
Systems Pharmacology and Network Analysis to Advance Pharmacogenomics and Precision Medicine Decisions in Type-2 Diabetes Therapy
 , and
, and
Abstract
:1. Introduction
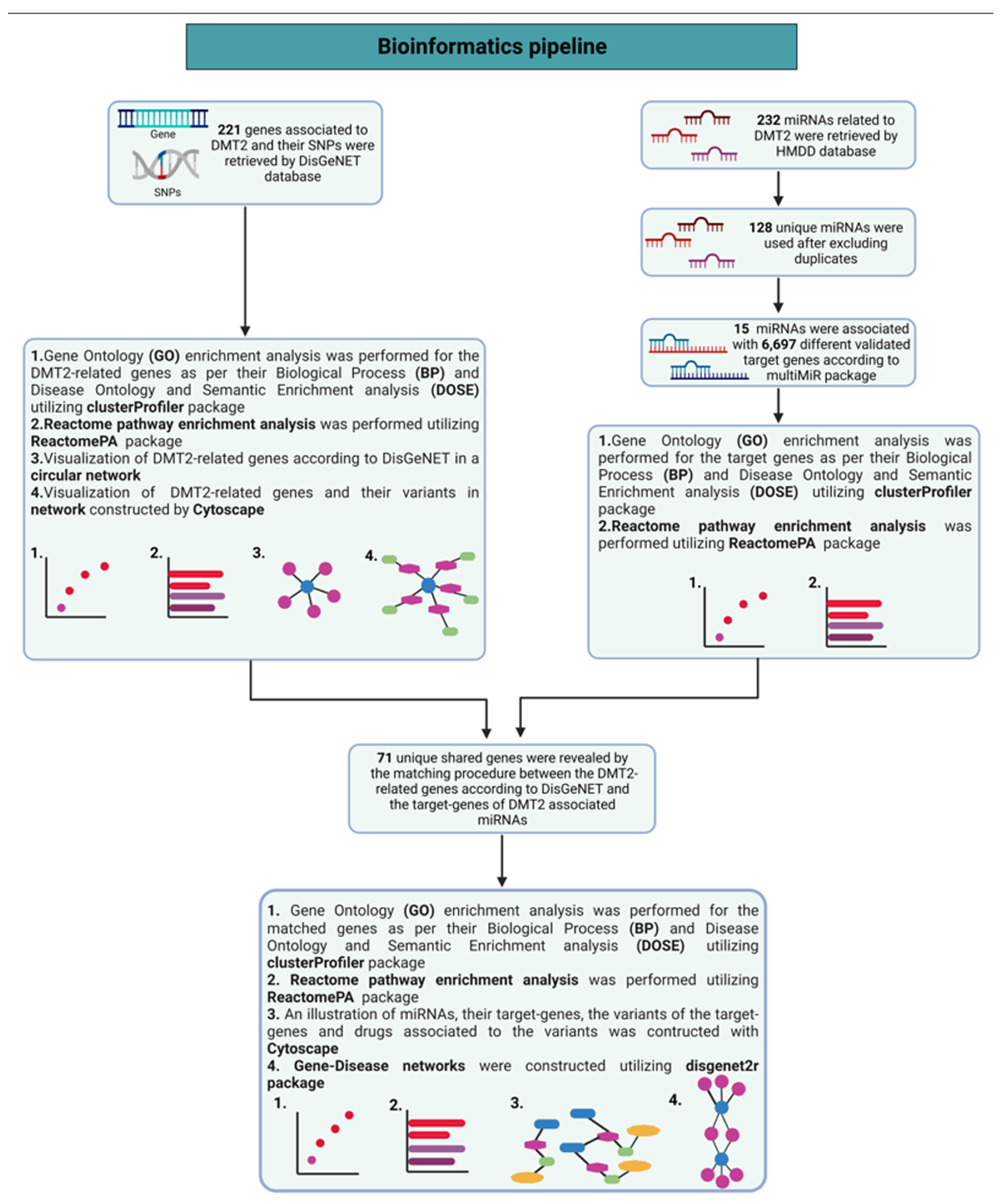
2. Materials and Methods
Methodology to Perform Network Analysis of Pharmacogenomics Data Related to DMT2
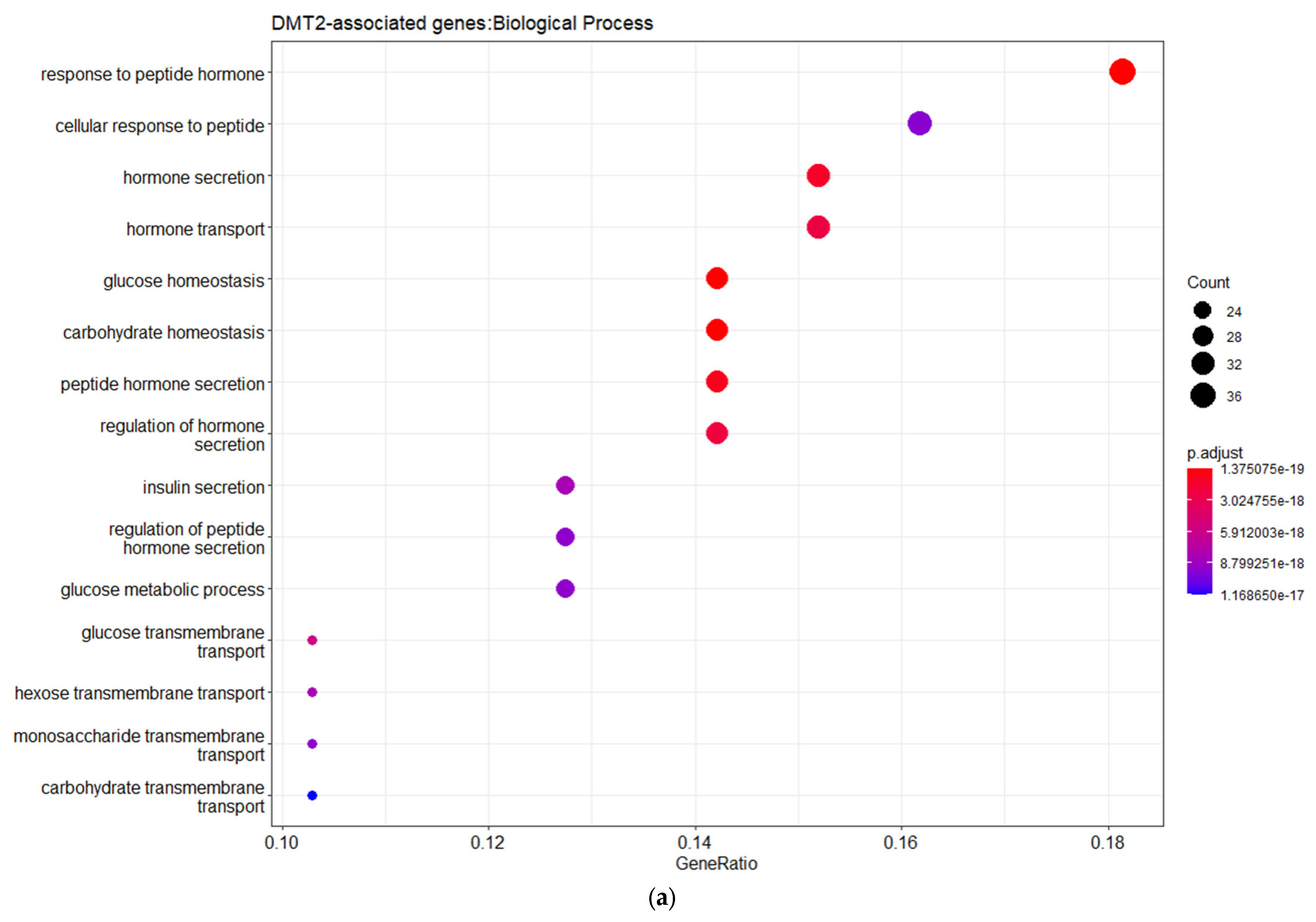
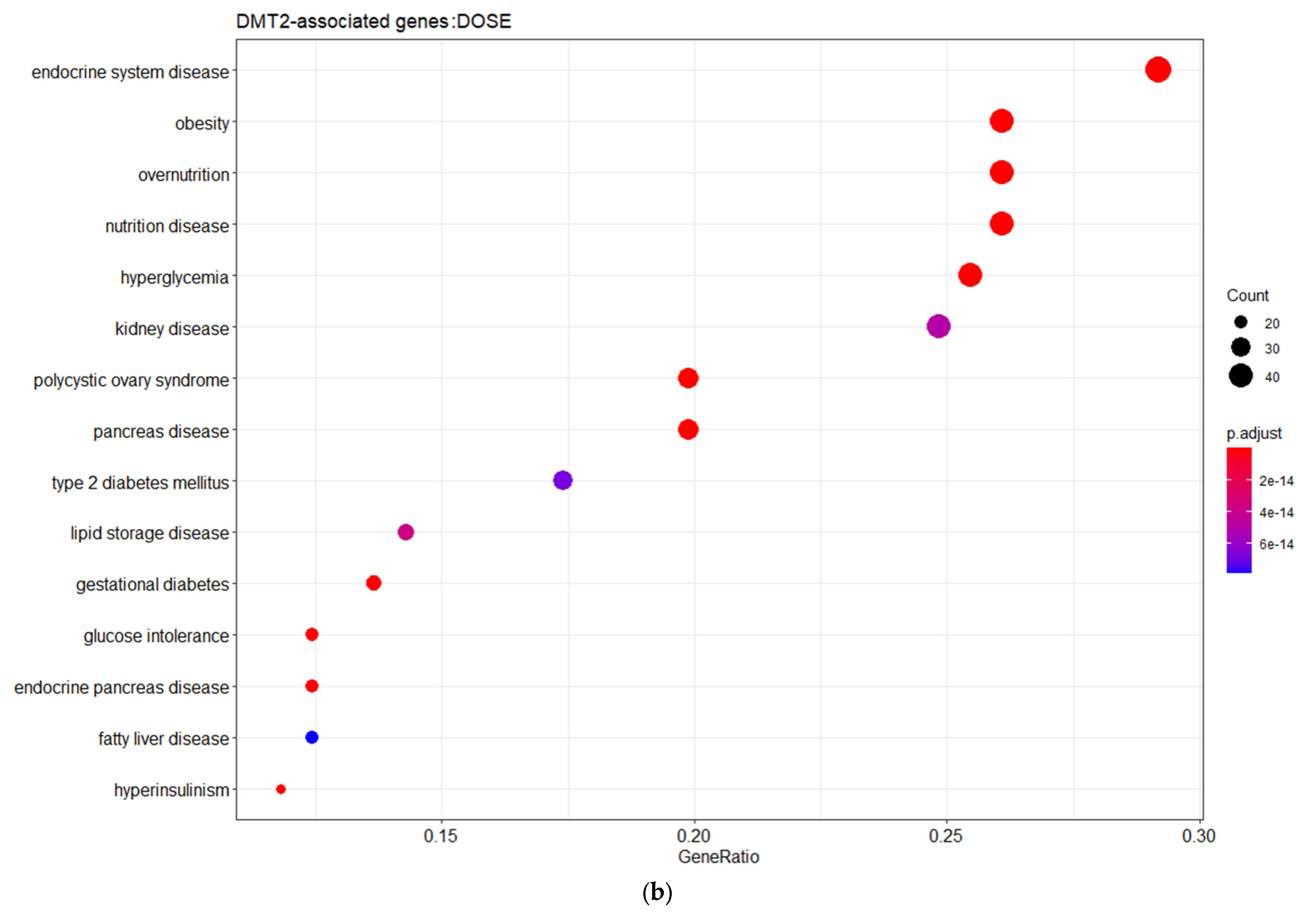
3. Results and Discussion
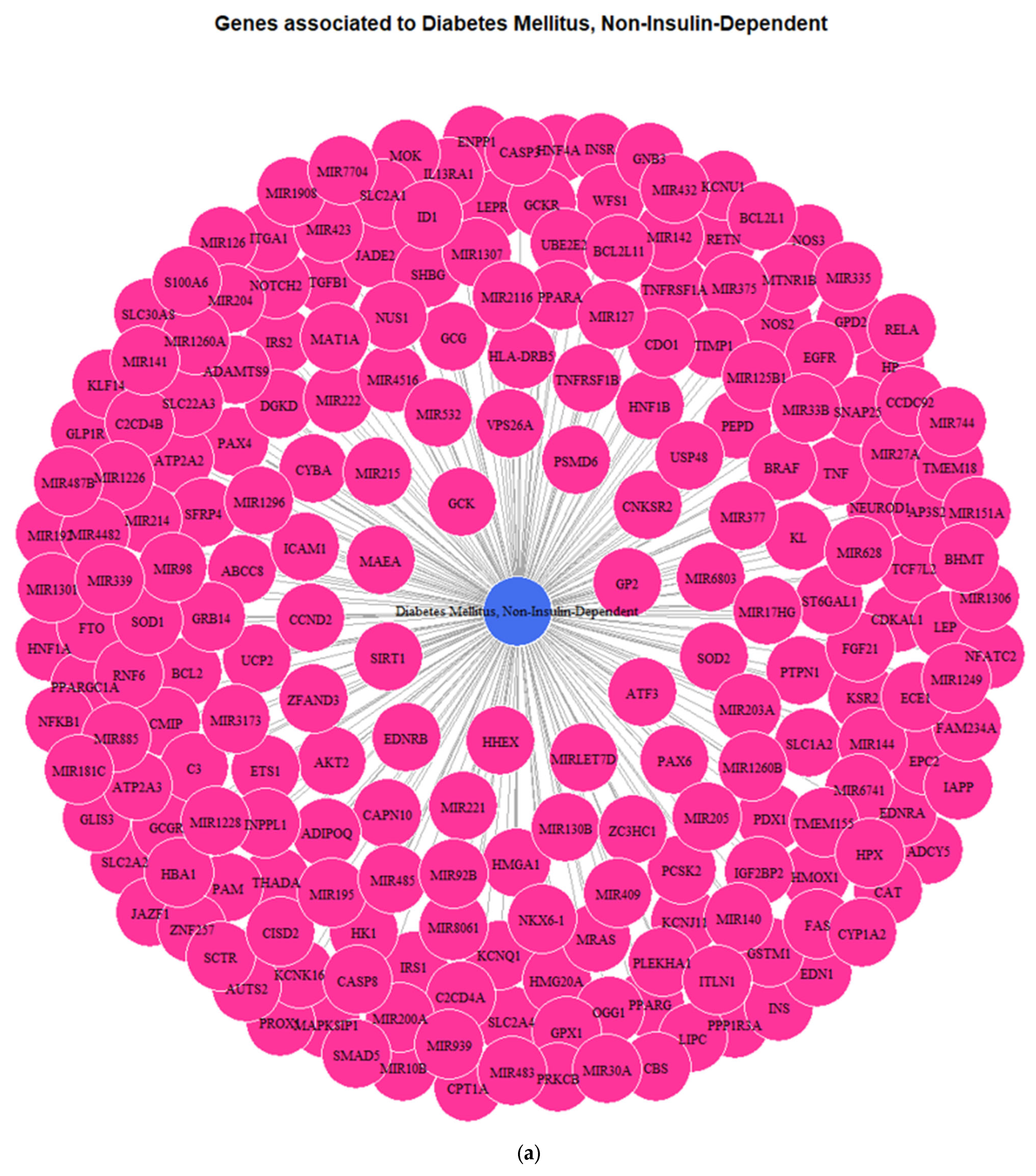
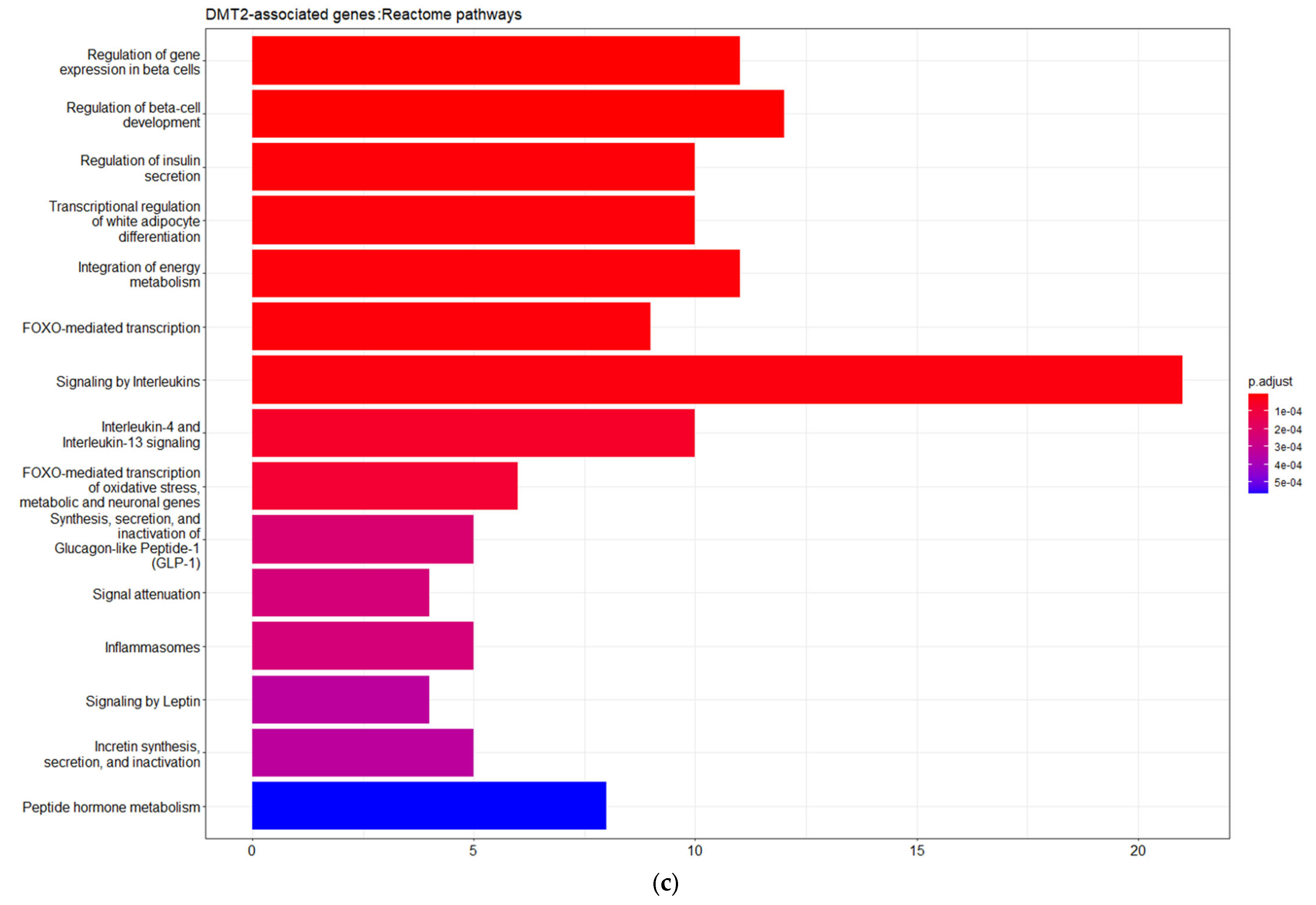
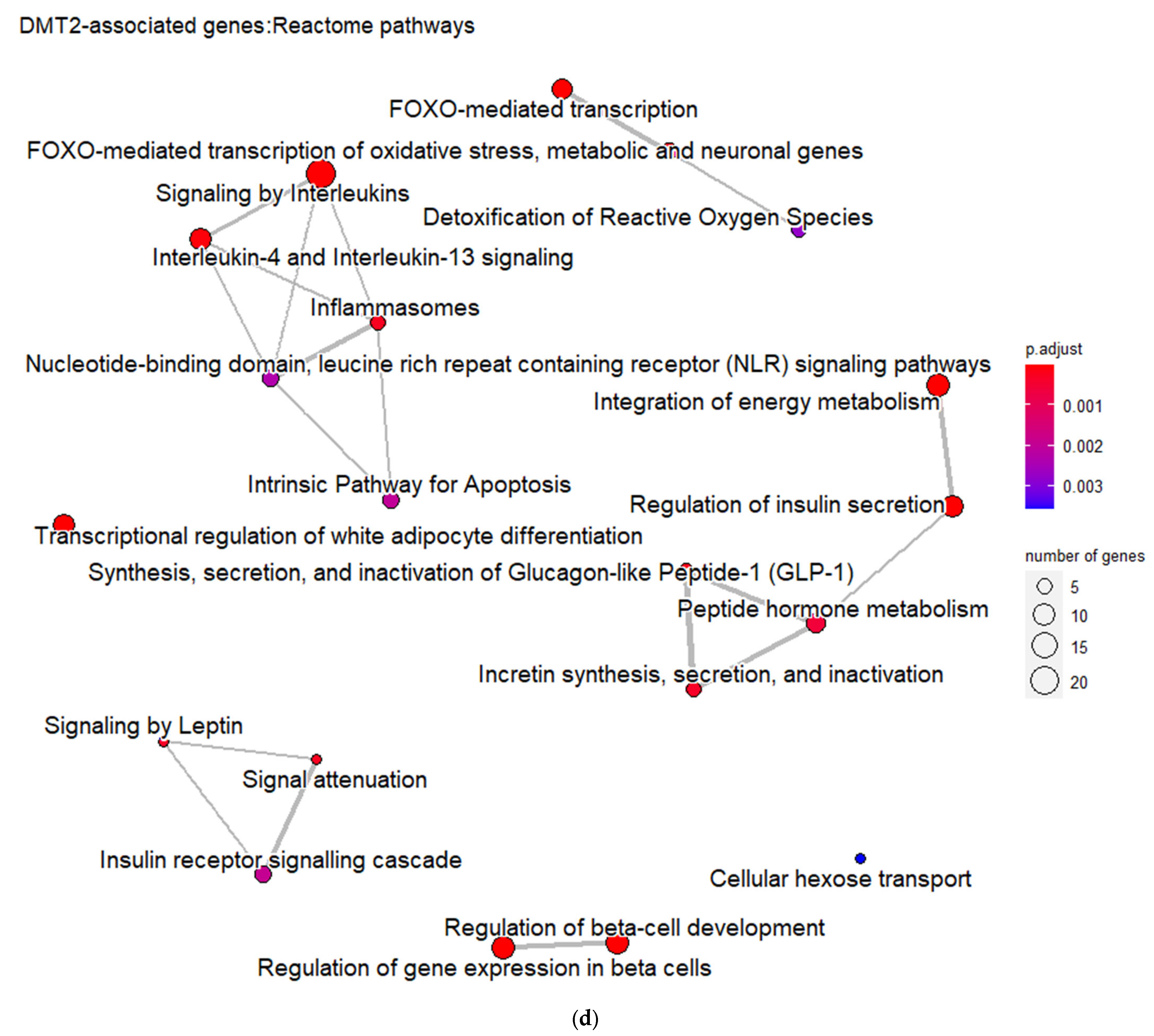
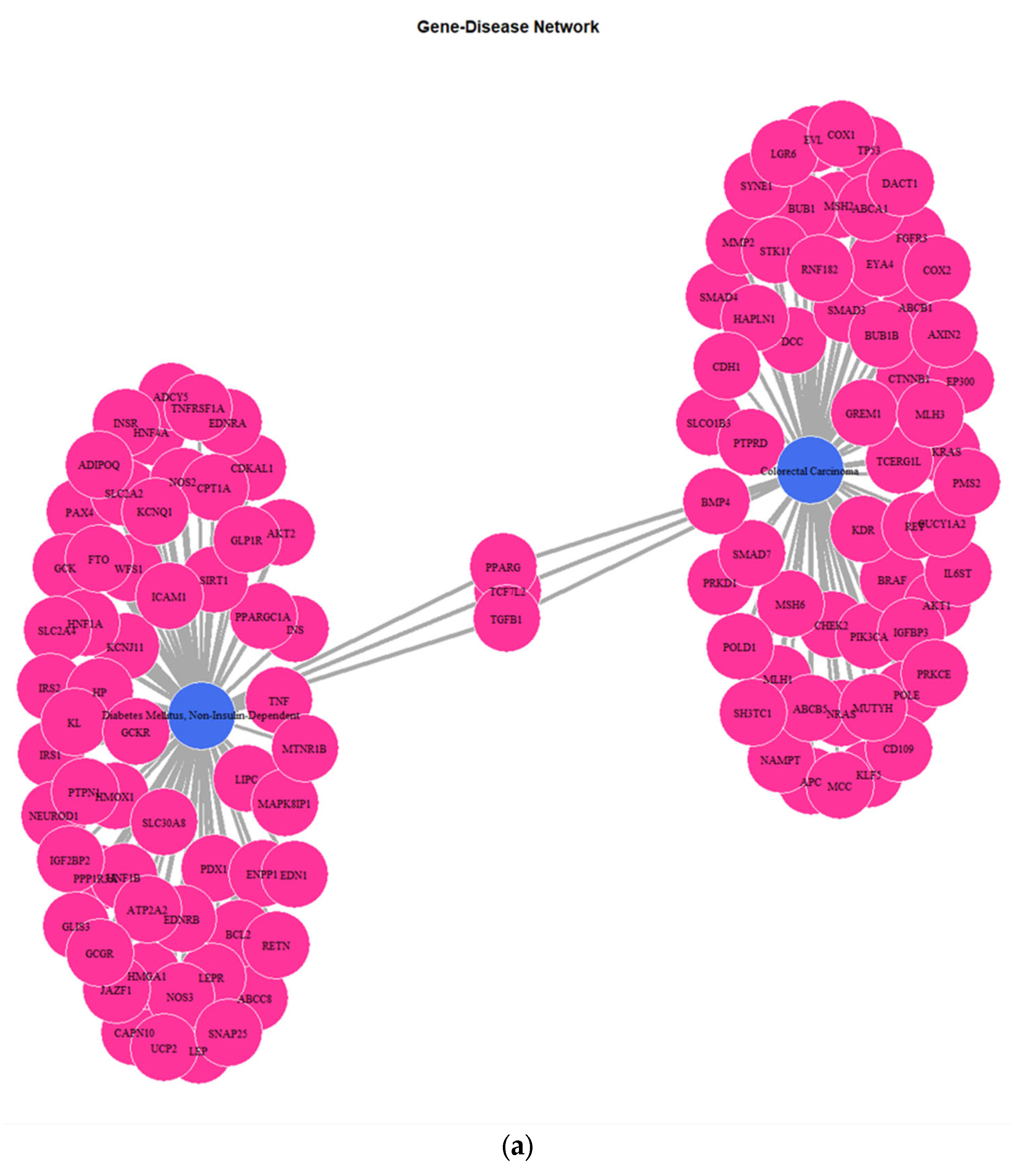
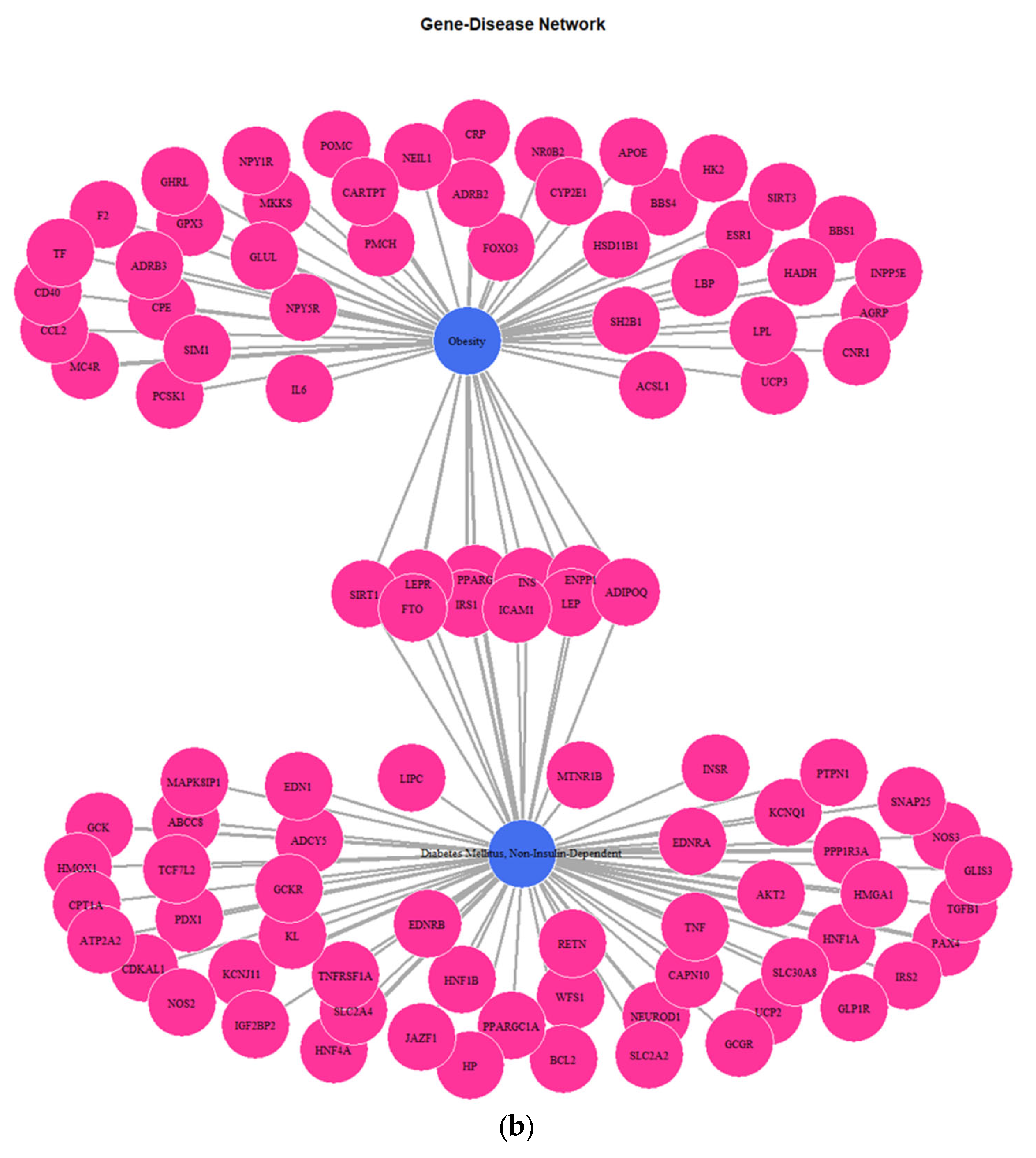
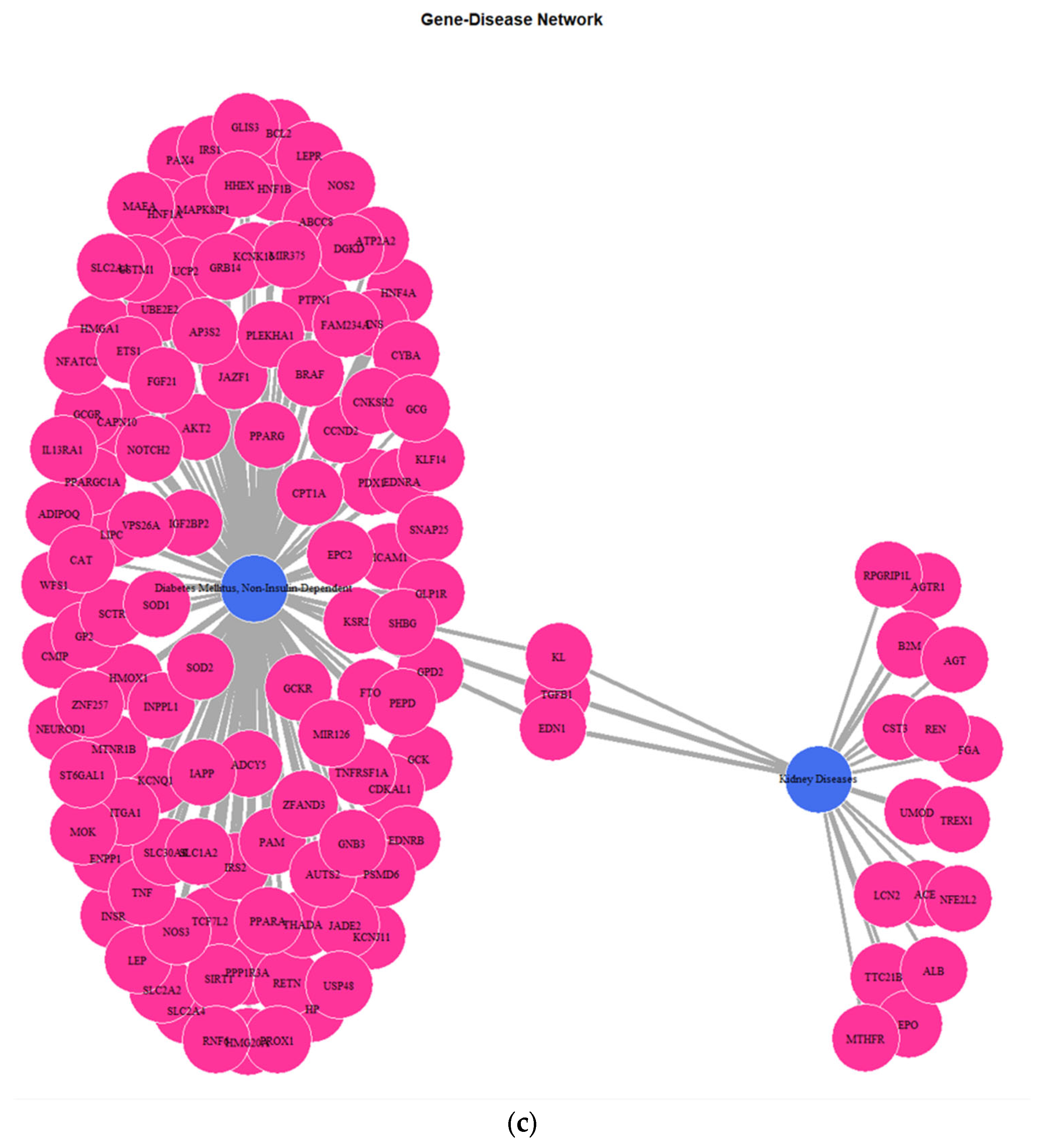
3.1. Genes Implicated in DMT2 Disease According to DisGeNET Database
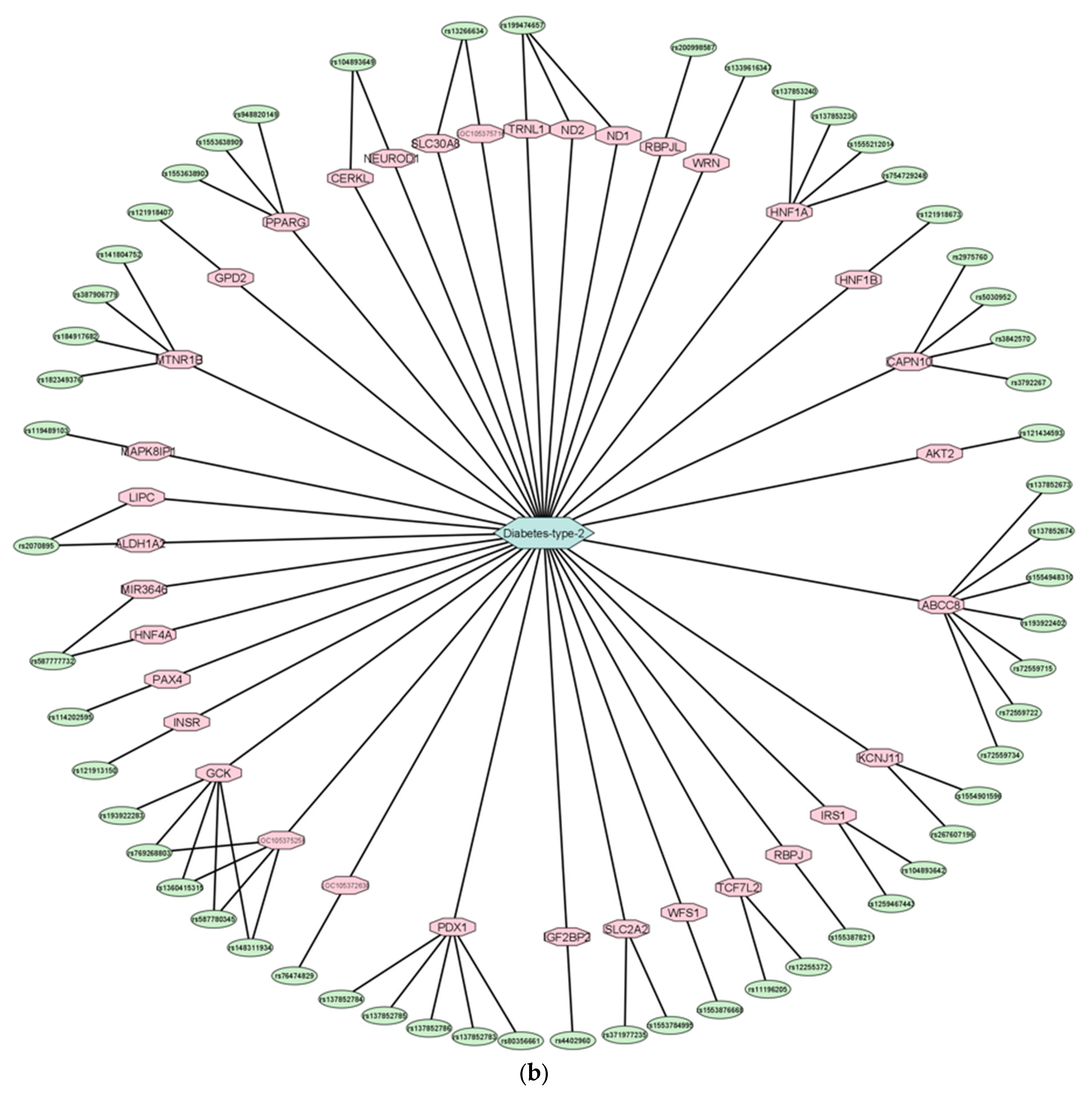
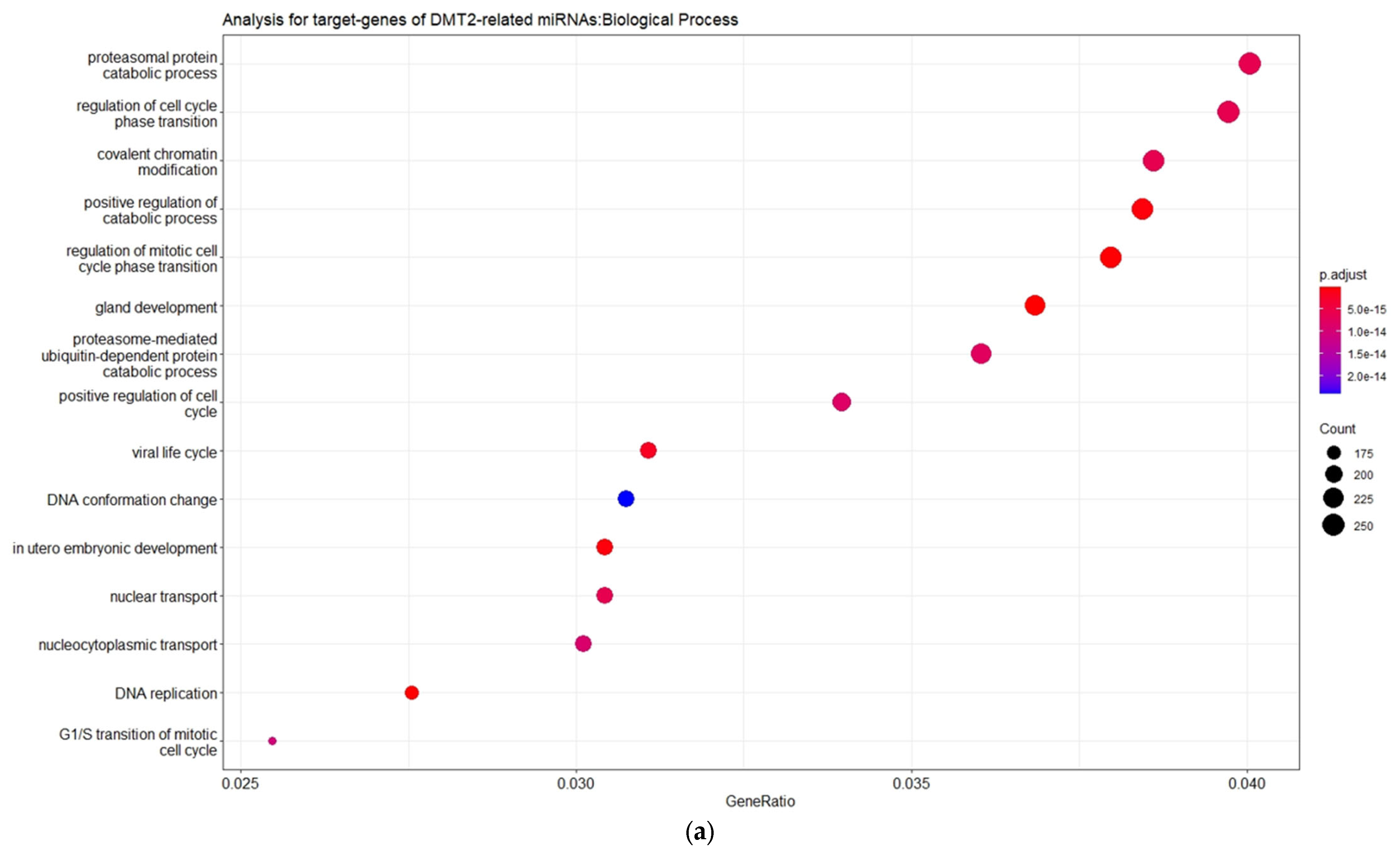
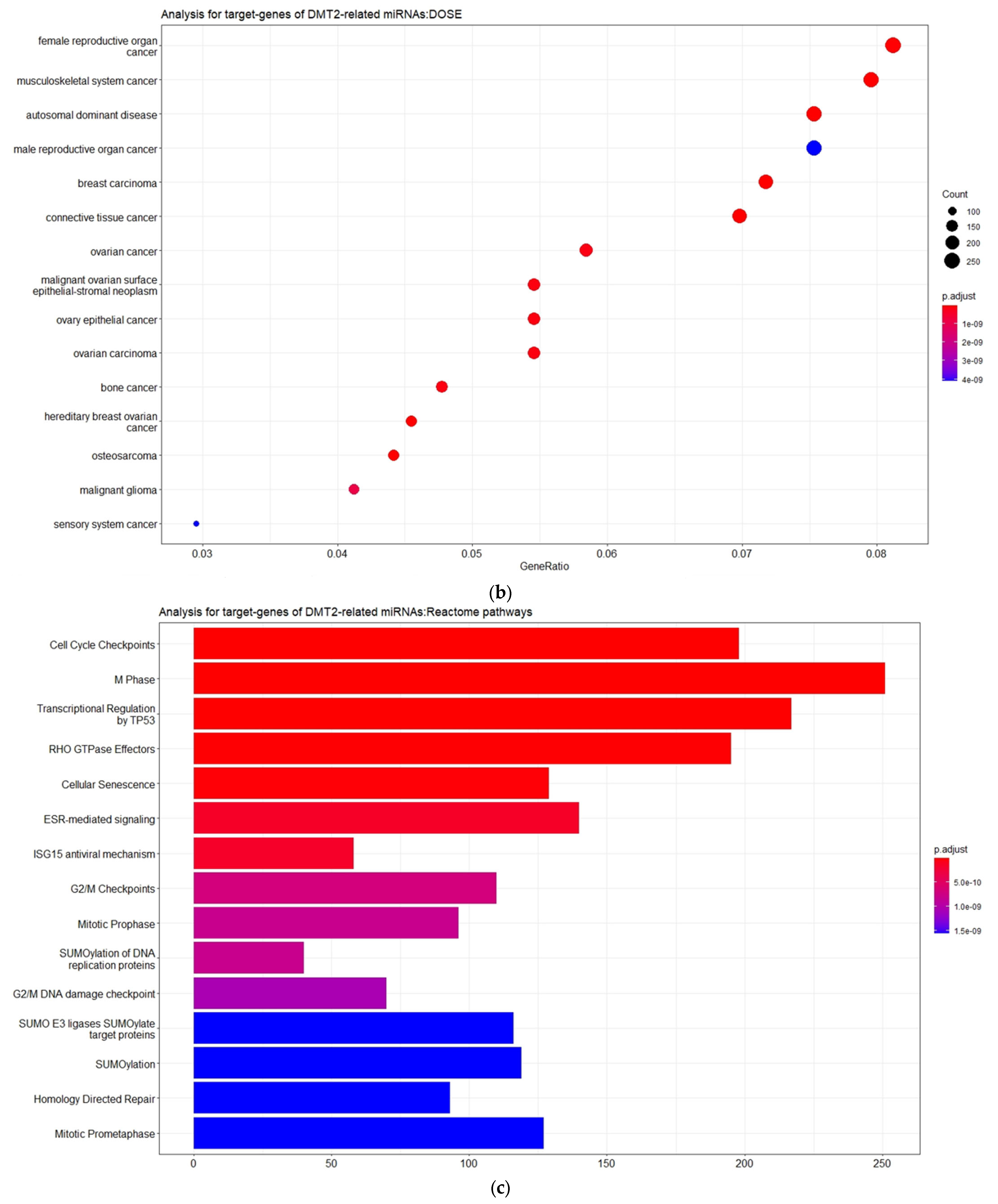
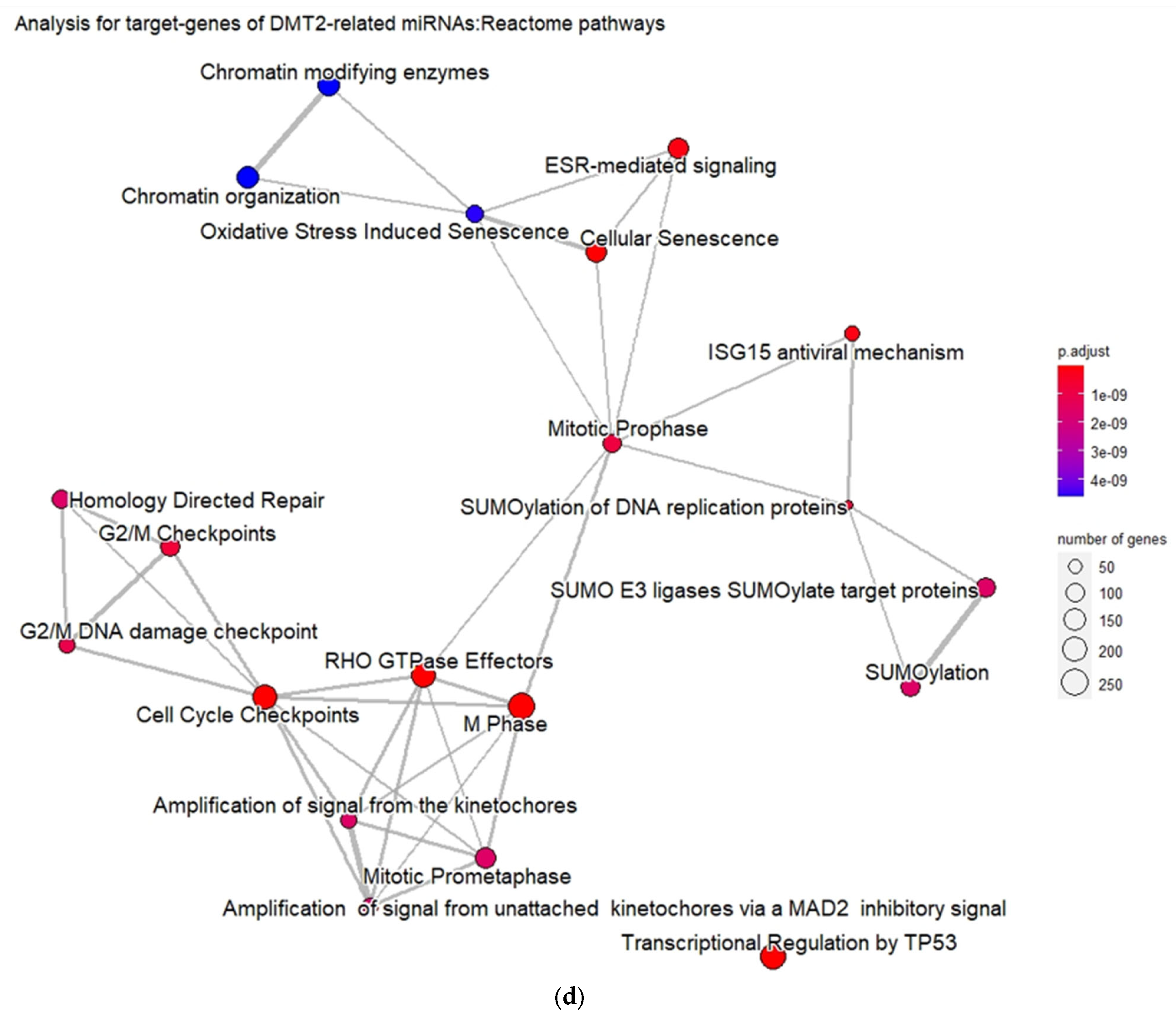
3.2. Experimentally Validated Target Genes of DMT2-Related miRNAs
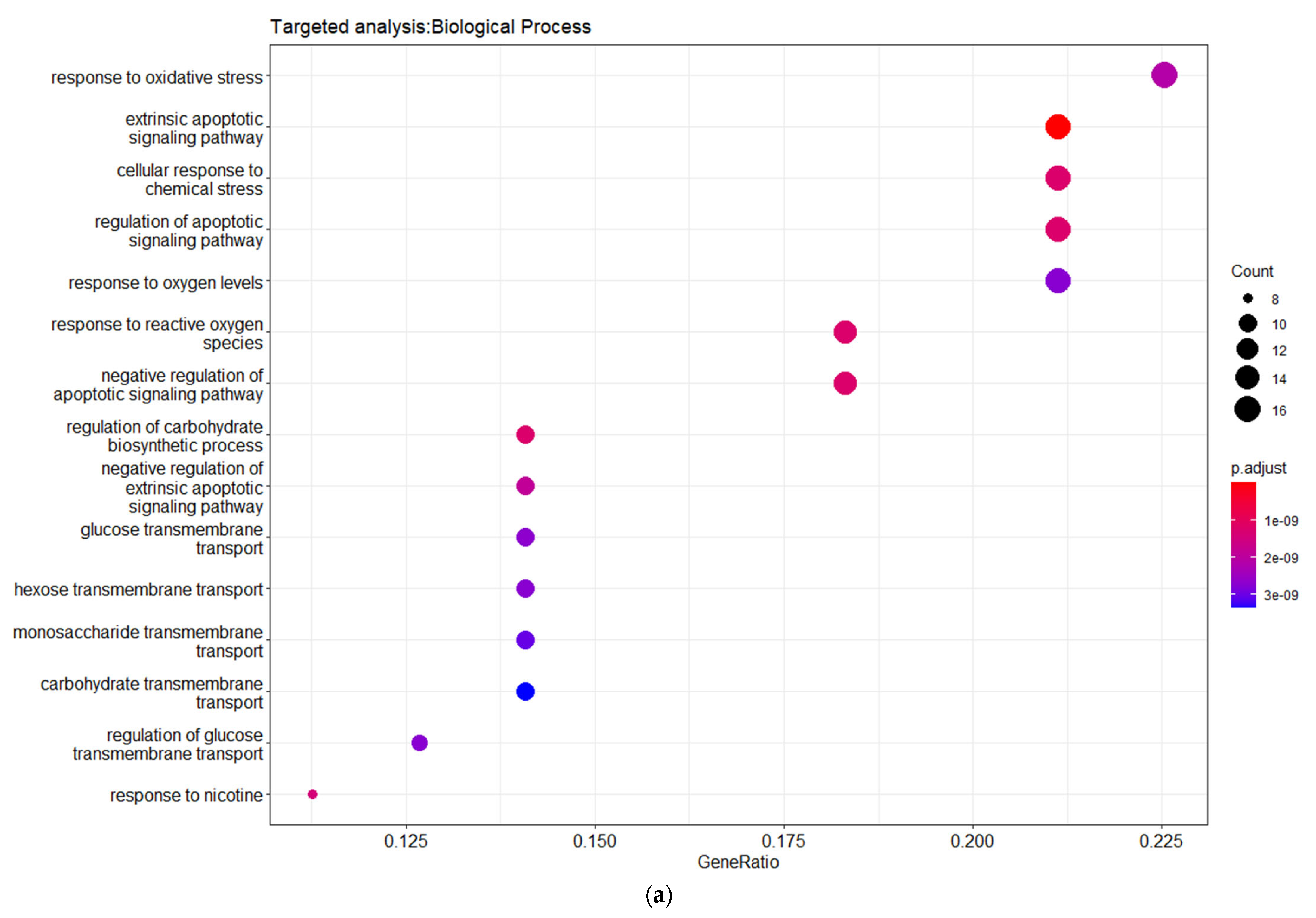
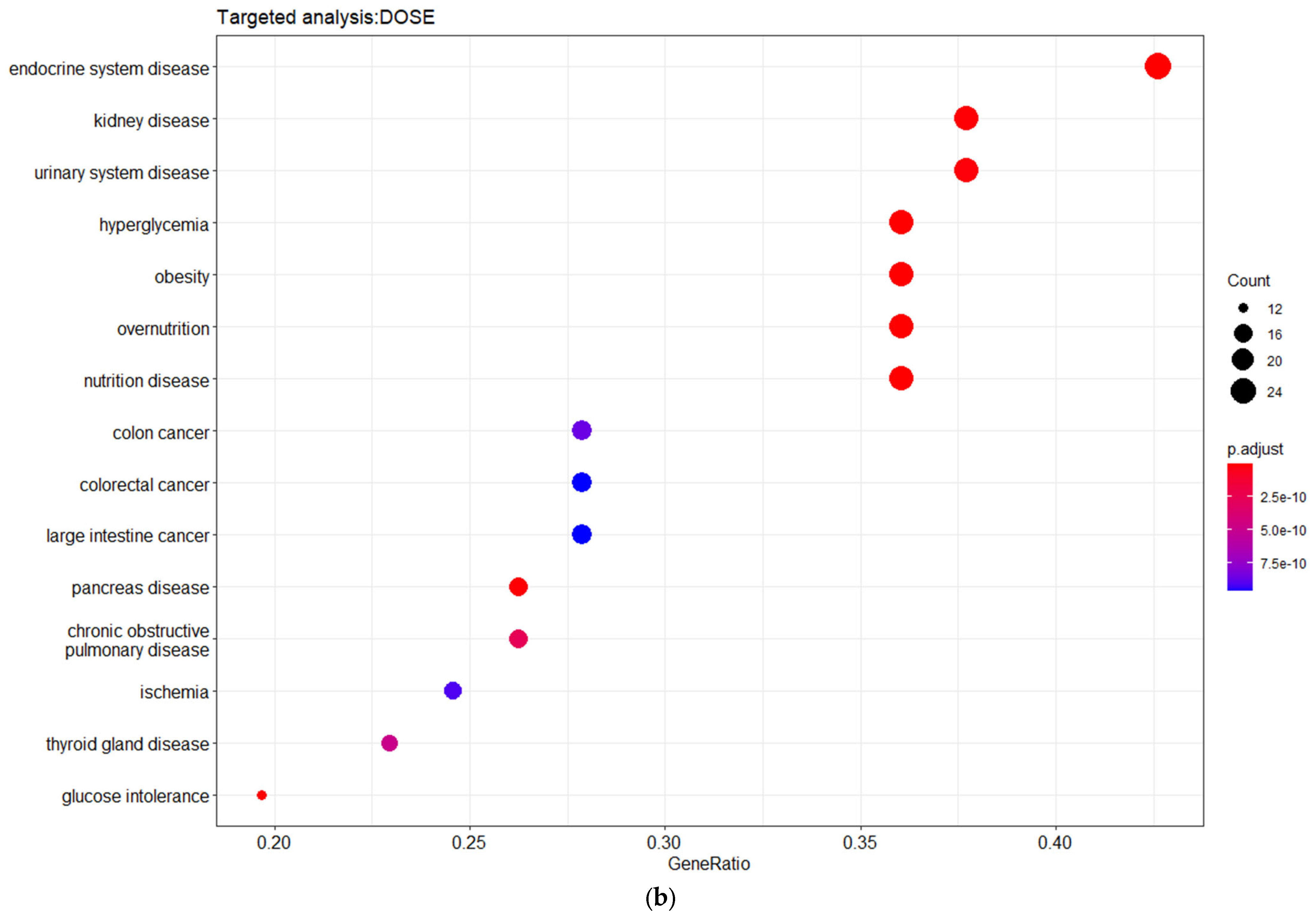
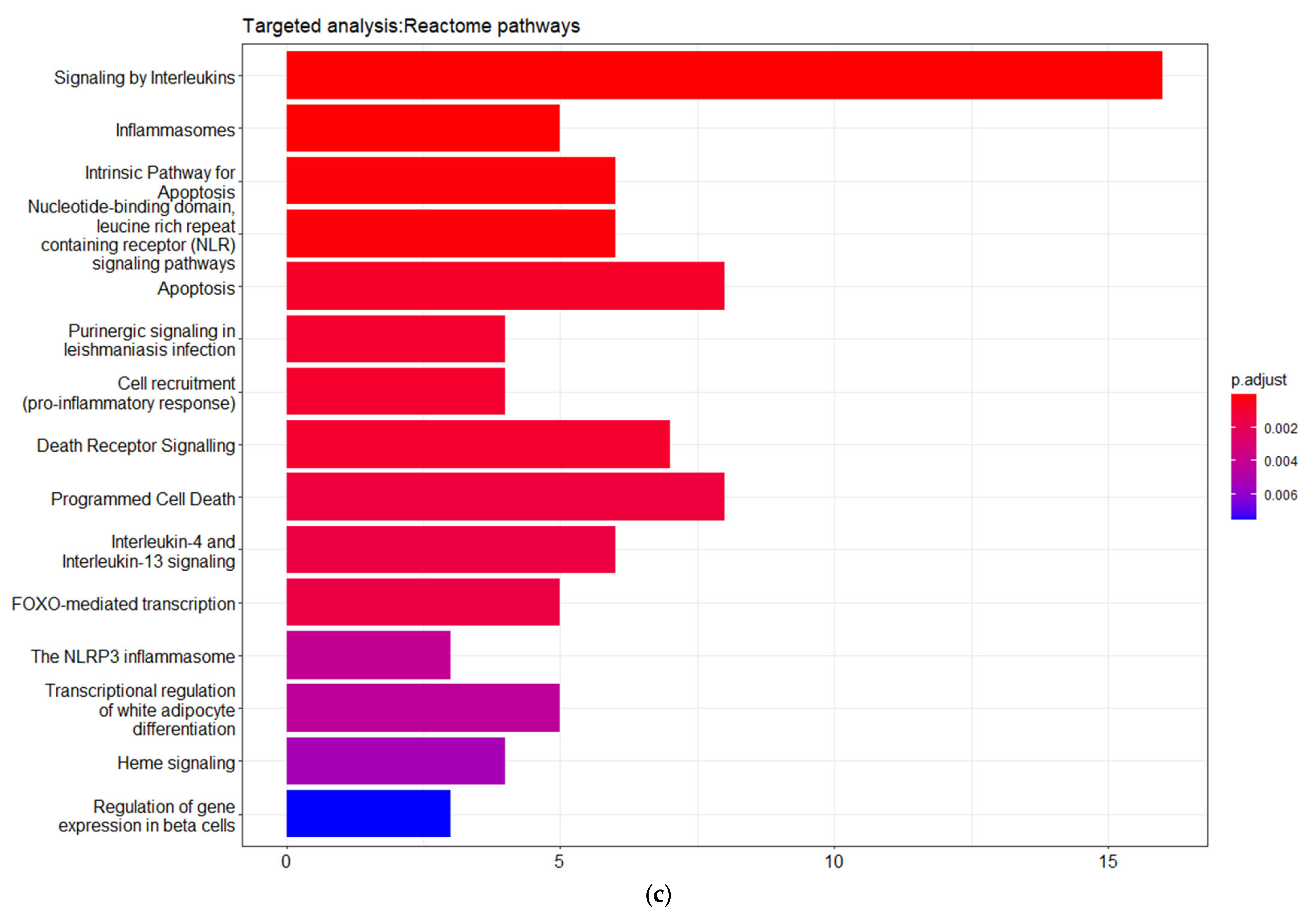
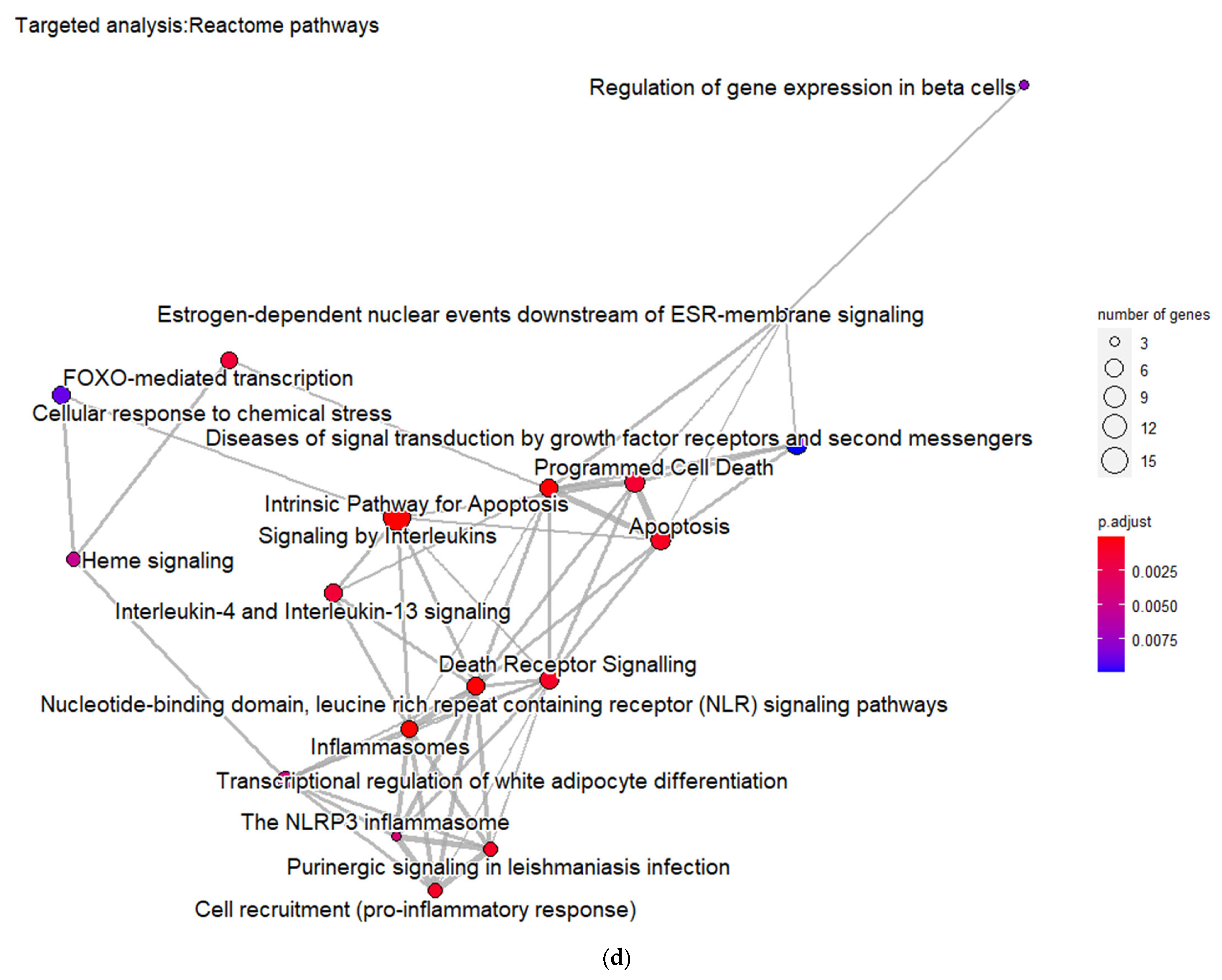
3.3. Targeted Analysis to Reveal Unique Shared Genes between the Experimentally Validated Target Genes of DMT2-Related miRNAs and the Genes Implicated in DMT2 Pathogenesis Relevant to Pharmacogenomics Analysis
3.4. Assessment of Drug Interactions of Various Pharmacological Classes of Antidiabetic Drugs
3.4.1. Sulfonylureas and Glinides

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
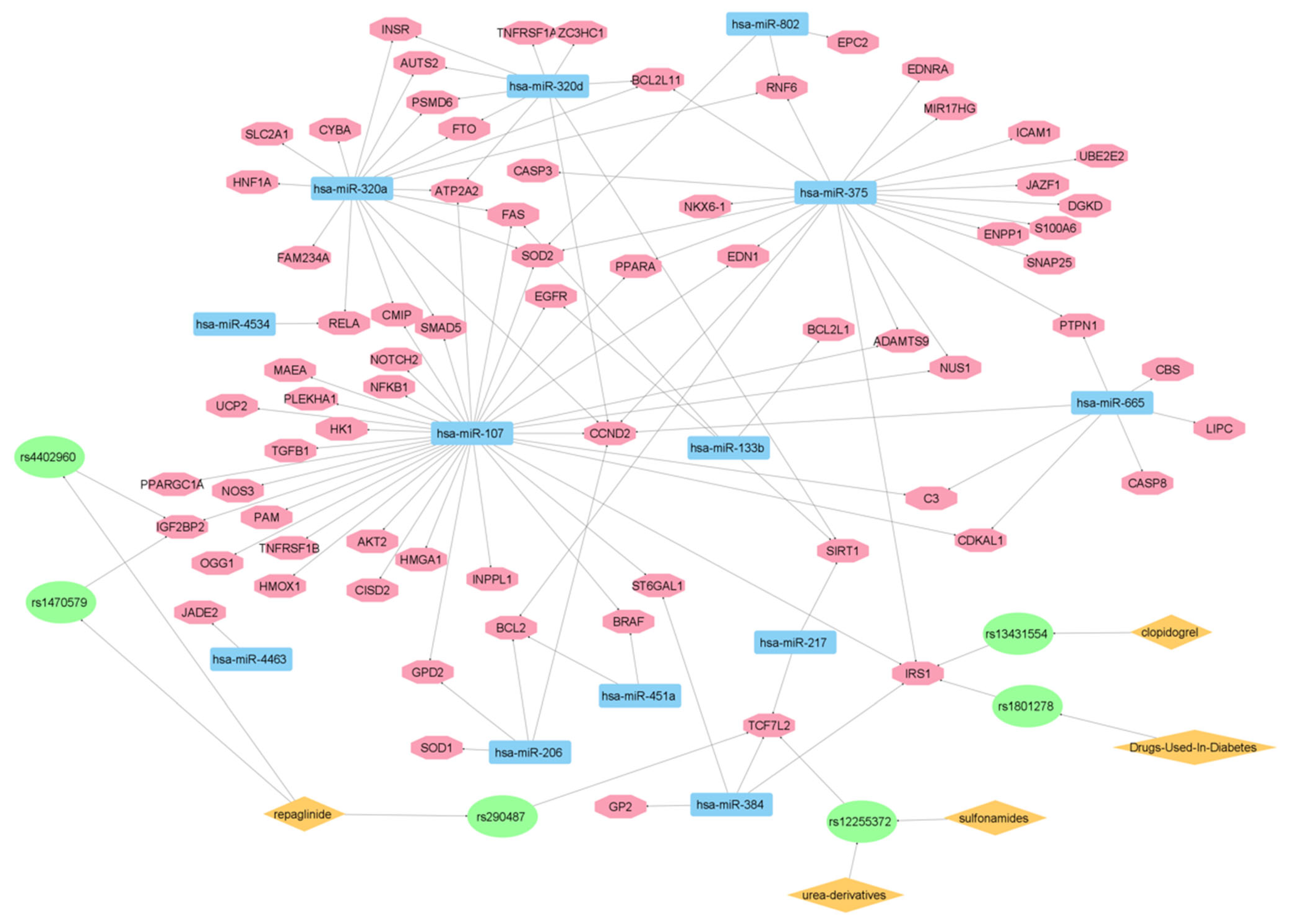{kind=link}
{kind=link}
| Drugs | CO-Administered Drugs | Effect | References |
|---|---|---|---|
| Glibenclamide | Statins | Cmax and AUC of glibenclamide increased by up to 20% | [70] |
| SUs | Inducers of CYP2C9 (carbamazepine, phenobarbital, rifampicin, ritonavir, St John’s wort) | Increased elimination rate of SUs | [70] |
| SUs | Inhibitors of CYP2C9 (amiodarone, cimetidine, ranitidine, trimethoprim, fluconazole, ketoconazole, voriconazole, fluoxetine, leflunomide, metronidazole | Prolong SU effect | [70] |
| Glibenclamide | Clarithromycin | Increased levels of glibenclamide | [70] |
| Glibenclamide | Verapamil | Increased levels of glibenclamide | [70] |
| SUs | Ethanol | Increased risk of hypoglycemia | [78] |
| SUs | Antiacids | Increased risk of hypoglycemia | [79] |
| SUs | Cholestyramine | Decreased absorption of SUs | [79] |
| Repaglinide | Gemfibrozil | Increased plasma concentration of repaglinide | [80] |
| Repaglinide | Cyclosporin | Increased plasma concentration of repaglinide | [80] |
| Repaglinide | Inducers of CYP enzymes (carbamazepine, phenytoin, St John’s wort, rifampicin) | Reduced plasma concentrations of repaglinide | [81] |
| Repaglinide | Ketoconazole (inhibitor of CYP3A4) | Increased AUC by 15% and the mean Cmax by 8% for repaglinide | [82] |
| Repaglinide | Macrolide antibiotics (clarithromycin) | The effect of repaglinide is increased | [83] |
| Repaglinide | Clopidogrel | Increased risk of hypoglycemia | [84] |
3.4.2. Metformin
3.4.3. Thiazolidinediones
| Drug | Co-Administered Drug | Effect | References |
|---|---|---|---|
| Pioglitazone | Gemfibrozil | Increased plasma concentration of pioglitazone | [106] |
| Pioglitazone | Trimethoprim | Increased plasma concentration of pioglitazone | [73] |
| Pioglitazone | Clopidogrel | Increased levels of pioglitazone | [84] |
| Pioglitazone | Ketoconazole | Increased levels of pioglitazone | [108] |
| Pioglitazone | Rifampicin | Decreased efficacy of pioglitazone | [109] |
3.4.4. DPP-4 Inhibitors
| Drug | Co-Administered Drug | Effect | References |
|---|---|---|---|
| Sitagliptin | Cyclosporin | Increased absorption of sitagliptin | [115] |
| Sitagliptin | Simvastatin | Risk of rhabdomyolysis | [117] |
| Vildagliptin | ACE Inhibitors | Increased risk of angioedema | [121] |
| Saxagliptin | Ketoconazole | Increased plasma concentration of saxagliptin | [124] |
| Saxagliptin | Diltiazem | Increased plasma concentration of saxagliptin | [124] |
| Saxagliptin | Macrolide antibiotics | Increased plasma concentration of saxagliptin | [70] |
3.4.5. GLP-1 Receptor Agonists (GLP-1RA)
3.4.6. SGLT2 Inhibitors
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- International Diabetes Federation. IDF Diabetes Atlas, 10th ed.; International Diabetes Federation: Brussels, Belgium, 2021; Available online: http://www.diabetesatlas.org/ (accessed on 10 February 2023).
- Kahn, S.E.; Cooper, M.E.; del Prato, S. Pathophysiology and treatment of type 2 diabetes: Perspectives on the past, present, and future. Lancet 2014, 383, 1068–1083. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deshpande, A.D.; Harris-Hayes, M.; Schootman, M. Epidemiology of Diabetes and Diabetes-Related Complications. Phys Ther. 2008, 88, 1254–1264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robertson, R.P.; Zhou, H.; Zhang, T.; Harmon, J.S. Chronic oxidative stress as a mechanism for glucose toxicity of the beta cell in Type 2 diabetes. Cell Biochem. Biophys. 2007, 48, 139–146. [Google Scholar] [CrossRef] [PubMed]
- Pickup, J.C. Inflammation and Activated Innate Immunity in the Pathogenesis of Type 2 Diabetes. Diabetes Care 2004, 27, 813–823. [Google Scholar] [CrossRef] [Green Version]
- Gupta, A.; Jelinek, H.F.; Al-Aubaidy, H. Glucagon like peptide-1 and its receptor agonists: Their roles in management of Type 2 diabetes mellitus. Diabetes Metab. Syndr. Clin. Res. Rev. 2017, 11, 225–230. [Google Scholar] [CrossRef]
- Tiwari, P. Recent Trends in Therapeutic Approaches for Diabetes Management: A Comprehensive Update. J. Diabetes Res. 2015, 2015, 340838. [Google Scholar] [CrossRef] [Green Version]
- Hinton, W.; Feher, M.; Munro, N.; Walker, M.; Lusignan, S. Real-world prevalence of the inclusion criteria for the LEADER trial: Data from a national general practice network. Diabetes Obes. Metab. 2019, 21, 1661–1667. [Google Scholar] [CrossRef]
- McGovern, A.; Feher, M.; Munro, N.; de Lusignan, S. Sodium-Glucose Co-transporter 2 (SGLT2) Inhibitor: Comparing Trial Data and Real-World Use. Diabetes Ther. 2017, 8, 365–376. [Google Scholar] [CrossRef] [Green Version]
- Bell, J. Stratified medicines: Towards better treatment for disease. Lancet 2014, 383, S3–S5. [Google Scholar] [CrossRef]
- Dennis, J.M. Precision Medicine in Type 2 Diabetes: Using Individualized Prediction Models to Optimize Selection of Treatment. Diabetes 2020, 69, 2075–2085. [Google Scholar] [CrossRef]
- Shields, B.M.; Dennis, J.M.; Angwin, C.D.; Warren, F.; Henley, W.E.; Farmer, A.J.; Sattar, N.; Holman, R.R.; Jones, A.G.; Pearson, E.R.; et al. Patient stratification for determining optimal second-line and third-line therapy for type 2 diabetes: The TriMaster study. Nat. Med. 2022, 29, 376–383. [Google Scholar] [CrossRef]
- Xie, F.; Chan, J.C.; Ma, R.C. Precision medicine in diabetes prevention, classification and management. J. Diabetes Investig. 2018, 9, 998–1015. [Google Scholar] [CrossRef] [Green Version]
- Pearson, E.R. Diabetes: Is There a Future for Pharmacogenomics Guided Treatment? Clin. Pharmacol. Ther. 2019, 106, 329–337. [Google Scholar] [CrossRef] [Green Version]
- Khoshnejat, M.; Kavousi, K.; Banaei-Moghaddam, A.M.; Moosavi-Mohavedi, A.A. Unraveling the molecular heterogeneity in type 2 diabetes: A potential subtype discovery followed by metabolic modeling. BMC Med Genomics 2020, 13, 119. [Google Scholar] [CrossRef] [PubMed]
- Dawed, A.Y.; Mari, A.; Brown, A.; McDonald, T.J.; Li, L.; Wang, S.; Hong, M.-G.; Sharma, S.; Robertson, N.R.; Mahajan, A.; et al. Pharmacogenomics of GLP-1 receptor agonists: A genome-wide analysis of observational data and large randomised controlled trials. Lancet Diabetes Endocrinol. 2023, 11, 33–41. [Google Scholar] [CrossRef] [PubMed]
- Whirl-Carrillo, M.; Huddart, R.; Gong, L.; Sangkuhl, K.; Thorn, C.F.; Whaley, R.; Klein, T.E. An Evidence-Based Framework for Evaluating Pharmacogenomics Knowledge for Personalized Medicine. Clin. Pharmacol. Ther. 2021, 110, 563–572. [Google Scholar] [CrossRef] [PubMed]
- Kahn, S.E.; Hull, R.L.; Utzschneider, K.M. Mechanisms linking obesity to insulin resistance and type 2 diabetes. Nature 2006, 444, 840–846. [Google Scholar] [CrossRef]
- Malone, J.I.; Hansen, B.C. Does obesity cause type 2 diabetes mellitus (T2DM)? Or is it the opposite? Pediatr. Diabetes 2019, 20, 5–9. [Google Scholar] [CrossRef] [Green Version]
- Akhtar, M.; Taha, N.M.; Nauman, A.; Mujeeb, I.B.; Al-Nabet, A.D.M.H. Diabetic Kidney Disease: Past and Present. Adv. Anat. Pathol. 2020, 27, 87–97. [Google Scholar] [CrossRef]
- Koye, D.N.; Magliano, D.J.; Nelson, R.G.; Pavkov, M.E. The Global Epidemiology of Diabetes and Kidney Disease. Adv. Chronic Kidney Dis. 2018, 25, 121–132. [Google Scholar] [CrossRef]
- Tanase, D.M.; Gosav, E.M.; Costea, C.F.; Ciocoiu, M.; Lacatusu, C.M.; Maranduca, M.A.; Ouatu, A.; Floria, M. The Intricate Relationship between Type 2 Diabetes Mellitus (T2DM), Insulin Resistance (IR), and Nonalcoholic Fatty Liver Disease (NAFLD). J. Diabetes Res. 2020, 2020, 3920196. [Google Scholar] [CrossRef] [PubMed]
- Muzica, C.M.; Sfarti, C.; Trifan, A.; Zenovia, S.; Cuciureanu, T.; Nastasa, R.; Huiban, L.; Cojocariu, C.; Singeap, A.-M.; Girleanu, I.; et al. Nonalcoholic Fatty Liver Disease and Type 2 Diabetes Mellitus: A Bidirectional Relationship. Can. J. Gastroenterol. Hepatol. 2020, 2020, 6638306. [Google Scholar] [CrossRef]
- Filippatos, T.D.; Alexakis, K.; Mavrikaki, V.; Mikhailidis, D.P. Nonalcoholic Fatty Pancreas Disease: Role in Metabolic Syndrome, “Prediabetes”, Diabetes and Atherosclerosis. Dig. Dis. Sci. 2022, 67, 26–41. [Google Scholar] [CrossRef]
- Livadas, S.; Anagnostis, P.; Bosdou, J.K.; Bantouna, D.; Paparodis, R. Polycystic ovary syndrome and type 2 diabetes mellitus: A state-of-the-art review. World J. Diabetes 2022, 13, 5–26. [Google Scholar] [CrossRef]
- Oguntibeju, O.O. Type 2 diabetes mellitus, oxidative stress and inflammation: Examining the links. Int. J. Physiol. Pathophysiol. Pharmacol. 2019, 11, 45–63. [Google Scholar]
- Galicia-Garcia, U.; Benito-Vicente, A.; Jebari, S.; Larrea-Sebal, A.; Siddiqi, H.; Uribe, K.; Ostolaza, H.; Martín, C. Pathophysiology of Type 2 Diabetes Mellitus. Int. J. Mol. Sci. 2020, 21, 6275. [Google Scholar] [CrossRef]
- Czyżyk, A.; Szczepanik, Z. Diabetes mellitus and cancer. Eur. J. Intern. Med. 2000, 11, 245–252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Larsson, S.C.; Mantzoros, C.S.; Wolk, A. Diabetes mellitus and risk of breast cancer: A meta-analysis. Int. J. Cancer 2007, 121, 856–862. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.F.; Liu, M.D.; Chen, P.; Chen, L.H.; Chang, Y.H.; Wen, P.C.; Li, C.Y. Risks of Breast and Endometrial Cancer in Women with Diabetes: A Population-Based Cohort Study. PLoS ONE 2013, 8, e67420. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Wang, L.; Zhang, J.; Wang, B.; Liu, H. Association between diabetes mellitus and subsequent ovarian cancer in women. Medicine 2017, 96, e6396. [Google Scholar] [CrossRef]
- Friedenreich, C.M. Review of anthropometric factors and breast cancer risk. Eur. J. Cancer Prev. 2001, 10, 15–32. [Google Scholar] [CrossRef] [PubMed]
- Risch, H.A. Hormonal Etiology of Epithelial Ovarian Cancer, with a Hypothesis Concerning the Role of Androgens and Progesterone. JNCI J. Natl. Cancer Inst. 1998, 90, 1774–1786. [Google Scholar] [CrossRef] [PubMed]
- Sciacca, L.; Costantino, A.; Pandini, G.; Mineo, R.; Frasca, F.; Scalia, P.; Sbraccia, P.; Goldfine, I.D.; Vigneri, R.; Belfiore, A. Insulin receptor activation by IGF-II in breast cancers: Evidence for a new autocrine/paracrine mechanism. Oncogene 1999, 18, 2471–2479. [Google Scholar] [CrossRef]
- Shen, M.-R.; Lin, A.-C.; Hsu, Y.-M.; Chang, T.-J.; Tang, M.-J.; Alper, S.L.; Ellory, J.C.; Chou, C.-Y. Insulin-like Growth Factor 1 Stimulates KCl Cotransport, Which Is Necessary for Invasion and Proliferation of Cervical Cancer and Ovarian Cancer Cells. J. Biol. Chem. 2004, 279, 40017–40025. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vona-Davis, L.; Rose, D.P. Type 2 Diabetes and Obesity Metabolic Interactions: Common Factors for Breast Cancer Risk and Novel Approaches to Prevention and Therapy. Curr. Diabetes Rev. 2012, 8, 116–130. [Google Scholar] [CrossRef] [PubMed]
- Kim, R.P.; Edelman, S.V.; Kim, D.D. Musculoskeletal Complications of Diabetes Mellitus. Clin. Diabetes 2001, 19, 132–135. [Google Scholar] [CrossRef] [Green Version]
- Davey, J.S.; Carmichael, R.E.; Craig, T.J. Protein SUMOylation regulates insulin secretion at multiple stages. Sci. Rep. 2019, 9, 2895. [Google Scholar] [CrossRef] [Green Version]
- Strycharz, J.; Drzewoski, J.; Szemraj, J.; Sliwinska, A. Is p53 Involved in Tissue-Specific Insulin Resistance Formation? Oxid. Med. Cell. Longev. 2017, 2017, 9270549. [Google Scholar] [CrossRef] [Green Version]
- Shirakawa, J.; Fernandez, M.; Takatani, T.; el Ouaamari, A.; Jungtrakoon, P.; Okawa, E.R.; Zhang, W.; Yi, P.; Doria, A.; Kulkarni, R.N. Insulin Signaling Regulates the FoxM1/PLK1/CENP-A Pathway to Promote Adaptive Pancreatic β Cell Proliferation. Cell Metab. 2017, 25, 868–882.e5. [Google Scholar] [CrossRef] [Green Version]
- Zhao, L.; Zou, Y.; Liu, F. Transforming Growth Factor-Beta1 in Diabetic Kidney Disease. Front. Cell Dev. Biol. 2020, 8, 187. [Google Scholar] [CrossRef] [Green Version]
- Yamauchi, T.; Ohnaka, K.; Takayanagi, R.; Umeda, F.; Nawata, H. Enhanced secretion of endothelin-1 by elevated glucose levels from cultured bovine aortic endothelial cells. FEBS Lett. 1990, 267, 16–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anguiano, L.; Riera, M.; Pascual, J.; Soler, M. Endothelin Blockade in Diabetic Kidney Disease. J. Clin. Med. 2015, 4, 1171–1192. [Google Scholar] [CrossRef] [Green Version]
- Bego, T.; Čaušević, A.; Dujić, T.; Malenica, M.; Asimi, Z.V.; Prnjavorac, B.; Marc, J.; Nekvindová, J.; Palička, V.; Semiz, S. Association of FTO gene variant (rs8050136) with type 2 diabetes and markers of obesity, glycaemic control and inflammation. J. Med. Biochem. 2019, 38, 153–163. [Google Scholar] [CrossRef] [Green Version]
- Gutiérrez-Salmerón, M.; Lucena, S.R.; Chocarro-Calvo, A.; García-Martínez, J.M.; Martín Orozco, R.M.; García-Jiménez, C. Metabolic and hormonal remodeling of colorectal cancer cell signalling by diabetes. Endocr. Relat. Cancer 2021, 28, R191–R206. [Google Scholar] [CrossRef] [PubMed]
- Santinon, G.; Pocaterra, A.; Dupont, S. Control of YAP/TAZ Activity by Metabolic and Nutrient-Sensing Pathways. Trends Cell Biol. 2016, 26, 289–299. [Google Scholar] [CrossRef] [PubMed]
- Chocarro-Calvo, A.; García-Martínez, J.M.; Ardila-González, S.; De la Vieja, A.; García-Jiménez, C. Glucose-Induced β-Catenin Acetylation Enhances Wnt Signaling in Cancer. Mol. Cell 2013, 49, 474–486. [Google Scholar] [CrossRef] [Green Version]
- González, N.; Prieto, I.; del Puerto-Nevado, L.; Portal-Nuñez, S.; Ardura, J.A.; Corton, M.; Fernández-Fernández, B.; Aguilera, O.; Gomez-Guerrero, C.; Mas, S.; et al. 2017 update on the relationship between diabetes and colorectal cancer: Epidemiology, potential molecular mechanisms and therapeutic implications. Oncotarget 2017, 8, 18456–18485. [Google Scholar] [CrossRef] [Green Version]
- Mitroi, A.F.; Leopa, N.; Dumitru, E.; Brînzan, C.; Tocia, C.; Dumitru, A.; Popescu, R.C. Association of TCF7L2, CASC8 and GREM1 Polymorphisms in Patients with Colorectal Cancer and Type II Diabetes Mellitus. Genes 2022, 13, 1297. [Google Scholar] [CrossRef]
- Peng, S.; Zhu, Y.; Lü, B.; Xu, F.; Li, X.; Lai, M. TCF7L2 gene polymorphisms and type 2 diabetes risk: A comprehensive and updated meta-analysis involving 121 174 subjects. Mutagenesis 2013, 28, 25–37. [Google Scholar] [CrossRef] [Green Version]
- Sainz, J.; Rudolph, A.; Hoffmeister, M.; Frank, B.; Brenner, H.; Chang-Claude, J.; Hemminki, K.; Försti, A. Effect of Type 2 Diabetes Predisposing Genetic Variants on Colorectal Cancer Risk. J. Clin. Endocrinol. Metab. 2012, 97, E845–E851. [Google Scholar] [CrossRef] [Green Version]
- Cheng, I.; Caberto, C.P.; Lum-Jones, A.; Seifried, A.; Wilkens, L.R.; Schumacher, F.; Monroe, K.R.; Lim, U.; Tiirikainen, M.; Kolonel, L.N.; et al. Type 2 diabetes risk variants and colorectal cancer risk: The Multiethnic Cohort and PAGE studies. Gut 2011, 60, 1703–1711. [Google Scholar] [CrossRef] [PubMed]
- Huang, Q.; Yin, J.; Dai, X.; Pei, Q.; Dong, M.; Zhou, Z.; Huang, X.; Yu, M.; Zhou, H.; Liu, Z. IGF2BP2 variations influence repaglinide response and risk of type 2 diabetes in Chinese population. Acta Pharmacol. Sin. 2010, 31, 709–717. [Google Scholar] [CrossRef] [PubMed]
- Shokouhi, S.; Delpisheh, A.; Haghani, K.; Mahdizadeh, M.; Bakhtiyari, S. Association of rs7903146, rs12255372, and rs290487 Polymorphisms in TCF7L2 Gene with Type 2 Diabetes in an Iranian Kurdish Ethnic Group. Clin. Lab. 2014, 60, 1269–1276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Albegali, A.A.; Shahzad, M.; Mahmood, S.; Ullah, M.I. Genetic association of insulin receptor substrate-1 (IRS-1, rs1801278) gene with insulin resistant of type 2 diabetes mellitus in a Pakistani population. Mol. Biol. Rep. 2019, 46, 6065–6070. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Zhang, X.; Liu, D.; Liu, T.; Cai, W.; Yan, C.; Han, Y. Association between insulin receptor substrate-1 polymorphisms and high platelet reactivity with clopidogrel therapy in coronary artery disease patients with type 2 diabetes mellitus. Cardiovasc. Diabetol. 2016, 15, 50. [Google Scholar] [CrossRef] [Green Version]
- Esposito, D.L.; Aru, F.; Lattanzio, R.; Morgano, A.; Abbondanza, M.; Malekzadeh, R.; Bishehsari, F.; Valanzano, R.; Russo, A.; Piantelli, M.; et al. The Insulin Receptor Substrate 1 (Irs1) in Intestinal Epithelial Differentiation and in Colorectal Cancer. PLoS ONE 2012, 7, e36190. [Google Scholar] [CrossRef] [Green Version]
- Dearth, R.K.; Cui, X.; Kim, H.J.; Hadsell, D.L.; Lee, A.V. Oncogenic Transformation by the Signaling Adaptor Proteins Insulin Receptor Substrate (IRS)-1 and IRS-2. Cell Cycle 2007, 6, 705–713. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Zhang, B.; Wang, X.; Liu, Z.; Li, J.; Zhang, S.; Gu, X.; Jia, M.; Guo, H.; Feng, N.; et al. P53/PANK1/miR-107 signalling pathway spans the gap between metabolic reprogramming and insulin resistance induced by high-fat diet. J. Cell. Mol. Med. 2020, 24, 3611–3624. [Google Scholar] [CrossRef] [Green Version]
- De Gonzalo-Calvo, D.; van der Meer, R.W.; Rijzewijk, L.J.; Smit, J.W.A.; Revuelta-Lopez, E.; Nasarre, L.; Escola-Gil, J.C.; Lamb, H.J.; Llorente-Cortes, V. Serum microRNA-1 and microRNA-133a levels reflect myocardial steatosis in uncomplicated type 2 diabetes. Sci. Rep. 2017, 7, 47. [Google Scholar] [CrossRef] [PubMed]
- Di Mauro, V.; Crasto, S.; Colombo, F.S.; di Pasquale, E.; Catalucci, D. Wnt signalling mediates miR-133a nuclear re-localization for the transcriptional control of Dnmt3b in cardiac cells. Sci. Rep. 2019, 9, 9320. [Google Scholar] [CrossRef] [Green Version]
- Hua, Y.T.; Xu, W.X.; Li, H.; Xia, M. Emerging roles of MiR-133a in human cancers. J. Cancer 2021, 12, 198–206. [Google Scholar] [CrossRef] [PubMed]
- Li, X. miR-375, a microRNA related to diabetes. Gene 2014, 533, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Sedgeman, L.R.; Beysen, C.; Solano, M.A.R.; Michell, D.L.; Sheng, Q.; Zhao, S.; Turner, S.; Linton, M.F.; Vickers, K.C. Beta cell secretion of miR-375 to HDL is inversely associated with insulin secretion. Sci. Rep. 2019, 9, 3803. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, J.; Lu, Y.; Wang, R.; Xu, X.; Liu, Q.; He, S.; Pan, H.; Liu, X.; Yuan, B.; Ding, Y.; et al. MicroRNA-375: Potential cancer suppressor and therapeutic drug. Biosci. Rep. 2021, 41, BSR20211494. [Google Scholar] [CrossRef] [PubMed]
- Du, H.; Zhao, Y.; Yin, Z.; Wang, D.W.; Chen, C. The role of miR-320 in glucose and lipid metabolism disorder-associated diseases. Int. J. Biol. Sci. 2021, 17, 402–416. [Google Scholar] [CrossRef] [PubMed]
- Du, H.; Yin, Z.; Zhao, Y.; Li, H.; Dai, B.; Fan, J.; He, M.; Nie, X.; Wang, C.-Y.; Wang, D.W.; et al. miR-320a induces pancreatic β cells dysfunction in diabetes by inhibiting MafF. Mol. Ther. Nucleic Acids 2021, 26, 444–457. [Google Scholar] [CrossRef]
- Wan, C.; Wen, J.; Liang, X.; Xie, Q.; Wu, W.; Wu, M.; Liu, Z. Identification of miR-320 family members as potential diagnostic and prognostic biomarkers in myelodysplastic syndromes. Sci. Rep. 2021, 11, 183. [Google Scholar] [CrossRef]
- Al-Saleh, Y.; Sabico, S.; Al-Furqani, A.; Jayyousi, A.; Alromaihi, D.; Ba-Essa, E.; Alawadi, F.; Alkaabi, J.; Hassanein, M.; Al-Sifri, S.; et al. Sulfonylureas in the Current Practice of Type 2 Diabetes Management: Are They All the Same? Consensus from the Gulf Cooperation Council (GCC) Countries Advisory Board on Sulfonylureas. Diabetes Ther. 2021, 12, 2115–2132. [Google Scholar] [CrossRef]
- May, M.; Schindler, C. Clinically and pharmacologically relevant interactions of antidiabetic drugs. Ther. Adv. Endocrinol. Metab. 2016, 7, 69–83. [Google Scholar] [CrossRef] [Green Version]
- Holstein, A.; Beil, W.; Kovacs, P. CYP2C metabolism of oral antidiabetic drugs—Impact on pharmacokinetics, drug interactions and pharmacogenetic aspects. Expert Opin. Drug Metab. Toxicol. 2012, 8, 1549–1563. [Google Scholar] [CrossRef]
- Holstein, A.; Beil, W. Oral antidiabetic drug metabolism: Pharmacogenomics and drug interactions. Expert Opin. Drug Metab. Toxicol. 2009, 5, 225–241. [Google Scholar] [CrossRef] [PubMed]
- Tornio, A.; Niemi, M.; Neuvonen, P.J.; Backman, J.T. Drug interactions with oral antidiabetic agents: Pharmacokinetic mechanisms and clinical implications. Trends Pharmacol. Sci. 2012, 33, 312–322. [Google Scholar] [CrossRef] [PubMed]
- Parekh, T.M.; Raji, M.; Lin, Y.L.; Tan, A.; Kuo, Y.F.; Goodwin, J.S. Hypoglycemia After Antimicrobial Drug Prescription for Older Patients Using Sulfonylureas. JAMA Intern. Med. 2014, 174, 1605–1612. [Google Scholar] [CrossRef] [Green Version]
- Tirkkonen, T.; Heikkilä, P.; Huupponen, R.; Laine, K. Potential CYP2C9-mediated drug-drug interactions in hospitalized type 2 diabetes mellitus patients treated with the sulphonylureas glibenclamide, glimepiride or glipizide. J. Intern. Med. 2010, 268, 359–366. [Google Scholar] [CrossRef]
- Zu Schwabedissen, H.E.M.; Boettcher, K.; Steiner, T.; Schwarz, U.I.; Keiser, M.; Kroemer, H.K.; Siegmund, W. OATP1B3 is expressed in pancreatic β-islet cells and enhances the insulinotropic effect of the sulfonylurea derivative glibenclamide. Diabetes 2014, 63, 775–784. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scheen, A.J. Dipeptidylpeptidase-4 inhibitors (gliptins): Focus on drug-drug interactions. Clin. Pharmacokinet. 2010, 49, 573–588. [Google Scholar] [CrossRef] [PubMed]
- Lao, B.; Czyzyk, A.; Szutowski, M.; Szczepanik, Z. Alcohol tolerance in patients with non-insulin-dependent (type 2) diabetes treated with sulphonylurea derivatives. Arzneimittelforschung 1994, 44, 727–734. [Google Scholar]
- Amin, M.; Suksomboon, N. Pharmacotherapy of Type 2 Diabetes Mellitus: An Update on Drug–Drug Interactions. Drug Saf. 2014, 37, 903–919. [Google Scholar] [CrossRef]
- Pakkir Maideen, N.M.; Manavalan, G.; Balasubramanian, K. Drug interactions of meglitinide antidiabetics involving CYP enzymes and OATP1B1 transporter. Ther. Adv. Endocrinol. Metab. 2018, 9, 259–268. [Google Scholar] [CrossRef] [Green Version]
- Niemi, M.; Backman, J.T.; Neuvonen, M.; Neuvonen, P.J.; Kivistö, K.T. Rifampin decreases the plasma concentrations and effects of repaglinide. Clin. Pharmacol. Ther. 2000, 68, 495–500. [Google Scholar] [CrossRef]
- Hatorp, V.; Hansen, K.T.; Thomsen, M.S. Influence of Drugs Interacting with CYP3A4 on the Pharmacokinetics, Pharmacodynamics, and Safety of the Prandial Glucose Regulator Repaglinide. J. Clin. Pharmacol. 2003, 43, 649–660. [Google Scholar] [CrossRef]
- Niemi, M.; Neuvonen, P.J.; Kivistö, K.T. The cytochrome P4503A4 inhibitor clarithromycin increases the plasma concentrations and effects of repaglinide. Clin. Pharmacol. Ther. 2001, 70, 58–65. [Google Scholar] [CrossRef]
- Kim, S.-J.; Yoshikado, T.; Ieiri, I.; Maeda, K.; Kimura, M.; Irie, S.; Kusuhara, H.; Sugiyama, Y. Clarification of the Mechanism of Clopidogrel-Mediated Drug–Drug Interaction in a Clinical Cassette Small-dose Study and Its Prediction Based on In Vitro Information. Drug Metab. Dispos. 2016, 44, 1622–1632. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Milner, Z.; Akhondi, H. Repaglinide. In xPharm: The Comprehensive Pharmacology Reference; StatPearls: Orlando, FL, USA, 2021; pp. 1–3. [Google Scholar]
- Rang, H.; Dale, M. Rang and Dale’s Pharmacology, 7th ed.; Elsevier Churchill Livingstone: Edinburgh, UK, 2012. [Google Scholar]
- Niemi, M.; Backman, J.T.; Neuvonen, M.; Neuvonen, P.J. Effects of gemfibrozil, itraconazole, and their combination on the pharmacokinetics and pharmacodynamics of repaglinide: Potentially hazardous interaction between gemfibrozil and repaglinide. Diabetologia 2003, 46, 347–351. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Chen, M.; Zhu, L.; Yu, L.; Zeng, S.; Xiang, M.; Zhou, Q. Pharmacokinetic drug interactions with clopidogrel: Updated review and risk management in combination therapy. Ther. Clin. Risk Manag. 2015, 11, 449–467. [Google Scholar] [PubMed] [Green Version]
- Scheen, A.J. Clinical Pharmacokinetics of Metformin. Clin. Pharmacokinet. 1996, 30, 359–371. [Google Scholar] [CrossRef] [PubMed]
- Rena, G.; Hardie, D.G.; Pearson, E.R. The mechanisms of action of metformin. Diabetologia 2017, 60, 1577–1585. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hibma, J.E.; Zur, A.A.; Castro, R.A.; Wittwer, M.B.; Keizer, R.J.; Yee, S.W.; Goswami, S.; Stocker, S.L.; Zhang, X.; Huang, Y.; et al. The Effect of Famotidine, a MATE1-Selective Inhibitor, on the Pharmacokinetics and Pharmacodynamics of Metformin. Clin. Pharmacokinet. 2016, 55, 711–721. [Google Scholar] [CrossRef] [Green Version]
- Seo, J.H.; Lee, D.Y.; Hong, C.W.; Lee, I.H.; Ahn, K.S.; Kang, G.W. Severe lactic acidosis and acute pancreatitis associated with cimetidine in a patient with type 2 diabetes mellitus taking metformin. Intern. Med. 2013, 52, 2245–2248. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.J.; Yin, O.Q.P.; Tomlinson, B.; Chow, M.S.S. OCT2 polymorphisms and in-vivo renal functional consequence: Studies with metformin and cimetidine. Pharmacogenet. Genom. 2008, 18, 637–645. [Google Scholar] [CrossRef]
- Ahlin, G.; Chen, L.; Lazorova, L.; Chen, Y.; Ianculescu, A.G.; Davis, R.L.; Giacomini, K.M.; Artursson, P. Genotype-dependent effects of inhibitors of the organic cation transporter, OCT1: Predictions of metformin interactions. Pharm. J. 2010, 11, 400–411. [Google Scholar] [CrossRef]
- Grün, B.; Kiessling, M.K.; Burhenne, J.; Riedel, K.-D.; Weiss, J.; Rauch, G.; Haefeli, W.E.; Czock, D. Trimethoprim–metformin interaction and its genetic modulation by OCT2 and MATE1 transporters. Br. J. Clin. Pharmacol. 2013, 76, 787–796. [Google Scholar] [CrossRef] [Green Version]
- Kim, A.; Chung, I.; Yoon, S.H.; Yu, K.-S.; Lim, K.S.; Cho, J.-Y.; Lee, H.; Jang, I.-J.; Chung, J.Y. Effects of proton pump inhibitors on metformin pharmacokinetics and pharmacodynamics. Drug Metab. Dispos. 2014, 42, 1174–1179. [Google Scholar] [CrossRef] [Green Version]
- Herrmann, W.; Obeid, R. Ursachen und frühzeitige diagnostik von vitamin-B12-mangel. Dtsch. Arztebl. 2008, 105, 680–685. [Google Scholar]
- Sheleme, T. Clinical Pharmacokinetics of Metformin. In Metformin—Pharmacology and Drug Interactions; IntechOpen: London, UK, 2021. [Google Scholar]
- Zack, J.; Berg, J.; Juan, A.; Pannacciulli, N.; Allard, M.; Gottwald, M.; Zhang, H.; Shao, Y.; Ben-Yehuda, O.; Jochelson, P. Pharmacokinetic drug-drug interaction study of ranolazine and metformin in subjects with type 2 diabetes mellitus. Clin. Pharmacol. Drug Dev. 2015, 4, 121–129. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.-R.; Shi, A.-X.; Qin, H.-Y.; Zhang, T.; Wu, Y.-F.; Zhang, G.-Q.; Wu, X.-A. Metoprolol decreases the plasma exposure of metformin via the induction of liver, kidney and muscle uptake in rats. Biopharm. Drug Dispos. 2016, 37, 511–521. [Google Scholar] [CrossRef] [PubMed]
- Maideen, N.M.P.; Jumale, A.; Balasubramaniam, R. Drug Interactions of Metformin Involving Drug Transporter Proteins. Adv. Pharm. Bull. 2017, 7, 501–505. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Long, A.N.; Atwell, C.L.; Yoo, W.; Solomon, S.S. Vitamin B12 Deficiency Associated with Concomitant Metformin and Proton Pump Inhibitor Use. Diabetes Care 2012, 35, e84. [Google Scholar] [CrossRef] [Green Version]
- Stage, T.B.; Brøsen, K.; Christensen, M.M.H. A Comprehensive Review of Drug–Drug Interactions with Metformin. Clin. Pharmacokinet. 2015, 54, 811–824. [Google Scholar] [CrossRef]
- Dujic, T.; Zhou, K.; Donnelly, L.A.; Tavendale, R.; Palmer, C.N.A.; Pearson, E.R. Association of Organic Cation Transporter 1 With Intolerance to Metformin in Type 2 Diabetes: A GoDARTS Study. Diabetes 2015, 64, 1786–1793. [Google Scholar] [CrossRef] [Green Version]
- Davidson, M.A.; Mattison, D.R.; Azoulay, L.; Krewski, D. Thiazolidinedione drugs in the treatment of type 2 diabetes mellitus: Past, present and future. Crit. Rev. Toxicol. 2017, 48, 52–108. [Google Scholar] [CrossRef]
- Jaakkola, T.; Backman, J.T.; Neuvonen, M.; Neuvonen, P.J. Effects of Gemfibrozil, Itraconazole, and Their Combination on the Pharmacokinetics of Pioglitazone. Clin. Pharmacol. Ther. 2005, 77, 404–414. [Google Scholar] [CrossRef]
- Ledl, M.; Hohenecker, J.; Francesconi, C.; Roots, I.; Bauer, M.F.; Roden, M. Acute myopathy in a type 2 diabetic patient on combination therapy with metformin, fenofibrate and rosiglitazone. Diabetologia 2005, 48, 1996–1998. [Google Scholar] [CrossRef] [Green Version]
- Park, J.Y.; Kim, K.A.; Shin, J.G.; Lee, K.Y. Effect of ketoconazole on the pharmacokinetics of rosiglitazone in healthy subjects. Br. J. Clin. Pharmacol. 2004, 58, 397–402. [Google Scholar] [CrossRef] [Green Version]
- Jaakkola, T.; Backman, J.T.; Neuvonen, M.; Laitila, J.; Neuvonen, P.J. Effect of rifampicin on the pharmacokinetics of pioglitazone. Br. J. Clin. Pharmacol. 2006, 61, 70–78. [Google Scholar] [CrossRef] [PubMed]
- Park, J.Y.; Kim, K.A.; Kang, M.H.; Kim, S.L.; Shin, J.G. Effect of Rifampin on the Pharmacokinetics of Rosiglitazone in Healthy Subjects. Clin. Pharmacol. Ther. 2004, 75, 157–162. [Google Scholar] [CrossRef]
- Omar, B.A.; Vikman, J.; Winzell, M.S.; Voss, U.; Ekblad, E.; Foley, J.E.; Ahrén, B. Enhanced beta cell function and anti-inflammatory effect after chronic treatment with the dipeptidyl peptidase-4 inhibitor vildagliptin in an advanced-aged diet-induced obesity mouse model. Diabetologia 2013, 56, 1752–1760. [Google Scholar] [CrossRef] [PubMed]
- Drucker, D.J.; Nauck, M.A. The incretin system: Glucagon-like peptide-1 receptor agonists and dipeptidyl peptidase-4 inhibitors in type 2 diabetes. Lancet 2006, 368, 1696–1705. [Google Scholar] [CrossRef]
- American Diabetes Association. Standards of Medical Care in Diabetes—2010. Diabetes Care 2010, 33 (Suppl. S1), S11–S61. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zerilli, T.; Pyon, E.Y. Sitagliptin phosphate: A DPP-4 inhibitor for the treatment of type 2 diabetes mellitus. Clin. Ther. 2007, 29, 2614–2634. [Google Scholar] [CrossRef] [PubMed]
- Krishna, R.; Bergman, A.; Larson, P.; Cote, J.; Lasseter, K.; Dilzer, S.; Wang, A.; Zeng, W.; Chen, L.; Wagner, J.; et al. Effect of a single cyclosporine dose on the single-dose pharmacokinetics of sitagliptin (MK-0431), a dipeptidyl peptidase-4 inhibitor, in healthy male subjects. J. Clin. Pharmacol. 2007, 47, 165–174. [Google Scholar] [CrossRef]
- Mistry, G.C.; Bergman, A.J.; Zheng, W.; Hreniuk, D.; Zinny, M.A.; Gottesdiener, K.M.; Wagner, J.A.; Herman, G.A.; Ruddy, M. Sitagliptin, an dipeptidyl peptidase-4 inhibitor, does not alter the pharmacokinetics of the sulphonylurea, glyburide, in healthy subjects. Br. J. Clin. Pharmacol. 2008, 66, 36–42. [Google Scholar] [CrossRef] [Green Version]
- Kao, D.P.; Kohrt, H.E.; Kugler, J. Renal failure and rhabdomyolysis associated with sitagliptin and simvastatin use. Diabet. Med. 2008, 25, 1229–1230. [Google Scholar] [CrossRef] [PubMed]
- Serra, D.; He, Y.L.; Bullock, J.; Riviere, G.J.; Balez, S.; Schwartz, S.; Wang, Y.; Ligueros-Saylan, M.; Jarugula, V.; Dole, W.P. Evaluation of pharmacokinetic and pharmacodynamic interaction between the dipeptidyl peptidase IV inhibitor vildagliptin, glyburide and pioglitazone in patients with Type 2 diabetes. Int. J. Clin. Pharmacol. Ther. 2008, 46, 349–364. [Google Scholar] [CrossRef]
- Ayalasomayajula, S.; Dole, K.; He, Y.-L.; Ligueros-Saylan, M.; Wang, Y.; Campestrini, J.; Humbert, H.; Sunkara, G. Evaluation of the potential for steady-state pharmacokinetic interaction between vildagliptin and simvastatin in healthy subjects. Curr. Med. Res. Opin. 2007, 23, 2913–2920. [Google Scholar] [CrossRef]
- He, Y.-L.; Sabo, R.; Sunkara, G.; Bizot, M.-N.; Riviere, G.-J.; Leon, S.; Ligueros-Saylan, M.; Dole, W.P.; Howard, D. Evaluation of Pharmacokinetic Interactions Between Vildagliptin and Digoxin in Healthy Volunteers. J. Clin. Pharmacol. 2007, 47, 998–1004. [Google Scholar] [CrossRef] [PubMed]
- Brown, N.J.; Byiers, S.; Carr, D.; Maldonado, M.; Warner, B.A. Dipeptidyl peptidase-IV inhibitor use associated with increased risk of ACE inhibitor-associated angioedema. Hypertension 2009, 54, 516–523. [Google Scholar] [CrossRef] [Green Version]
- Graefe-Mody, E.; Brand, T.; Ring, A.; Withopf, B.; Stangier, J.; Iovino, M.; Woerle, H.-J. Effect of linagliptin on the pharmacokinetics and pharmacodynamics of warfarin in healthy volunteers. Int. J. Clin. Pharmacol. Ther. 2011, 49, 300–310. [Google Scholar] [CrossRef]
- Teng, R.; Butler, K. Effect of the CYP3A inhibitors, diltiazem and ketoconazole, on ticagrelor pharmacokinetics in healthy volunteers. J. Drug Assess. 2013, 2, 30–39. [Google Scholar] [CrossRef] [Green Version]
- Patel, C.G.; Li, L.; Girgis, S.; Kornhauser, D.M.; Frevert, E.U.; Boulton, D.W. Two-way pharmacokinetic interaction studies between saxagliptin and cytochrome P450 substrates or inhibitors: Simvastatin, diltiazem extended-release, and ketoconazole. Clin. Pharmacol. 2011, 3, 13–25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drucker, D.J. Mechanisms of Action and Therapeutic Application of Glucagon-like Peptide-1. Cell Metab 2018, 27, 740–756. [Google Scholar] [CrossRef] [Green Version]
- Davies, M.J.; Aroda, V.R.; Collins, B.S.; Gabbay, R.A.; Green, J.; Maruthur, N.M.; Rosas, S.E.; Del Prato, S.; Mathieu, C.; Mingrone, G.; et al. Management of Hyperglycemia in Type 2 Diabetes, 2022. A Consensus Report by the American Diabetes Association (ADA) and the European Association for the Study of Diabetes (EASD). Diabetes Care 2022, 45, 2753–2786. [Google Scholar] [CrossRef] [PubMed]
- Kalra, S.; Baruah, M.P.; Sahay, R.K.; Unnikrishnan, A.G.; Uppal, S.; Adetunji, O. Glucagon-like peptide-1 receptor agonists in the treatment of type 2 diabetes: Past, present, and future. Indian J. Endocrinol. Metab. 2016, 20, 254–267. [Google Scholar] [PubMed]
- Garber, A.J. Long-Acting Glucagon-Like Peptide 1 Receptor Agonists: A review of their efficacy and tolerability. Diabetes Care 2011, 34 (Suppl. S2), S279–S284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hinnen, D. Glucagon-Like Peptide 1 Receptor Agonists for Type 2 Diabetes. Diabetes Spectr. 2017, 30, 202–210. [Google Scholar] [CrossRef] [Green Version]
- Saraiva, F.K.; Sposito, A.C. Cardiovascular effects of glucagon-like peptide 1 (GLP-1) receptor agonists. Cardiovasc. Diabetol. 2014, 13, 142. [Google Scholar] [CrossRef] [Green Version]
- Htike, Z.Z.; Zaccardi, F.; Papamargaritis, D.; Webb, D.R.; Khunti, K.; Davies, M.J. Efficacy and safety of glucagon-like peptide-1 receptor agonists in type 2 diabetes: A systematic review and mixed-treatment comparison analysis. Diabetes Obes. Metab. 2017, 19, 524–536. [Google Scholar] [CrossRef] [Green Version]
- Singh, S.; Chang, H.Y.; Richards, T.M.; Weiner, J.P.; Clark, J.M.; Segal, J.B. Glucagonlike peptide 1-based therapies and risk of hospitalization for acute pancreatitis in type 2 diabetes mellitus: A population-based matched case-control study. JAMA Intern. Med. 2013, 173, 534–539. [Google Scholar] [CrossRef] [Green Version]
- Blase, E.; Taylor, K.; Gao, H.Y.; Wintle, M.; Fineman, M. Pharmacokinetics of an oral drug (acetaminophen) administered at various times in relation to subcutaneous injection of exenatide (exendin-4) in healthy subjects. J. Clin. Pharmacol. 2005, 45, 570–577. [Google Scholar] [CrossRef]
- Hurren, K.M.; Pinelli, N.R. Drug-drug interactions with glucagon-like peptide-1 receptor agonists. Ann. Pharmacother. 2012, 46, 710–717. [Google Scholar] [CrossRef]
- Kapitza, C.; Zdravkovic, M.; Hindsberger, C.; Flint, A. The effect of the once-daily human glucagon-like peptide 1 analog liraglutide on the pharmacokinetics of acetaminophen. Adv. Ther. 2011, 28, 650–660. [Google Scholar] [CrossRef] [PubMed]
- Hausner, H.; Karsbøl, J.D.; Holst, A.G.; Jacobsen, J.B.; Wagner, F.-D.; Golor, G.; Anderson, T.W. Effect of Semaglutide on the Pharmacokinetics of Metformin, Warfarin, Atorvastatin and Digoxin in Healthy Subjects. Clin. Pharmacokinet. 2017, 56, 1391–1401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Filippatos, T.D.; Panagiotopoulou, T.V.; Elisaf, M.S. Adverse Effects of GLP-1 Receptor Agonists. Rev. Diabet. Stud. 2014, 11, 202–230. [Google Scholar] [CrossRef] [Green Version]
- Malm-Erjefält, M.; Ekblom, M.; Vouis, J.; Zdravkovic, M.; Lennernäs, H. Effect on the Gastrointestinal Absorption of Drugs from Different Classes in the Biopharmaceutics Classification System, When Treating with Liraglutide. Mol. Pharm. 2015, 12, 4166–4173. [Google Scholar] [CrossRef] [PubMed]
- De la Peña, A.; Cui, X.; Geiser, J.; Loghin, C. No Dose Adjustment is Recommended for Digoxin, Warfarin, Atorvastatin or a Combination Oral Contraceptive When Coadministered with Dulaglutide. Clin. Pharmacokinet. 2017, 56, 1415–1427. [Google Scholar] [CrossRef]
- Petersen, A.B.; Knop, F.K.; Christensen, M. Lixisenatide for the treatment of type 2 diabetes. Drugs Today 2013, 49, 537–553. [Google Scholar] [CrossRef]
- Hsia, D.S.; Grove, O.; Cefalu, W.T. An Update on SGLT2 Inhibitors for the Treatment of Diabetes Mellitus. Curr. Opin. Endocrinol. Diabetes Obes. 2017, 24, 73–79. [Google Scholar]
- Joshi, S.S.; Singh, T.; Newby, D.E.; Singh, J.; Shruti, D.; Joshi, S. Sodium-glucose co-transporter 2 inhibitor therapy: Mechanisms of action in heart failure. Heart 2021, 107, 1032–1038. [Google Scholar] [CrossRef]
- Tentolouris, A.; Vlachakis, P.; Tzeravini, E.; Eleftheriadou, I.; Tentolouris, N. SGLT2 Inhibitors: A Review of Their Antidiabetic and Cardioprotective Effects. Int. J. Environ. Res. Public Health 2019, 16, 2965. [Google Scholar] [CrossRef] [Green Version]
- Kasichayanula, S.; Liu, X.; Shyu, W.C.; Zhang, W.; Pfister, M.; Griffen, S.C.; Li, T.; LaCreta, F.P.; Boulton, D.W. Lack of pharmacokinetic interaction between dapagliflozin, a novel sodium-glucose transporter 2 inhibitor, and metformin, pioglitazone, glimepiride or sitagliptin in healthy subjects. Diabetes Obes. Metab. 2011, 13, 47–54. [Google Scholar] [CrossRef]
- Strojek, K.; Yoon, K.H.; Hruba, V.; Elze, M.; Langkilde, A.M.; Parikh, S. Effect of dapagliflozin in patients with type 2 diabetes who have inadequate glycaemic control with glimepiride: A randomized, 24-week, double-blind, placebo-controlled trial. Diabetes Obes. Metab. 2011, 13, 928–938. [Google Scholar] [CrossRef] [PubMed]
- Kasichayanula, S.; Liu, X.; Griffen, S.C.; Lacreta, F.P.; Boulton, D.W. Effects of rifampin and mefenamic acid on the pharmacokinetics and pharmacodynamics of dapagliflozin. Diabetes Obes. Metab. 2013, 15, 280–283. [Google Scholar] [CrossRef]
- Kasichayanula, S.; Chang, M.; Liu, X.; Shyu, W.-C.; Griffen, S.C.; LaCreta, F.P.; Boulton, D.W. Lack of pharmacokinetic interactions between dapagliflozin and simvastatin, valsartan, warfarin, or digoxin. Adv. Ther. 2012, 29, 163–177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Devineni, D.; Manitpisitkul, P.; Vaccaro, N.; Bernard, A.; Skee, D.; Mamidi, R.N.; Tian, H.; Weiner, S.; Stieltjes, H.; Sha, S.; et al. Effect of canagliflozin, a sodium glucose co-transporter 2 inhibitor, on the pharmacokinetics of oral contraceptives, warfarin, and digoxin in healthy participants. Int. J. Clin. Pharmacol. Ther. 2015, 53, 41–53. [Google Scholar] [CrossRef] [PubMed]
- Devineni, D.; Vaccaro, N.; Murphy, J.; Curtin, C.; Mamidi, R.N.; Weiner, S.; Wang, S.-S.; Ariyawansa, J.; Stieltjes, H.; Wajs, E.; et al. Effects of rifampin, cyclosporine A, and probenecid on the pharmacokinetic profile of canagliflozin, a sodium glucose co-transporter 2 inhibitor, in healthy participants. Int. J. Clin. Pharmacol. Ther. 2015, 53, 115–128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scheen, A.J. Drug-drug interactions with sodium-glucose cotransporters type 2 (SGLT2) inhibitors, new oral glucose-lowering agents for the management of type 2 diabetes mellitus. Clin. Pharmacokinet. 2014, 53, 295–304. [Google Scholar] [CrossRef] [Green Version]
- Brand, T.; MacHa, S.; Mattheus, M.; Pinnetti, S.; Woerle, H.J. Pharmacokinetics of empagliflozin, a sodium glucose cotransporter-2 (SGLT-2) inhibitor, coadministered with sitagliptin in healthy volunteers. Adv. Ther. 2012, 29, 889–899. [Google Scholar] [CrossRef]
- Elkinson, S.; Scott, L.J. Canagliflozin: First global approval. Drugs 2013, 73, 979–988. [Google Scholar] [CrossRef]
- Devineni, D.; Curtin, C.R.; Polidori, D.; Gutierrez, M.J.; Murphy, J.; Rusch, S.; Rothenberg, P.L. Pharmacokinetics and pharmacodynamics of canagliflozin, a sodium glucose co-transporter 2 inhibitor, in subjects with type 2 diabetes mellitus. J. Clin. Pharmacol. 2013, 53, 601–610. [Google Scholar] [CrossRef] [PubMed]
- Macha, S.; Mattheus, M.; Pinnetti, S.; Seman, L.; JWoerle, H. Pharmacokinetics of Empagliflozin, a Sodium Glucose Cotransporter 2 Inhibitor, and Glimepiride Following Co-administration in Healthy Volunteers: A Randomised, Open-label, Crossover Study. J. Diabetes Res. Clin. Metab. 2012, 1, 14. [Google Scholar] [CrossRef] [Green Version]
- Macha, S.; Sennewald, R.; Rose, P.; Schoene, K.; Pinnetti, S.; Woerle, H.J.; Broedl, U.C. Lack of clinically relevant drug-drug interaction between empagliflozin, a sodium glucose cotransporter 2 inhibitor, and verapamil, ramipril, or digoxin in healthy volunteers. Clin. Ther. 2013, 35, 226–235. [Google Scholar] [CrossRef] [PubMed]
- Macha, S.; Mattheus, M.; Pinnetti, S.; Woerle, H.J.; Broedl, U.C. Effect of Empagliflozin on the Steady-State Pharmacokinetics of Ethinylestradiol and Levonorgestrel in Healthy Female Volunteers. Clin. Drug Investig. 2013, 33, 351–357. [Google Scholar] [CrossRef] [PubMed]
- Vizirianakis, I.S. Clinical Translation of Genotyping and Haplotyping Data. Clin. Pharmacokinet. 2007, 46, 807–824. [Google Scholar] [CrossRef] [PubMed]


















| Drug | Co-Administered Drug | Effect | References |
|---|---|---|---|
| Metformin | Iodinated contrast agents | Risk of lactic acidosis | [79] |
| Metformin | Ranitidine | Decreased renal clearance of metformin | [91] |
| Metformin | Cimetidine | Increased exposure of metformin | [92] |
| Metformin | PPIs | Increased exposure of metformin | [96] |
| Metformin | Vitamin B12 | Decreased absorption of B12 | [97] |
| Metformin | Trimethoprim | Decreased elimination of metformin | [95] |
| Metformin | Verapamil | Decreased effect of metformin | [98] |
| Metformin | Ranolazine | Decreased elimination | [99] |
| Metformin | Metoprolol | Decreased plasma concentration of metformin | [100] |
| Metformin | Vandetanib | Increased plasma concentration | [101] |
| Metformin | Anticancer drugs | Reduced elimination of metformin | [101] |
| Drug | Co-Administered Drug | Effect | References |
|---|---|---|---|
| Exenatide | Acetaminophen | Delayed absorption of acetaminophen | [133] |
| Lovastatin | Decreased AUC and Cmax of lovastatin | [134] | |
| Liraglutide | Acetaminophen | Delayed absorption of acetaminophen | [135] |
| Digoxin | Delayed Tmax of digoxin | [136] | |
| Sulfonylureas | Increased risk of hypoglycemia | [137] | |
| Atorvastatin | Delayed Tmax of atorvastatin | [138] | |
| Lisinopril | Delayed Tmax of lisinopril | [138] | |
| Semaglutide | Digoxin | Delayed Tmax of digoxin | [136] |
| Warfarin | Delayed absorption of warfarin | [136] | |
| Atorvastatin | Prolonged absorption and lowered Cmax of atorvastatin | [136] | |
| Dulaglutide | Digoxin | Delayed Tmax of digoxin | [139] |
| Warfarin | Delayed absorption of warfarin | [139] | |
| Atorvastatin | Reduced AUC of atorvastatin | [139] | |
| Lixisenatide | Ramipril | Delayed Tmax of ramipril | [140] |
| Drug | Co-Administered Drug | Effect | References |
|---|---|---|---|
| Dapagliflozin | Simvastatin | Increased AUC of simvastatin | [147] |
| Rifampicin | Decreased AUC of dapagliflozin | [146] | |
| Mefenamic Acid | Increased AUC of dapagliflozin | [146] | |
| Canagliflozin | Digoxin | Increased exposure of digoxin | [148] |
| Rifampicin | Decreased Cmax of canagliflozin | [149] | |
| Probenecid Acid | Increased Cmax of canagliflozin | [149] | |
| Cyclosporine | Increased AUC of canagliflozin | [149] | |
| Empagliflozin | Glimepiride | Decreased exposure of empagliflozin | [150] |
| Sitagliptin | Increased exposure of empagliflozin | [151] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Saiti, A.; Giannopoulos-Dimitriou, A.; Kazakos, I.; Galatou, E.; Vizirianakis, I.S. Systems Pharmacology and Network Analysis to Advance Pharmacogenomics and Precision Medicine Decisions in Type-2 Diabetes Therapy. Future Pharmacol. 2023, 3, 329-363. https://doi.org/10.3390/futurepharmacol3010021
Saiti A, Giannopoulos-Dimitriou A, Kazakos I, Galatou E, Vizirianakis IS. Systems Pharmacology and Network Analysis to Advance Pharmacogenomics and Precision Medicine Decisions in Type-2 Diabetes Therapy. Future Pharmacology. 2023; 3(1):329-363. https://doi.org/10.3390/futurepharmacol3010021
Chicago/Turabian StyleSaiti, Aikaterini, Alexandros Giannopoulos-Dimitriou, Ioannis Kazakos, Eleftheria Galatou, and Ioannis S. Vizirianakis. 2023. "Systems Pharmacology and Network Analysis to Advance Pharmacogenomics and Precision Medicine Decisions in Type-2 Diabetes Therapy" Future Pharmacology 3, no. 1: 329-363. https://doi.org/10.3390/futurepharmacol3010021