Anzalone, A.V.; Koblan, L.W.; Liu, D.R. Genome editing with CRISPR-Cas nucleases, base editors, transposases and prime editors. Nat. Biotechnol. 2020, 38, 824–844. [Google Scholar] [CrossRef]
- Gaj, T.; Gersbach, C.A.; Barbas, C.F., 3rd. ZFN, TALEN, and CRISPR/Cas-based methods for genome engineering. Trends Biotechnol. 2013, 31, 397–405. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.; Kim, J.S. A guide to genome engineering with programmable nucleases. Nat. Rev. Genet. 2014, 15, 321–334. [Google Scholar] [CrossRef]
- Chandrasegaran, S.; Carroll, D. Origins of Programmable Nucleases for Genome Engineering. J. Mol. Biol. 2016, 428, 963–989. [Google Scholar] [CrossRef] [Green Version]
- Kropocheva, E.V.; Lisitskaya, L.A.; Agapov, A.A.; Musabirov, A.A.; Kulbachinskiy, A.V.; Esyunina, D.M. Prokaryotic Argonaute Proteins as a Tool for Biotechnology. Mol. Biol. 2022, 1–20. [Google Scholar] [CrossRef]
- Kim, Y.G.; Chandrasegaran, S. Chimeric restriction endonuclease. Proc. Natl. Acad. Sci. USA 1994, 91, 883–887. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hafez, M.; Hausner, G. Homing endonucleases: DNA scissors on a mission. Genome 2012, 55, 553–569. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Urnov, F.D.; Rebar, E.J.; Holmes, M.C.; Zhang, H.S.; Gregory, P.D. Genome editing with engineered zinc finger nucleases. Nat. Rev. Genet. 2010, 11, 636–646. [Google Scholar] [CrossRef] [PubMed]
- Joung, J.K.; Sander, J.D. TALENs: A widely applicable technology for targeted genome editing. Nat. Rev. Mol. Cell Biol. 2013, 14, 49–55. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boch, J.; Scholze, H.; Schornack, S.; Landgraf, A.; Hahn, S.; Kay, S.; Lahaye, T.; Nickstadt, A.; Bonas, U. Breaking the code of DNA binding specificity of TAL-type III effectors. Science 2009, 326, 1509–1512. [Google Scholar] [CrossRef]
- Bhardwaj, A.; Nain, V. TALENs-an indispensable tool in the era of CRISPR: A mini review. J. Genet. Eng. Biotechnol. 2021, 19, 125. [Google Scholar] [CrossRef]
- Nussenzweig, P.M.; Marraffini, L.A. Molecular Mechanisms of CRISPR-Cas Immunity in Bacteria. Annu. Rev. Genet. 2020, 54, 93–120. [Google Scholar] [CrossRef]
- Koonin, E.V.; Makarova, K.S. Evolutionary plasticity and functional versatility of CRISPR systems. PLoS Biol. 2022, 20, e3001481. [Google Scholar] [CrossRef]
- Chiruvella, K.K.; Liang, Z.; Wilson, T.E. Repair of double-strand breaks by end joining. Cold Spring Harb. Perspect. Biol. 2013, 5, a012757. [Google Scholar] [CrossRef]
- Yamagata, M.; Sanes, J.R. CRISPR-mediated Labeling of Cells in Chick Embryos Based on Selectively Expressed Genes. Bio Protoc. 2021, 11, e4105. [Google Scholar] [CrossRef]
- Kampmann, M. CRISPRi and CRISPRa Screens in Mammalian Cells for Precision Biology and Medicine. ACS Chem. Biol. 2018, 13, 406–416. [Google Scholar] [CrossRef] [PubMed]
- Knott, G.J.; Doudna, J.A. CRISPR-Cas guides the future of genetic engineering. Science 2018, 361, 866–869. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakamura, M.; Gao, Y.; Dominguez, A.A.; Qi, L.S. CRISPR technologies for precise epigenome editing. Nat. Cell Biol. 2021, 23, 11–22. [Google Scholar] [CrossRef]
- Zetsche, B.; Gootenberg, J.S.; Abudayyeh, O.O.; Slaymaker, I.M.; Makarova, K.S.; Essletzbichler, P.; Volz, S.E.; Joung, J.; van der Oost, J.; Regev, A.; et al. Cpf1 is a single RNA-guided endonuclease of a class 2 CRISPR-Cas system. Cell 2015, 163, 759–771. [Google Scholar] [CrossRef] [Green Version]
- Safari, F.; Zare, K.; Negahdaripour, M.; Barekati-Mowahed, M.; Ghasemi, Y. CRISPR Cpf1 proteins: Structure, function and implications for genome editing. Cell Biosci. 2019, 9, 36. [Google Scholar] [CrossRef] [Green Version]
- Teng, F.; Cui, T.; Feng, G.; Guo, L.; Xu, K.; Gao, Q.; Li, T.; Li, J.; Zhou, Q.; Li, W. Repurposing CRISPR-Cas12b for mammalian genome engineering. Cell Discov. 2018, 4, 63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Strecker, J.; Jones, S.; Koopal, B.; Schmid-Burgk, J.; Zetsche, B.; Gao, L.; Makarova, K.S.; Koonin, E.V.; Zhang, F. Engineering of CRISPR-Cas12b for human genome editing. Nat. Commun. 2019, 10, 212. [Google Scholar] [CrossRef] [Green Version]
- Abudayyeh, O.O.; Gootenberg, J.S.; Konermann, S.; Joung, J.; Slaymaker, I.M.; Cox, D.B.; Shmakov, S.; Makarova, K.S.; Semenova, E.; Minakhin, L.; et al. C2c2 is a single-component programmable RNA-guided RNA-targeting CRISPR effector. Science 2016, 353, aaf5573. [Google Scholar] [CrossRef] [Green Version]
- O’Connell, M.R. Molecular Mechanisms of RNA Targeting by Cas13-containing Type VI CRISPR-Cas Systems. J. Mol. Biol. 2019, 431, 66–87. [Google Scholar] [CrossRef]
- Tang, T.; Han, Y.; Wang, Y.; Huang, H.; Qian, P. Programmable System of Cas13-Mediated RNA Modification and Its Biological and Biomedical Applications. Front. Cell Dev. Biol. 2021, 9, 677587. [Google Scholar] [CrossRef]
- Kaminski, M.M.; Abudayyeh, O.O.; Gootenberg, J.S.; Zhang, F.; Collins, J.J. CRISPR-based diagnostics. Nat. Biomed. Eng. 2021, 5, 643–656. [Google Scholar] [CrossRef] [PubMed]
- Karvelis, T.; Druteika, G.; Bigelyte, G.; Budre, K.; Zedaveinyte, R.; Silanskas, A.; Kazlauskas, D.; Venclovas, Č.; Siksnys, V. Transposon-associated TnpB is a programmable RNA-guided DNA endonuclease. Nature 2021, 599, 692–696. [Google Scholar] [CrossRef] [PubMed]
- Altae-Tran, H.; Kannan, S.; Demircioglu, F.E.; Oshiro, R.; Nety, S.P.; McKay, L.J.; Dlakic, M.; Inskeep, W.P.; Makarova, K.S.; Macrae, R.K.; et al. The widespread IS200/IS605 transposon family encodes diverse programmable RNA-guided endonucleases. Science 2021, 374, 57–65. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Yang, J.; Cho, W.C.; Zheng, Y. Argonaute proteins: Structural features, functions and emerging roles. J. Adv. Res. 2020, 24, 317–324. [Google Scholar] [CrossRef]
- Cao, Y.; Sun, W.; Wang, J.; Sheng, G.; Xiang, G.; Zhang, T.; Shi, W.; Li, C.; Wang, Y.; Zhao, F.; et al. Argonaute proteins from human gastrointestinal bacteria catalyze DNA-guided cleavage of single- and double-stranded DNA at 37 °C. Cell Discov. 2019, 5, 38. [Google Scholar] [CrossRef] [Green Version]
- Škrlec, K.; Štrukelj, B.; Berlec, A. Non-immunoglobulin scaffolds: A focus on their targets. Trends Biotechnol. 2015, 33, 408–418. [Google Scholar] [CrossRef]
- Yu, X.; Yang, Y.P.; Dikici, E.; Deo, S.K.; Daunert, S. Beyond Antibodies as Binding Partners: The Role of Antibody Mimetics in Bioanalysis. Annu. Rev. Anal. Chem. 2017, 10, 293–320. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Richards, D.A. Exploring alternative antibody scaffolds: Antibody fragments and antibody mimics for targeted drug delivery. Drug Discov. Today Technol. 2018, 30, 35–46. [Google Scholar] [CrossRef]
- Gebauer, M.; Skerra, A. Engineered Protein Scaffolds as Next-Generation Therapeutics. Annu. Rev. Pharmacol. Toxicol. 2020, 60, 391–415. [Google Scholar] [CrossRef]
- Lu, R.M.; Hwang, Y.C.; Liu, I.J.; Lee, C.C.; Tsai, H.Z.; Li, H.J.; Wu, H.C. Development of therapeutic antibodies for the treatment of diseases. J. Biomed. Sci. 2020, 27, 1. [Google Scholar] [CrossRef]
- Lipovsek, D.; Plückthun, A. In-vitro protein evolution by ribosome display and mRNA display. J. Immunol. Methods 2004, 290, 51–67. [Google Scholar] [CrossRef] [PubMed]
- Cherf, G.M.; Cochran, J.R. Applications of Yeast Surface Display for Protein Engineering. Methods Mol. Biol. 2015, 1319, 155–175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Packer, M.S.; Liu, D.R. Methods for the directed evolution of proteins. Nat. Rev. Genet. 2015, 16, 379–394. [Google Scholar] [CrossRef] [PubMed]
- Almagro, J.C.; Pedraza-Escalona, M.; Arrieta, H.I.; Pérez-Tapia, S.M. Phage Display Libraries for Antibody Therapeutic Discovery and Development. Antibodies 2019, 8, 44. [Google Scholar] [CrossRef] [Green Version]
- Alfaleh, M.A.; Alsaab, H.O.; Mahmoud, A.B.; Alkayyal, A.A.; Jones, M.L.; Mahler, S.M.; Hashem, A.M. Phage Display Derived Monoclonal Antibodies: From Bench to Bedside. Front. Immunol. 2020, 11, 1986. [Google Scholar] [CrossRef]
- Chauhan, V.M.; Pantazes, R.J. MutDock: A computational docking approach for fixed-backbone protein scaffold design. Front. Mol. Biosci. 2022, 9, 933400. [Google Scholar] [CrossRef]
- Urvoas, A.; Guellouz, A.; Valerio-Lepiniec, M.; Graille, M.; Durand, D.; Desravines, D.C.; van Tilbeurgh, H.; Desmadril, M.; Minard, P. Design, production and molecular structure of a new family of artificial alpha-helicoidal repeat proteins (αRep) based on thermostable HEAT-like repeats. J. Mol. Biol. 2010, 404, 307–327. [Google Scholar] [CrossRef]
- Lee, S.C.; Park, K.; Han, J.; Lee, J.J.; Kim, H.J.; Hong, S.; Heu, W.; Kim, Y.J.; Ha, J.S.; Lee, S.G.; et al. Design of a binding scaffold based on variable lymphocyte receptors of jawless vertebrates by module engineering. Proc. Natl. Acad. Sci. USA 2012, 109, 3299–3304. [Google Scholar] [CrossRef] [Green Version]
- Grabulovski, D.; Kaspar, M.; Neri, D. A novel, non-immunogenic Fyn SH3-derived binding protein with tumor vascular targeting properties. J. Biol. Chem. 2007, 282, 3196–3204. [Google Scholar] [CrossRef] [Green Version]
- Tiede, C.; Bedford, R.; Heseltine, S.J.; Smith, G.; Wijetunga, I.; Ross, R.; AlQallaf, D.; Roberts, A.P.; Balls, A.; Curd, A.; et al. Affimer proteins are versatile and renewable affinity reagents. eLife 2017, 6, e24903. [Google Scholar] [CrossRef]
- Lorey, S.; Fiedler, E.; Kunert, A.; Nerkamp, J.; Lange, C.; Fiedler, M.; Bosse-Doenecke, E.; Meysing, M.; Gloser, M.; Rundfeldt, C.; et al. Novel ubiquitin-derived high affinity binding proteins with tumor targeting properties. J. Biol. Chem. 2014, 289, 8493–8507. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Silverman, J.; Liu, Q.; Bakker, A.; To, W.; Duguay, A.; Alba, B.M.; Smith, R.; Rivas, A.; Li, P.; Le, H.; et al. Multivalent avimer proteins evolved by exon shuffling of a family of human receptor domains. Nat. Biotechnol. 2005, 23, 1556–1561. [Google Scholar] [CrossRef] [PubMed]
- Hamers-Casterman, C.; Atarhouch, T.; Muyldermans, S.; Robinson, G.; Hamers, C.; Songa, E.B.; Bendahman, N.; Hamers, R. Naturally occurring antibodies devoid of light chains. Nature 1993, 363, 446–448. [Google Scholar] [CrossRef]
- Greenberg, A.S.; Hughes, A.L.; Guo, J.; Avila, D.; McKinney, E.C.; Flajnik, M.F. A novel “chimeric” antibody class in cartilaginous fish: IgM may not be the primordial immunoglobulin. Eur. J. Immunol. 1996, 26, 1123–1129. [Google Scholar] [CrossRef]
- Matz, H.; Munir, D.; Logue, J.; Dooley, H. The immunoglobulins of cartilaginous fishes. Dev. Comp. Immunol. 2021, 115, 103873. [Google Scholar] [CrossRef] [PubMed]
- Muyldermans, S. Applications of Nanobodies. Annu. Rev. Anim. Biosci. 2021, 9, 401–421. [Google Scholar] [CrossRef]
- Muyldermans, S. A guide to: Generation and design of nanobodies. FEBS J. 2021, 288, 2084–2102. [Google Scholar] [CrossRef]
- Ingram, J.R.; Schmidt, F.I.; Ploegh, H.L. Exploiting Nanobodies’ Singular Traits. Annu. Rev. Immunol. 2018, 36, 695–715. [Google Scholar] [CrossRef]
- Yamagata, M.; Sanes, J.R. Reporter-nanobody fusions (RANbodies) as versatile, small, sensitive immunohistochemical reagents. Proc. Natl. Acad. Sci. USA 2018, 115, 2126–2131. [Google Scholar] [CrossRef] [Green Version]
- Cheloha, R.W.; Harmand, T.J.; Wijne, C.; Schwartz, T.U.; Ploegh, H.L. Exploring cellular biochemistry with nanobodies. J. Biol. Chem. 2020, 295, 15307–15327. [Google Scholar] [CrossRef]
- Arbabi-Ghahroudi, M. Camelid Single-Domain Antibodies: Promises and Challenges as Lifesaving Treatments. Int. J. Mol. Sci. 2022, 23, 5009. [Google Scholar] [CrossRef] [PubMed]
- Plückthun, A. Designed ankyrin repeat proteins (DARPins): Binding proteins for research, diagnostics, and therapy. Annu. Rev. Pharmacol. Toxicol. 2015, 55, 489–511. [Google Scholar] [CrossRef] [PubMed]
- Schilling, J.; Jost, C.; Ilie, I.M.; Schnabl, J.; Buechi, O.; Eapen, R.S.; Truffer, R.; Caflisch, A.; Forrer, P. Thermostable designed ankyrin repeat proteins (DARPins) as building blocks for innovative drugs. J. Biol. Chem. 2022, 298, 101403. [Google Scholar] [CrossRef] [PubMed]
- Binz, H.K.; Stumpp, M.T.; Forrer, P.; Amstutz, P.; Plückthun, A. Designing repeat proteins: Well-expressed, soluble and stable proteins from combinatorial libraries of consensus ankyrin repeat proteins. J. Mol. Biol. 2003, 332, 489–503. [Google Scholar] [CrossRef]
- Veesler, D.; Dreier, B.; Blangy, S.; Lichière, J.; Tremblay, D.; Moineau, S.; Spinelli, S.; Tegoni, M.; Plückthun, A.; Campanacci, V.; et al. Crystal structure and function of a DARPin neutralizing inhibitor of lactococcal phage TP901-1: Comparison of DARP in and camelid VHH binding mode. J. Biol. Chem. 2009, 284, 30718–30726. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sha, F.; Salzman, G.; Gupta, A.; Koide, S. Monobodies and other synthetic binding proteins for expanding protein science. Protein Sci. 2017, 26, 910–924. [Google Scholar] [CrossRef] [Green Version]
- Akkapeddi, P.; Teng, K.W.; Koide, S. Monobodies as tool biologics for accelerating target validation and druggable site discovery. RSC Med. Chem. 2021, 12, 1839–1853. [Google Scholar] [CrossRef]
- Lipovsek, D. Adnectins: Engineered target-binding protein therapeutics. Protein Eng. Des. Sel. 2011, 24, 3–9. [Google Scholar] [CrossRef] [Green Version]
- Gross, G.G.; Junge, J.A.; Mora, R.J.; Kwon, H.B.; Olson, C.A.; Takahashi, T.T.; Liman, E.R.; Ellis-Davies, G.C.; McGee, A.W.; Sabatini, B.L.; et al. Recombinant probes for visualizing endogenous synaptic proteins in living neurons. Neuron 2013, 78, 971–985. [Google Scholar] [CrossRef] [Green Version]
- Nygren, P.A. Alternative binding proteins: Affibody binding proteins developed from a small three-helix bundle scaffold. FEBS J. 2008, 275, 2668–2676. [Google Scholar] [CrossRef]
- Ståhl, S.; Gräslund, T.; Eriksson Karlström, A.; Frejd, F.Y.; Nygren, P.Å.; Löfblom, J. Affibody Molecules in Biotechnological and Medical Applications. Trends Biotechnol. 2017, 35, 691–712. [Google Scholar] [CrossRef] [PubMed]
- Gebauer, M.; Skerra, A. Anticalins small engineered binding proteins based on the lipocalin scaffold. Methods Enzymol. 2012, 503, 157–188. [Google Scholar] [CrossRef] [PubMed]
- Rothe, C.; Skerra, A. Anticalin® Proteins as Therapeutic Agents in Human Diseases. BioDrugs 2018, 32, 233–243. [Google Scholar] [CrossRef] [Green Version]
- Griffin, M.E.; Hsieh-Wilson, L.C. Tools for mammalian glycoscience research. Cell 2022, 185, 2657–2677. [Google Scholar] [CrossRef] [PubMed]
- Arnaud, J.; Audfray, A.; Imberty, A. Binding sugars: From natural lectins to synthetic receptors and engineered neolectins. Chem. Soc. Rev. 2013, 42, 4798–4813. [Google Scholar] [CrossRef] [PubMed]
- Tommasone, S.; Allabush, F.; Tagger, Y.K.; Norman, J.; Köpf, M.; Tucker, J.H.R.; Mendes, P.M. The challenges of glycan recognition with natural and artificial receptors. Chem. Soc. Rev. 2019, 48, 5488–5505. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ward, E.M.; Kizer, M.E.; Imperiali, B. Strategies and Tactics for the Development of Selective Glycan-Binding Proteins. ACS Chem. Biol. 2021, 16, 1795–1813. [Google Scholar] [CrossRef]
- Cummings, R.D.; Etzler, M.E. Antibodies and Lectins in Glycan Analysis. In Essentials of Glycobiology, 2nd ed.; Varki, A., Cummings, R.D., Esko, J.D., Freeze, H.H., Hart, G.W., Etzler, M.E., Eds.; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 2009; Chapter 45. Available online: https://www.ncbi.nlm.nih.gov/books/NBK1919/ (accessed on 27 October 2022).
- Tsaneva, M.; Van Damme, E.J.M. 130 years of Plant Lectin Research. Glycoconj. J. 2020, 37, 533–551. [Google Scholar] [CrossRef]
- Lundstrøm, J.; Korhonen, E.; Lisacek, F.; Bojar, D. LectinOracle: A Generalizable Deep Learning Model for Lectin-Glycan Binding Prediction. Adv. Sci. 2022, 9, e2103807. [Google Scholar] [CrossRef]
- Armenta, S.; Moreno-Mendieta, S.; Sánchez-Cuapio, Z.; Sánchez, S.; Rodríguez-Sanoja, R. Advances in molecular engineering of carbohydrate-binding modules. Proteins 2017, 85, 1602–1617. [Google Scholar] [CrossRef]
- Warkentin, R.; Kwan, D.H. Resources and Methods for Engineering “Designer” Glycan-Binding Proteins. Molecules 2021, 26, 380. [Google Scholar] [CrossRef] [PubMed]
- Yabe, R.; Itakura, Y.; Nakamura-Tsuruta, S.; Iwaki, J.; Kuno, A.; Hirabayashi, J. Engineering a versatile tandem repeat-type alpha2-6sialic acid-binding lectin. Biochem. Biophys. Res. Commun. 2009, 384, 204–209. [Google Scholar] [CrossRef]
- Hu, D.; Tateno, H.; Sato, T.; Narimatsu, H.; Hirabayashi, J. Tailoring GalNAcα1-3Galβ-specific lectins from a multi-specific fungal galectin: Dramatic change of carbohydrate specificity by a single amino-acid substitution. Biochem. J. 2013, 453, 261–270. [Google Scholar] [CrossRef] [PubMed]
- Norton, P.; Comunale, M.A.; Herrera, H.; Wang, M.; Houser, J.; Wimmerova, M.; Romano, P.R.; Mehta, A. Development and application of a novel recombinant Aleuria aurantia lectin with enhanced core fucose binding for identification of glycoprotein biomarkers of hepatocellular carcinoma. Proteomics 2016, 16, 3126–3136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hirabayashi, J.; Arai, R. Lectin engineering: The possible and the actual. Interface Focus 2019, 9, 20180068. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Notova, S.; Bonnardel, F.; Lisacek, F.; Varrot, A.; Imberty, A. Structure and engineering of tandem repeat lectins. Curr. Opin. Struct. Biol. 2020, 62, 39–47. [Google Scholar] [CrossRef] [Green Version]
- Katoch, R.; Tripathi, A. Research advances and prospects of legume lectins. J. Biosci. 2021, 46, 104. [Google Scholar] [CrossRef]
- Mattox, D.E.; Bailey-Kellogg, C. Comprehensive analysis of lectin-glycan interactions reveals determinants of lectin specificity. PLoS Comput. Biol. 2021, 17, e1009470. [Google Scholar] [CrossRef]
- Montalbán-López, M.; Scott, T.A.; Ramesh, S.; Rahman, I.R.; van Heel, A.J.; Viel, J.H.; Bandarian, V.; Dittmann, E.; Genilloud, O.; Goto, Y.; et al. New developments in RiPP discovery, enzymology and engineering. Nat. Prod. Rep. 2021, 38, 130–239. [Google Scholar] [CrossRef]
- Hwang, S.; Lee, N.; Cho, S.; Palsson, B.; Cho, B.K. Repurposing Modular Polyketide Synthases and Non-ribosomal Peptide Synthetases for Novel Chemical Biosynthesis. Front. Mol. Biosci. 2020, 7, 87. [Google Scholar] [CrossRef]
- Wu, C.; van der Donk, W.A. Engineering of new-to-nature ribosomally synthesized and post-translationally modified peptide natural products. Curr. Opin. Biotechnol. 2021, 69, 221–231. [Google Scholar] [CrossRef] [PubMed]
- Yi, D.; Bayer, T.; Badenhorst, C.P.S.; Wu, S.; Doerr, M.; Höhne, M.; Bornscheuer, U.T. Recent trends in biocatalysis. Chem. Soc. Rev. 2021, 50, 8003–8049. [Google Scholar] [CrossRef] [PubMed]
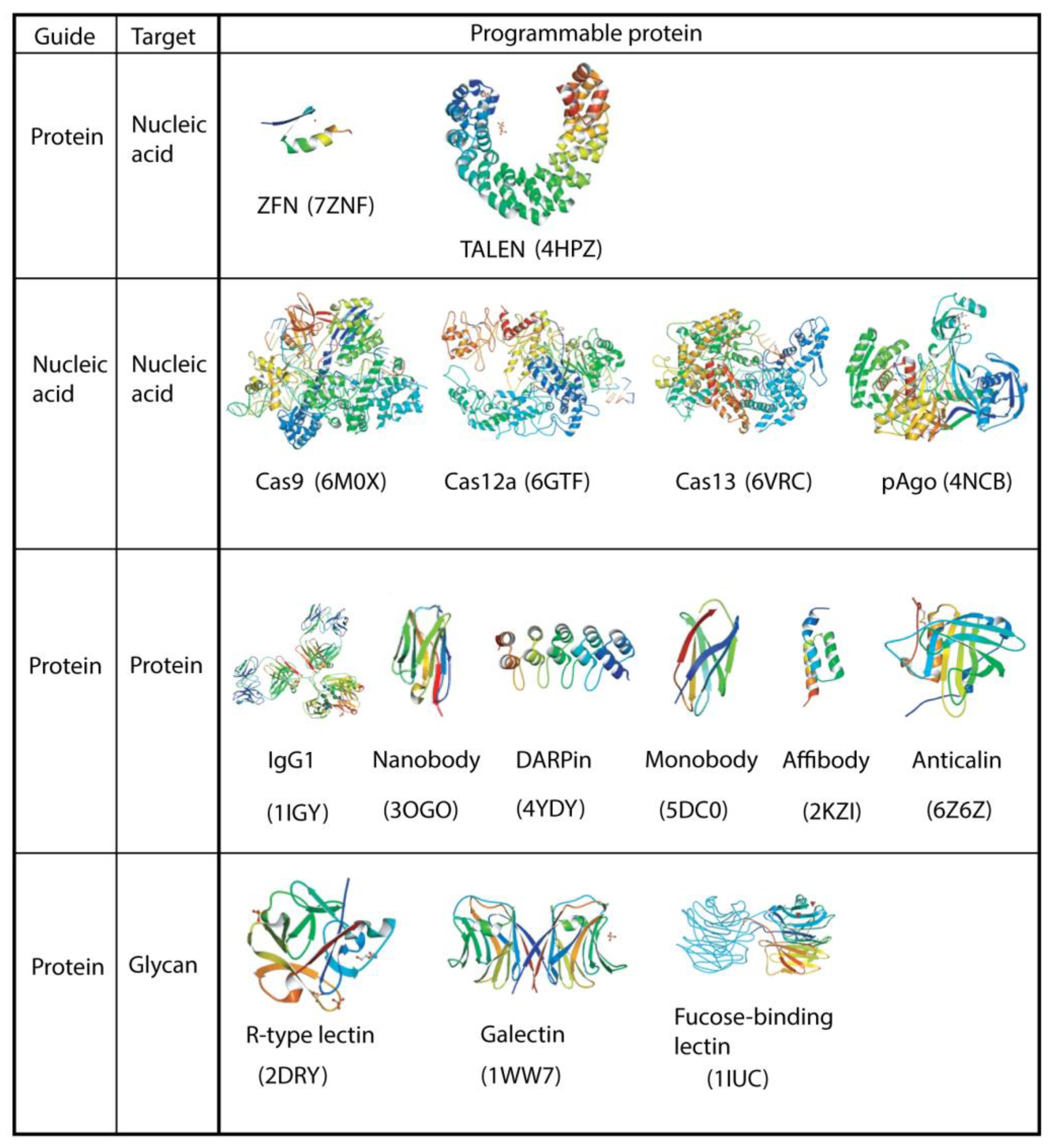
Figure 1.
Programmable proteins are categorized into two major types based on their guide moiety: nucleic acid and protein. They can also be classified based on the target they interact with: nucleic acid (Section 2), protein (Section 3), and glycan (Section 4). IgG1 is an intact monoclonal antibody for phenobarbital. Galectin (β-galactoside-binding lectin) from Agrocybe cylindracea (ACG) and fucose-binding lectin from Aleuria aurantia (AAL) are shown as dimers. Ribbon drawings (PDB ID) are based on https://pdbj.org (accessed on 17 October 2022). The size of each protein is arbitrary.
Figure 1.
Programmable proteins are categorized into two major types based on their guide moiety: nucleic acid and protein. They can also be classified based on the target they interact with: nucleic acid (Section 2), protein (Section 3), and glycan (Section 4). IgG1 is an intact monoclonal antibody for phenobarbital. Galectin (β-galactoside-binding lectin) from Agrocybe cylindracea (ACG) and fucose-binding lectin from Aleuria aurantia (AAL) are shown as dimers. Ribbon drawings (PDB ID) are based on https://pdbj.org (accessed on 17 October 2022). The size of each protein is arbitrary.

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Yamagata, M. Programmable Proteins: Target Specificity, Programmability and Future Directions. SynBio 2023, 1, 65-76. https://doi.org/10.3390/synbio1010005
AMA Style
Yamagata M. Programmable Proteins: Target Specificity, Programmability and Future Directions. SynBio. 2023; 1(1):65-76. https://doi.org/10.3390/synbio1010005
Chicago/Turabian StyleYamagata, Masahito. 2023. "Programmable Proteins: Target Specificity, Programmability and Future Directions" SynBio 1, no. 1: 65-76. https://doi.org/10.3390/synbio1010005