
Thermal Stability of Iron- and Silicon-Substituted Hydroxyapatite Prepared by Mechanochemical Method

Abstract
:1. Introduction
2. Materials and Methods
2.1. Mechanochemical Synthesis
2.2. Ex Situ Annealing
2.3. Sample Characterization
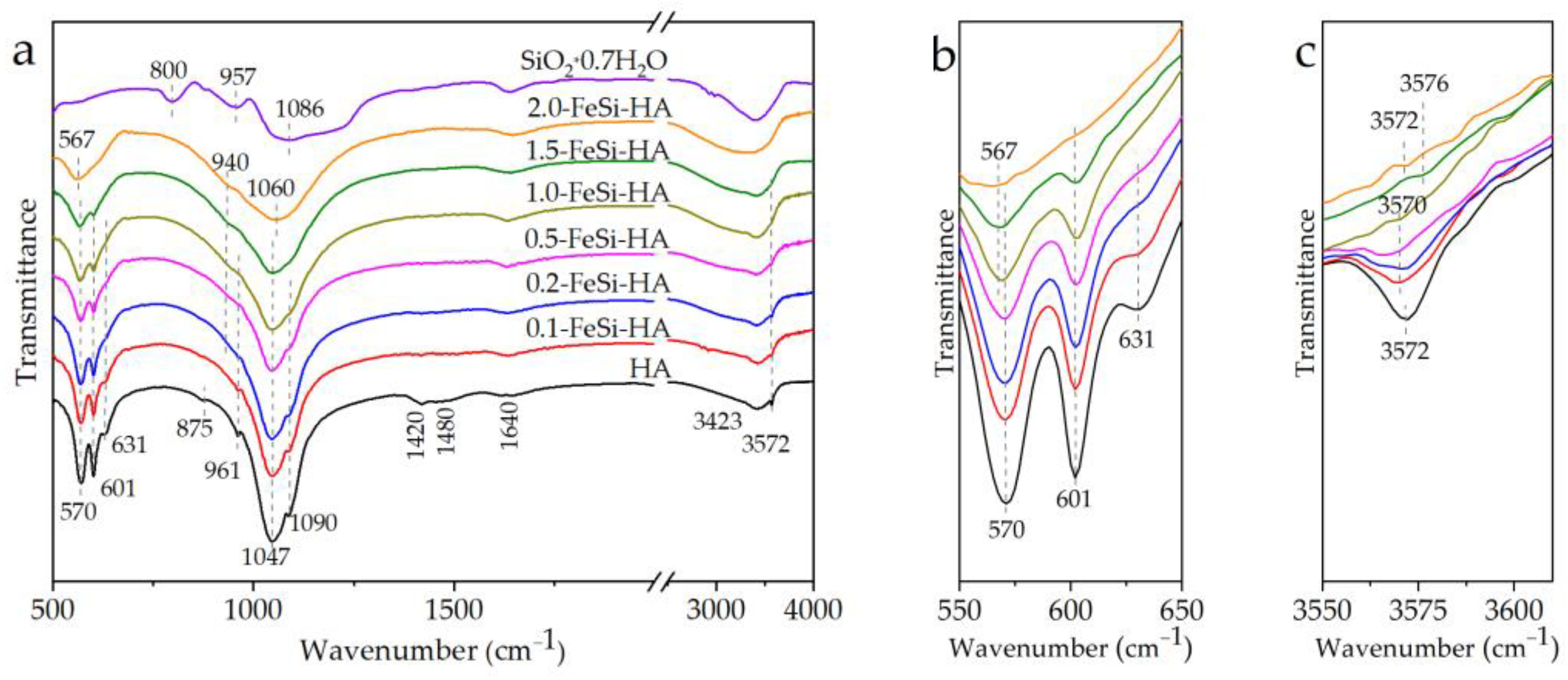
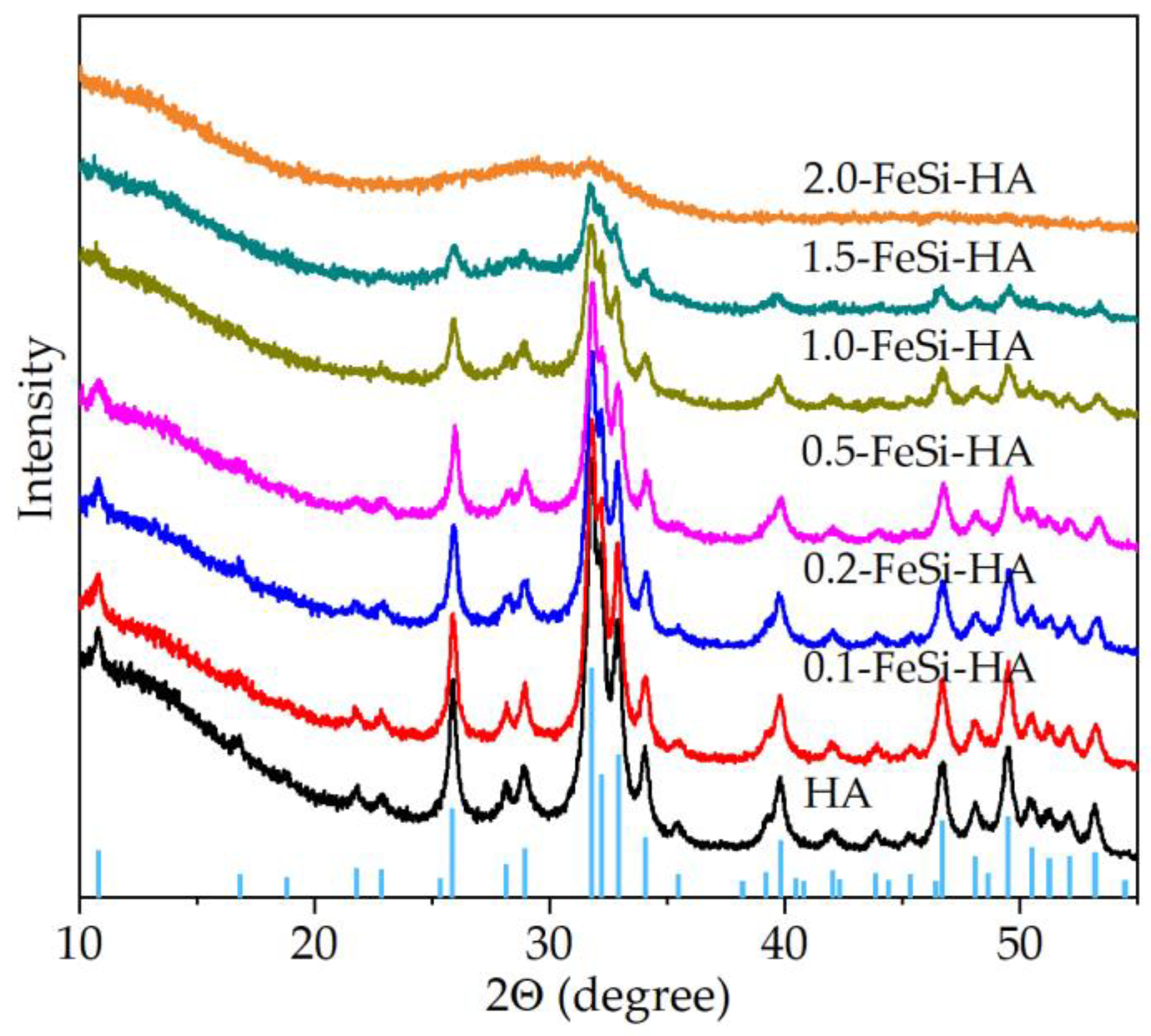
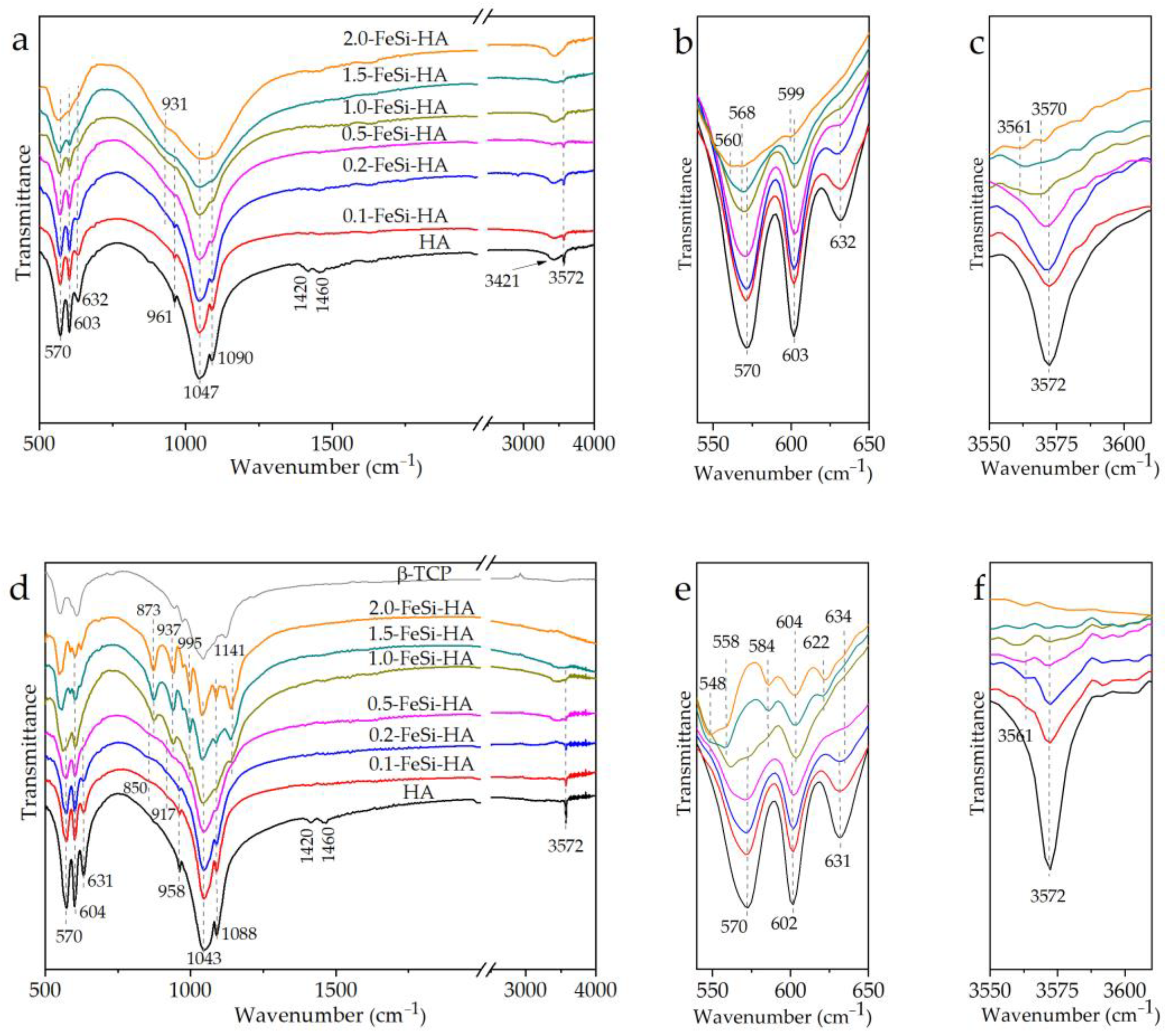
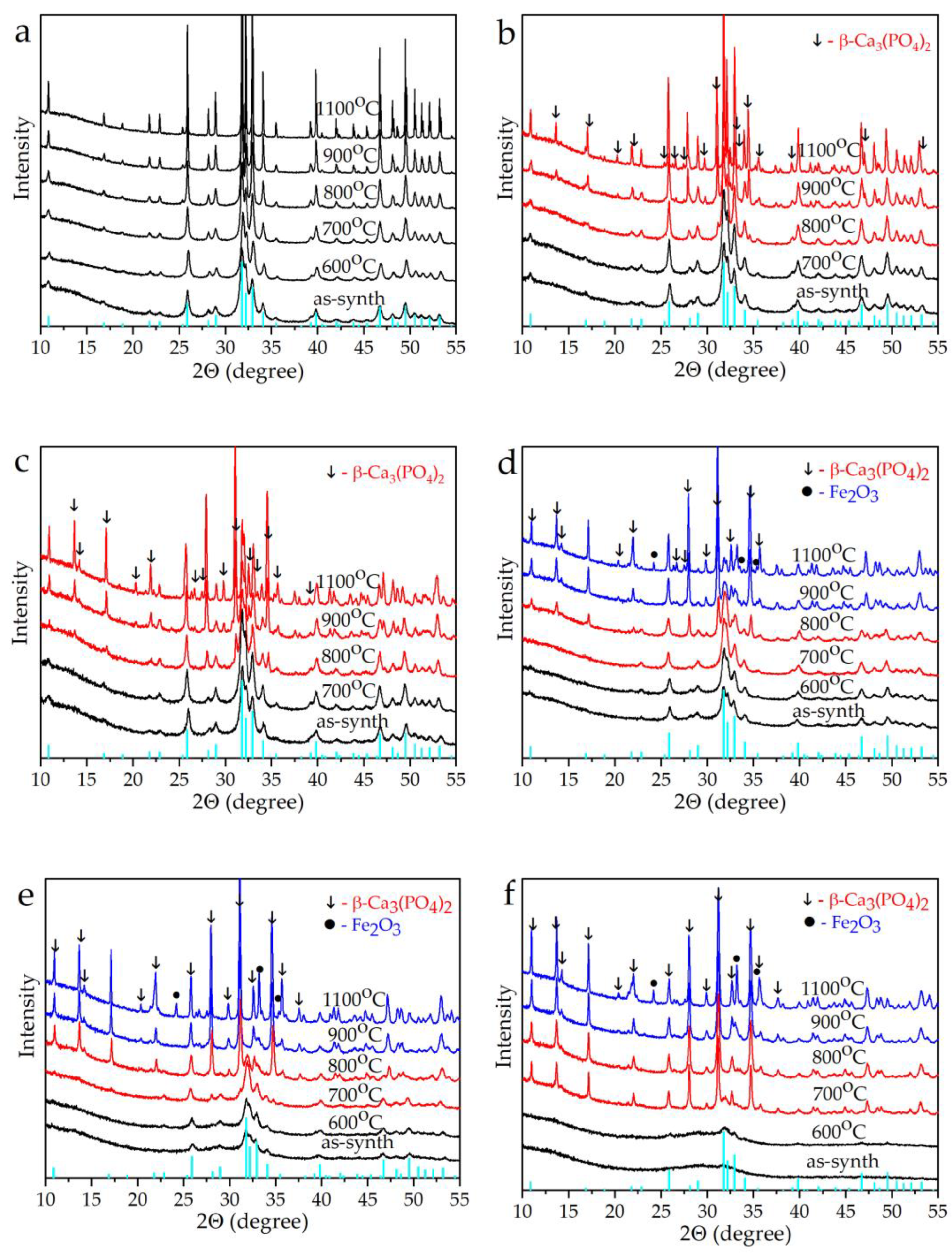
3. Results and Discussion
3.1. Analyses of the As-Prepared Samples
3.2. Thermal Properties
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lee, R.S.; Kayser, M.V.; Ali, S.Y. Calcium phosphate microcrystal deposition in the human intervertebral disc. J. Anat. 2006, 208, 13–19. [Google Scholar] [CrossRef] [PubMed]
- Dorozhkin, S.V. Calcium orthophosphates in nature, biology and medicine. Materials 2009, 2, 399–498. [Google Scholar] [CrossRef]
- Chaikina, M.V.; Bulina, N.V.; Vinokurova, O.B.; Gerasimov, K.B.; Prosanov, I.Y.; Kompankov, N.B.; Lapina, O.B.; Papulovskiy, E.S.; Ishchenko, A.V.; Makarova, S.V. Possibilities of Mechanochemical Synthesis of Apatites with Different Ca/P Ratios. Ceramics 2022, 5, 404–422. [Google Scholar] [CrossRef]
- Zakaria, S.M.; Zein, S.H.S.; Othman, M.R.; Yang, F.; Jansen, J.A. Nanophase hydroxyapatite as a biomaterial in advanced hard tissue engineering: A review. Tissue Eng. B Rev. 2013, 19, 431–441. [Google Scholar] [CrossRef]
- Kolmas, J.; Groszyk, E.; Kwiatkowska-Rózycka, D. Substituted hydroxyapatites with antibacterial properties. Biomed Res. Int. 2014, 2014, 178123. [Google Scholar] [CrossRef]
- Elliott, J.C. Structure and Chemistry of Apatite and Other Calcium Orthophosphates; Studies in Inorganic Chemistry; Elsevier: Amsterdam, The Netherlands, 1994; pp. 1–404. ISBN 0-444-81582-1. [Google Scholar]
- Bigi, A.; Cojazzi, G.; Panzavolta, S.; Ripamonti, A.; Roveri, N.; Romanello, M.; Noris Suarez, K.; Moro, L. Chemical and structural characterization of the mineral phase from cortical and trabecular bone. J. Inorg. Biochem. 1997, 68, 45–51. [Google Scholar] [CrossRef]
- Šupová, M. Substituted hydroxyapatites for biomedical applications: A review. Ceram. Int. 2015, 41, 9203–9231. [Google Scholar] [CrossRef]
- Carlisle, E.M. Silicon: A requirement in bone formation independent of vitamin D1. Calcif. Tissue Int. 1981, 33, 27–34. [Google Scholar] [CrossRef]
- Patel, N.; Best, S.M.; Bonfield, W.; Gibson, I.R.; Hing, K.A.; Damien, E.; Revell, P.A. A comparative study on the in vivo behavior of hydroxyapatite and silicon substituted hydroxyapatite granules. J. Mater. Sci. Mater. Med. 2002, 13, 1199–1206. [Google Scholar] [CrossRef]
- Hing, K.A.; Revell, P.A.; Smith, N.; Buckland, T. Effect of silicon level on rate, quality and progression of bone healing within silicate-substituted porous hydroxyapatite scaffolds. Biomaterials 2006, 27, 5014–5026. [Google Scholar] [CrossRef]
- Balas, F.; Pérez-Pariente, J.; Vallet-Regí, M. In vitro bioactivity of silicon-substituted hydroxyapatites. J. Biomed. Mater. Res. A 2003, 66, 364–375. [Google Scholar] [CrossRef] [PubMed]
- Camaioni, A.; Cacciotti, I.; Campagnolo, L.; Bianco, A. Silicon-Substituted Hydroxyapatite for Biomedical Applications. In Hydroxyapatite (Hap) for Biomedical Applications; Mucalo, M.R., Ed.; Elsevier: Amsterdam, The Netherlands, 2015; Volume 95, pp. 343–373. [Google Scholar] [CrossRef]
- Kim, S.R.; Lee, J.H.; Kim, Y.T.; Riu, D.H.; Jung, S.J.; Lee, Y.J.; Chung, S.C.; Kim, Y.H. Synthesis of Si, Mg substituted hydroxyapatites and their sintering behaviors. Biomaterials 2003, 24, 1389–1398. [Google Scholar] [CrossRef] [PubMed]
- Chang, Q.; Chen, D.L.; Ru, H.Q.; Yue, X.Y.; Yu, L.; Zhang, C.P. Toughening mechanism in iron-containing hydroxyapatite/titanium composite. Biomaterials 2010, 31, 1493–1501. [Google Scholar] [CrossRef] [PubMed]
- Pon-On, W.; Charoenphandhu, N.; Teeraporpuntakit, J.; Thongbunchoo, J.; Krishnamra, N.; Tang, I.M. Physicochemical and biochemical properties of ironloaded silicon substituted hydroxyapatite (FeSiHAp). Mater. Chem. Phys. 2013, 141, 850–860. [Google Scholar] [CrossRef]
- Kaygili, O.; Dorozhkin, S.V.; Ates, T.; Al-Ghamdi, A.A.; Yakuphanoglu, F. Dielectric properties of Fe doped hydroxyapatite prepared by sol gel method. Ceram. Int. 2016, 40, 9395–9402. [Google Scholar] [CrossRef]
- Panseri, S.; Cunha, C.; D’Alessandro, T.; Sandri, M.; Giavaresi, G.; Marcacci, M.; Hung, C.T.; Tampieri, A. Intrinsically superparamagnetic Fe-hydroxyapatite nanoparticles positively influence osteoblast-like behavior. J. Nanobiotechnol. 2012, 10, 1–10. [Google Scholar] [CrossRef]
- Iafisco, M.; Sandri, M.; Panseri, S.; Delgado-Lopez, J.M.; Gomez-Morales, J.; Tampieri, A. Magnetic bioactive and biodegradable hollow Fe-doped hydroxyapatite coated poly(L-lactic) acid micro-nanospheres. Chem. Mater. 2013, 25, 2610–2617. [Google Scholar] [CrossRef]
- Sarath Chandra, V.; Baskar, G.; Suganthi, R.V.; Elayaraja, K.; Ahymah Joshy, M.I.; Sofi Beaula, W.; Mythili, R.; Venkatraman, G.; Narayana Kalkura, S. Blood compatibility of iron-doped nanosize hydroxyapatite and its drug release. ACS Appl. Mater. Interfaces 2012, 4, 1200–1210. [Google Scholar] [CrossRef]
- Tampieri, A.; D’Alessandro, T.; Sandri, M.; Sprio, S.; Landi, E.; Bertinetti, L.; Panseri, S.; Pepponi, G.; Goettlicher, J.; Banobre-Lopez, M.; et al. Intrinsic magnetism and hyperthermia in bioactive Fe-doped hydroxyapatite. Acta Biomater. 2012, 8, 843–851. [Google Scholar] [CrossRef]
- Bulina, N.V.; Chaikina, M.V.; Andreev, A.S.; Lapina, O.B.; Ishchenko, A.V.; Prosanov, I.Y.; Gerasimov, K.B.; Solovyov, L.A. Mechanochemical synthesis of SiO44-substituted hydroxyapatite, part II—Reaction mechanism, structure, and substitution limit. Eur. J. Inorg. Chem. 2014, 2014, 4810–4825. [Google Scholar] [CrossRef]
- Marchat, D.; Zymelka, M.; Coelho, C.; Gremillard, L.; Joly-pottuz, L.; Babonneau, F.; Esnouf, C.; Chevalier, J.; Bernache-assollant, D. Accurate characterization of pure silicon-substituted hydroxyapatite powders synthesized by a new precipitation route. Acta. Biomater. 2013, 9, 6992–7004. [Google Scholar] [CrossRef] [PubMed]
- Glimcher, M.J. The Nature of the Mineral Phase in Bone: Biological and Clinical Implications. In Metabolic Bone Disease and Clinically Related Disorders, 3rd ed.; Avioli, L.V., Krane, S.M., Eds.; Academic Press: Camdridge, MA, USA, 1998; pp. 23–50. [Google Scholar] [CrossRef]
- Boskey, A. Bone mineral crystal size. Osteoporos. Int. 2003, 14, 16–21. [Google Scholar] [CrossRef]
- LeGeros, R.Z.; Bonel, G.; Legros, R. Types of “H2O” in human enamel and in precipitated apatites. Calcif. Tissue Res. 1978, 26, 111–118. [Google Scholar] [CrossRef] [PubMed]
- Eremina, N.V.; Makarova, S.V.; Isaev, D.D.; Bulina, N.V. Soft mechanochemical synthesis and thermal stability of hydroxyapatites with different types of substitution. Chim. Techno. Acta 2022, 9, 3–9. [Google Scholar] [CrossRef]
- Tõnsuaadu, K.; Gross, K.A.; Pluduma, L.; Veiderma, M. A review on the thermal stability of calcium apatites. J. Therm. Anal. Calorim. 2012, 110, 647–659. [Google Scholar] [CrossRef]
- Bulina, N.V.; Makarova, S.V.; Baev, S.G.; Matvienko, A.A.; Gerasimov, K.B.; Logutenko, O.A.; Bystrov, V.S. A study of thermal stability of hydroxyapatite. Minerals 2021, 11, 1310. [Google Scholar] [CrossRef]
- Singh, R.K.; Srivastava, M.; Prasad, N.K.; Awasthi, S.; Dhayalan, A.; Kannan, S. Iron doped β -Tricalcium phosphate: Synthesis, characterization, hyperthermia effect, biocompatibility and mechanical evaluation. Mater. Sci. Eng. C 2017, 78, 715–726. [Google Scholar] [CrossRef]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Degree of Substitution (x) | Sample Designation | Equations for Plausible Chemical Reactions |
|---|---|---|
| 0 | HA | 6.0CaHPO4 + 4.0CaO Ca10(PO4)6(OH)2 + 2H2O |
| 0.1 | 0.1−FeSi−HA | 5.8CaHPO4 + 4.1CaO + 0.1FePO4·2H2O + 0.1SiO2·0.7H2O Ca9.9Fe0.1(PO4)5.9(SiO4)0.1(OH)1.9O0.05 + 2.17H2O |
| 0.2 | 0.2−FeSi−HA | 5.6CaHPO4 + 4.2CaO + 0.2FePO4·2H2O + 0.2SiO2·0.7H2O Ca9.8Fe0.2(PO4)5.8(SiO4)0.2(OH)1.8O0.1 + 2.34H2O |
| 0.5 | 0.5−FeSi−HA | 5.0CaHPO4 + 4.5CaO + 0.5FePO4·2H2O + 0.5SiO2·0.7H2O Ca9.5Fe0.5(PO4)5.5(SiO4)0.5(OH)1.5O0.25 + 2.85H2O |
| 1.0 | 1.0−FeSi−HA | 4.0CaHPO4 + 5.0CaO + 1.0FePO4·2H2O + 1.0SiO2·0.7H2O Ca9.0Fe1.0(PO4)5.0(SiO4)1.0(OH)1.0O0.5 + 3.70H2O |
| 1.5 | 1.5−FeSi−HA | 3.0CaHPO4 + 5.5CaO + 1.5FePO4·2H2O + 1.5SiO2·0.7H2O Ca8.5Fe1.5(PO4)4.5(SiO4)1.5(OH)0.5O0.75 + 4.55H2O |
| 2.0 | 2.0−FeSi−HA | 2.0CaHPO4 + 6.0CaO + 2.0FePO4·2H2O + 2.0SiO2·0.7H2O Ca8.0Fe2.0(PO4)4.0(SiO4)2.0O + 5.40H2O |
| Sample | Concentration (at.%) | |||||||
|---|---|---|---|---|---|---|---|---|
| Composition of Synthesized Materials | Expected Composition | |||||||
| Ca | Fe | P | Si | Ca | Fe | P | Si | |
| HA | 66 | - | 34 | - | 63 | - | 37 | - |
| 0.5−FeSi−HA | 59 | 3 | 35 | 3 | 59 | 3 | 35 | 3 |
| 1.0−FeSi−HA | 58 | 8 | 29 | 5 | 57 | 6 | 31 | 6 |
| 2.0−FeSi−HA | 44 | 11 | 33 | 12 | 50 | 12 | 26 | 12 |
| Sample | Phase | 700 °C | 800 °C | 900 °C | 1000 °C | 1100 °C |
|---|---|---|---|---|---|---|
| HA | HA | 100 | 100 | 100 | 100 | 100 |
| 0.1−FeSi−HA | HA | 100 | 100 | 92 | 89 | 87 |
| β−TCP | 0 | 0 | 8 | 11 | 13 | |
| 0.2−FeSi−HA | HA | 100 | 95 | 76 | 70 | 66 |
| β−TCP | 0 | 5 | 24 | 30 | 34 | |
| 0.5−FeSi−HA | HA | 100 | 85 | 57 | 43 | 37 |
| β−TCP | 0 | 15 | 43 | 57 | 63 | |
| 1.0−FeSi−HA | HA | 97 | 67 | 32 | 14 | 12 |
| β−TCP | 3 | 33 | 67 | 81 | 83 | |
| Fe2O3 | 0 | 0 | 1 | 5 | 6 | |
| 1.5−FeSi−HA | HA | 79 | 31 | 7 | 2 | 1 |
| β−TCP | 21 | 69 | 93 | 90 | 90 | |
| Fe2O3 | 0 | 0 | 1 | 8 | 9 | |
| 2.0−FeSi−HA | HA | 15 | 6 | 7 | 3 | 1 |
| β−TCP | 85 | 94 | 91 | 89 | 89 | |
| Fe2O3 | 0 | 0 | 1 | 8 | 10 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Makarova, S.V.; Bulina, N.V.; Vinokurova, O.B.; Ishchenko, A.V. Thermal Stability of Iron- and Silicon-Substituted Hydroxyapatite Prepared by Mechanochemical Method. Powders 2023, 2, 372-386. https://doi.org/10.3390/powders2020022
Makarova SV, Bulina NV, Vinokurova OB, Ishchenko AV. Thermal Stability of Iron- and Silicon-Substituted Hydroxyapatite Prepared by Mechanochemical Method. Powders. 2023; 2(2):372-386. https://doi.org/10.3390/powders2020022
Chicago/Turabian StyleMakarova, Svetlana V., Natalia V. Bulina, Olga B. Vinokurova, and Arcady V. Ishchenko. 2023. "Thermal Stability of Iron- and Silicon-Substituted Hydroxyapatite Prepared by Mechanochemical Method" Powders 2, no. 2: 372-386. https://doi.org/10.3390/powders2020022