
Role of Senescence-Resumed Proliferation in Keloid Pathogenesis


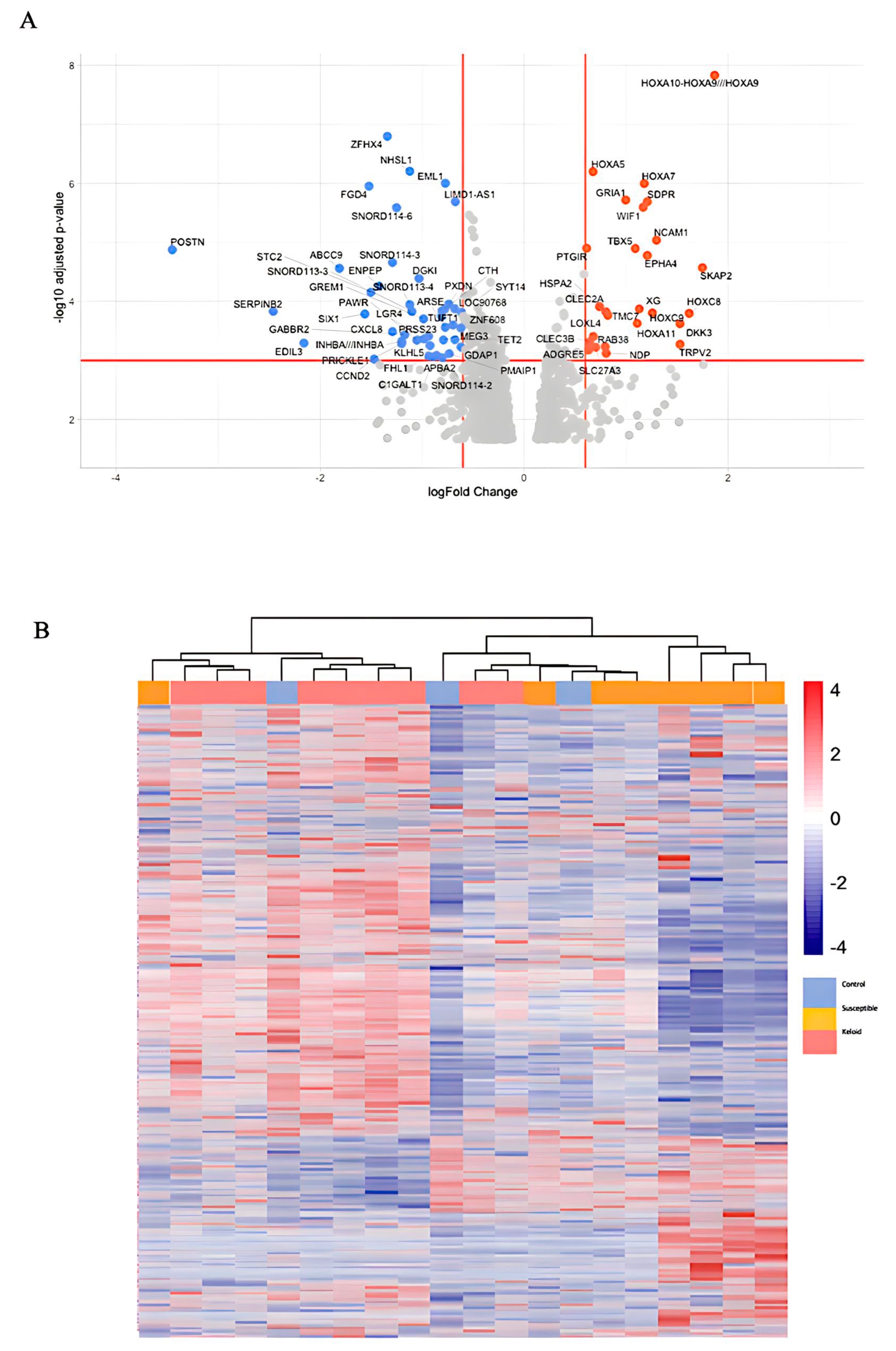{kind=link}
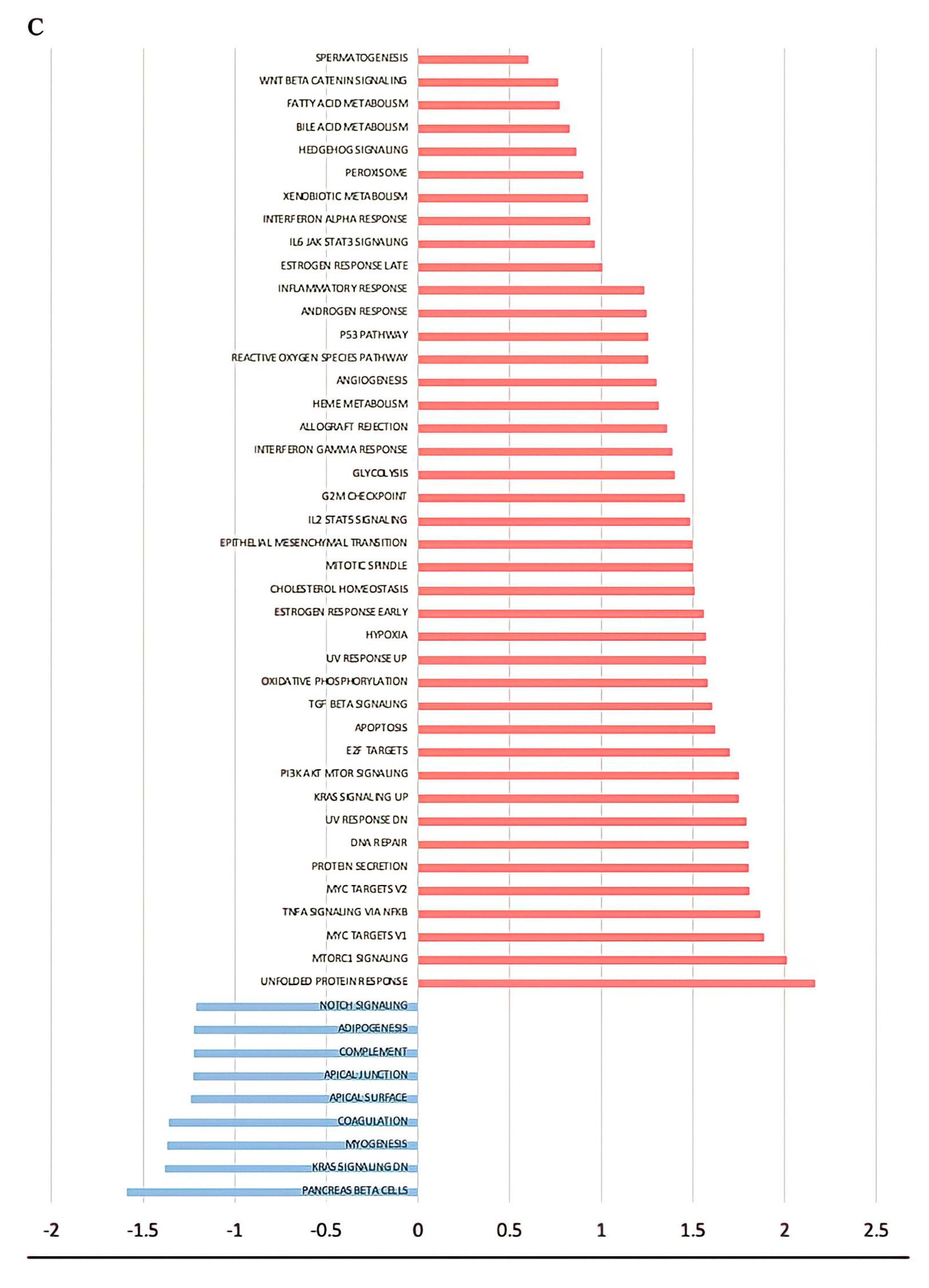{kind=link}
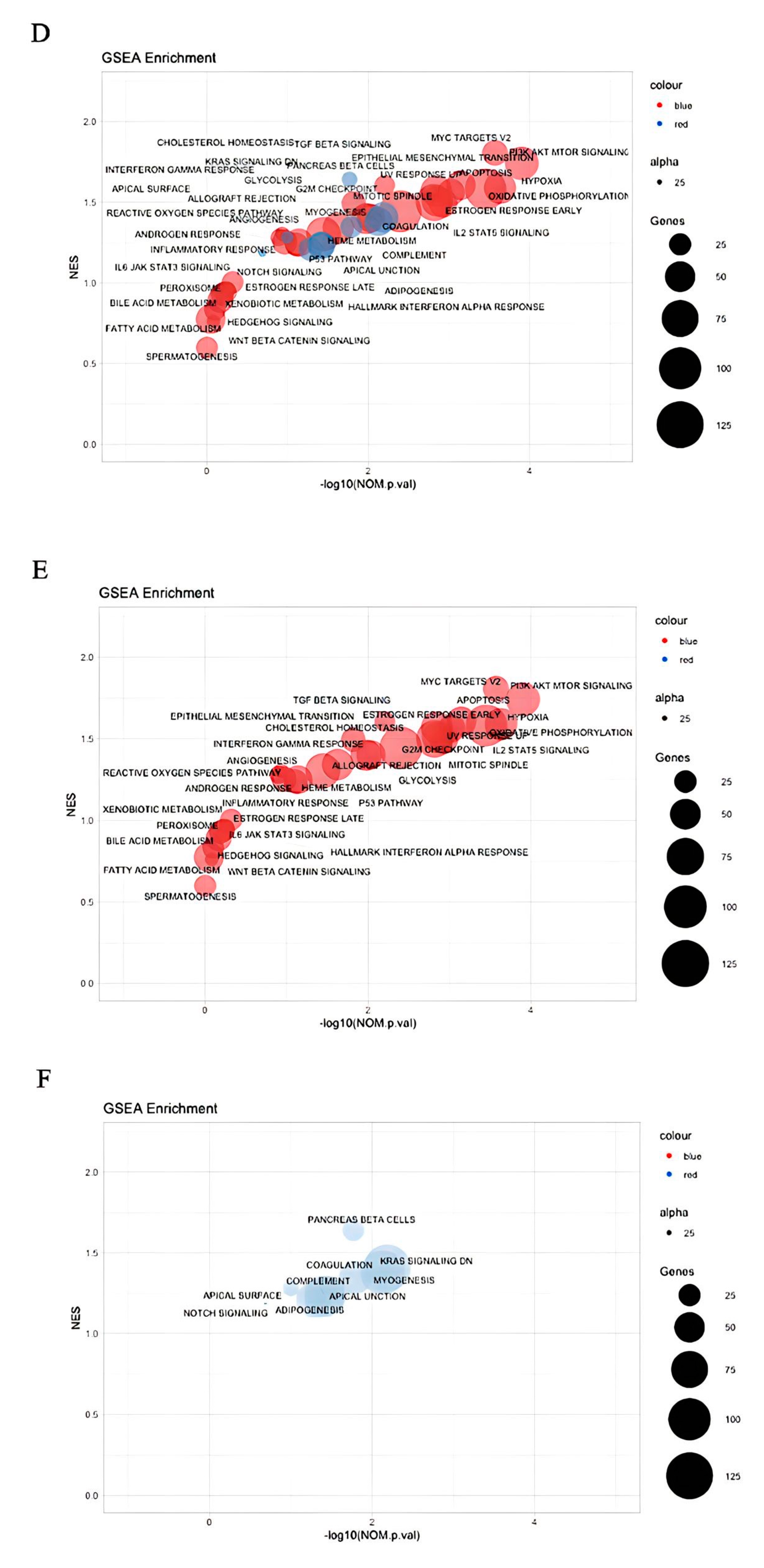{kind=link}
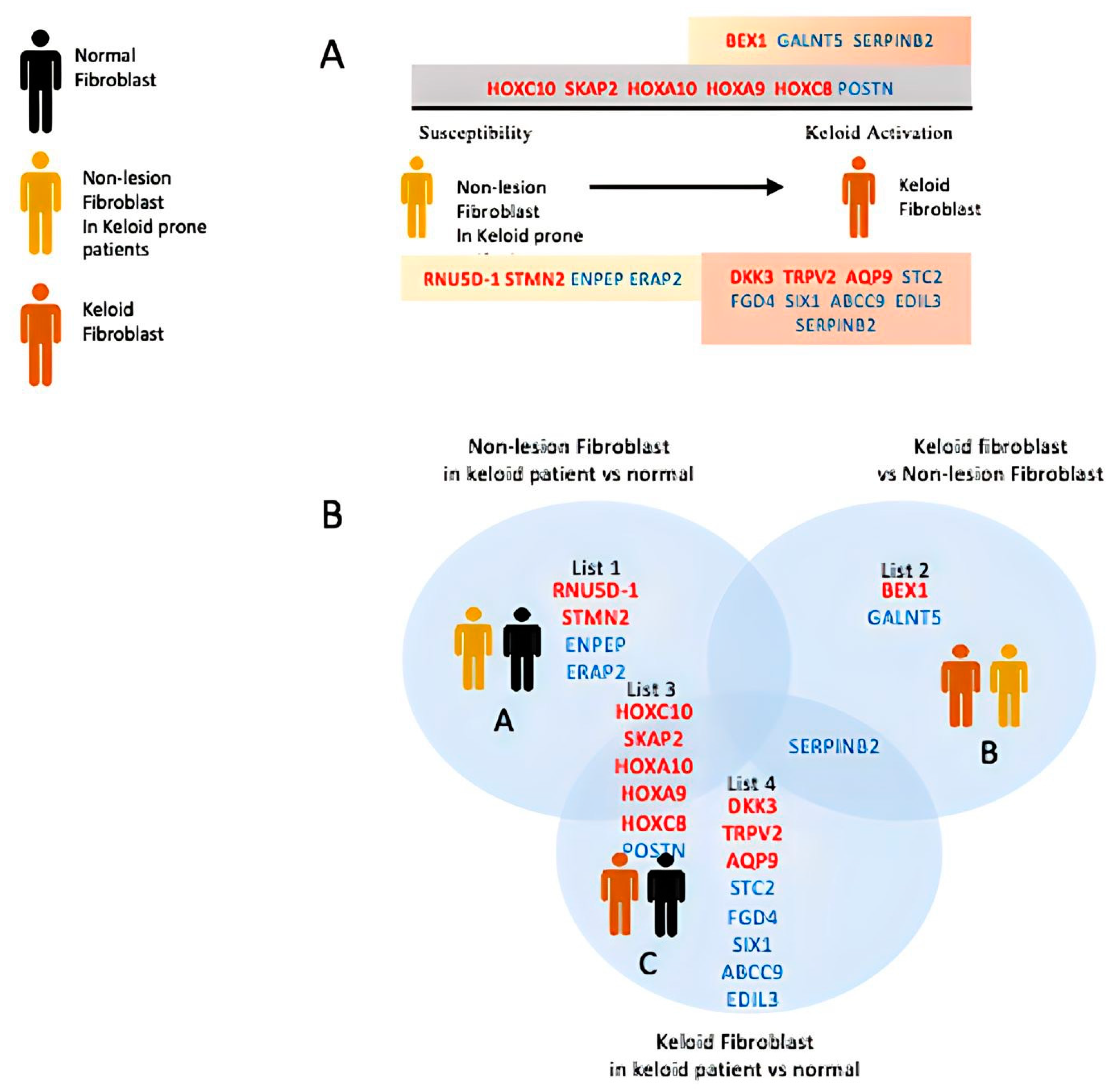{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
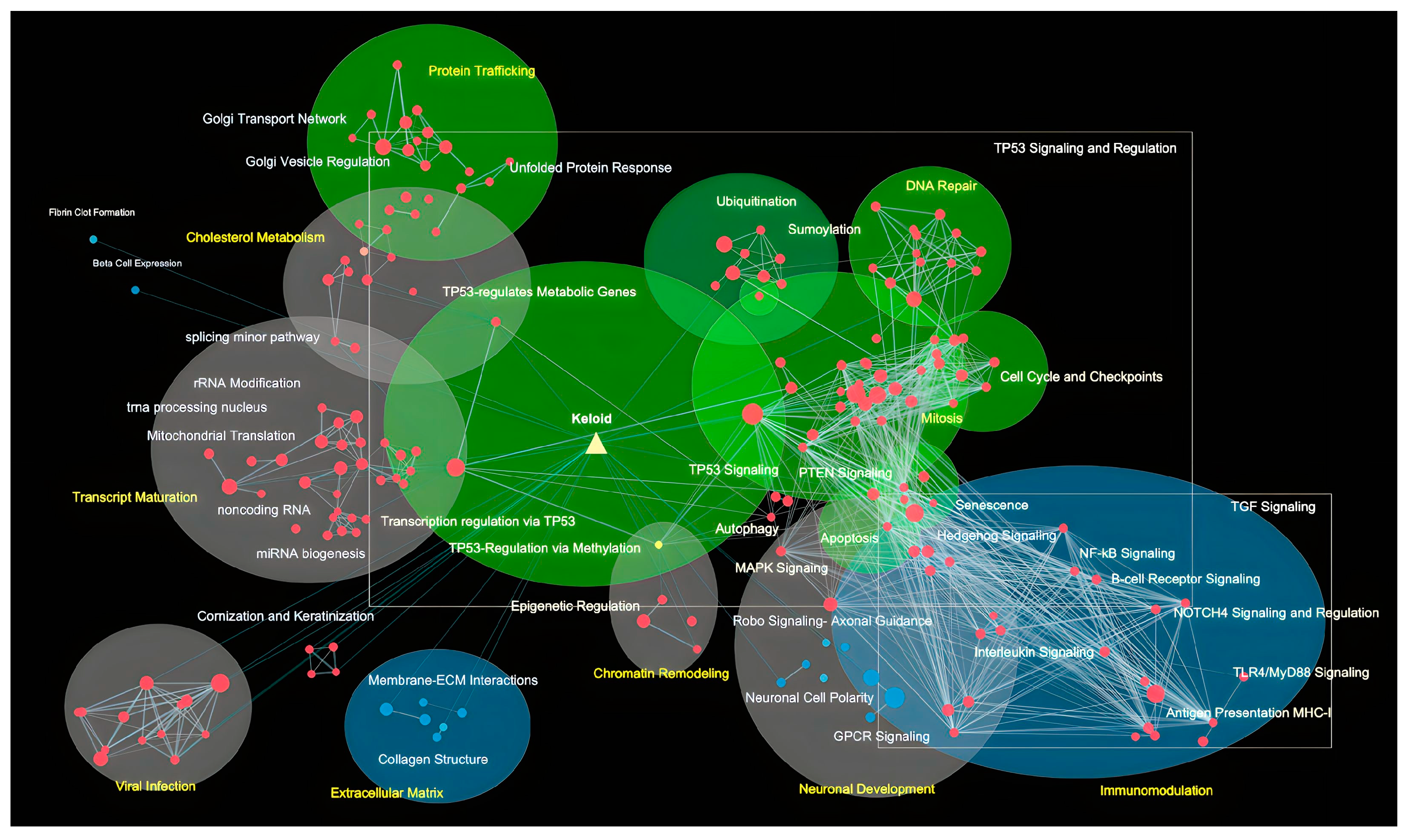
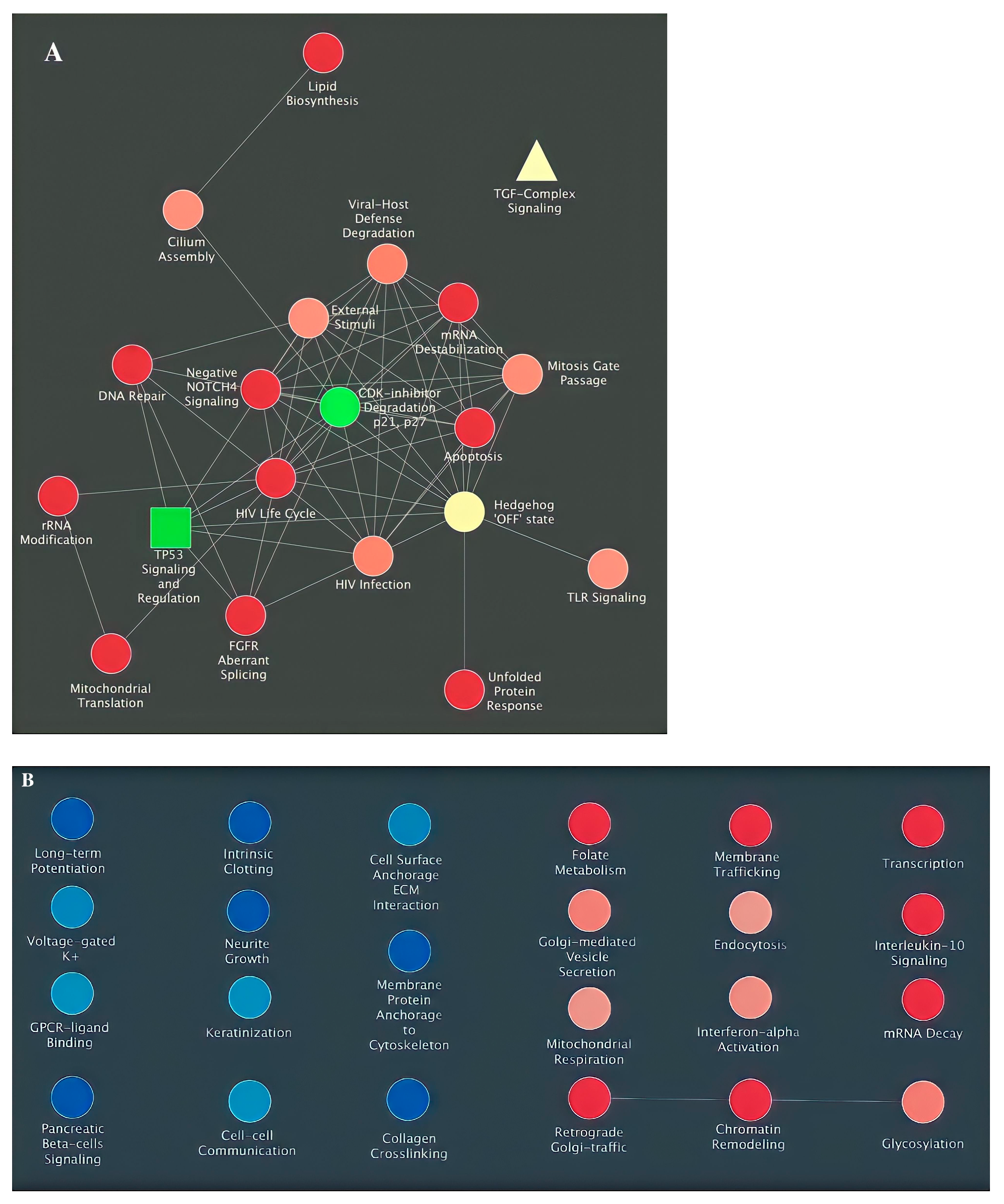
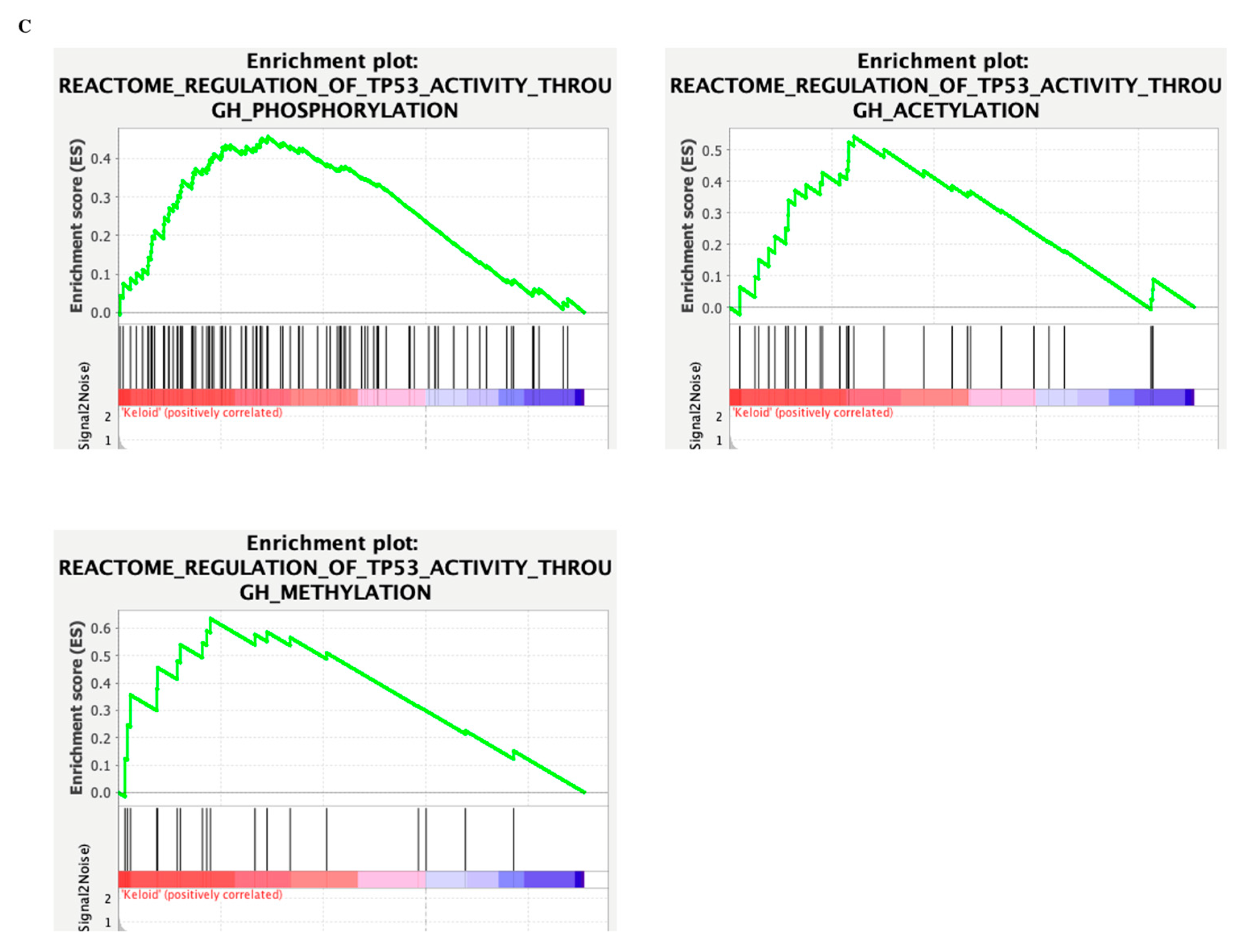
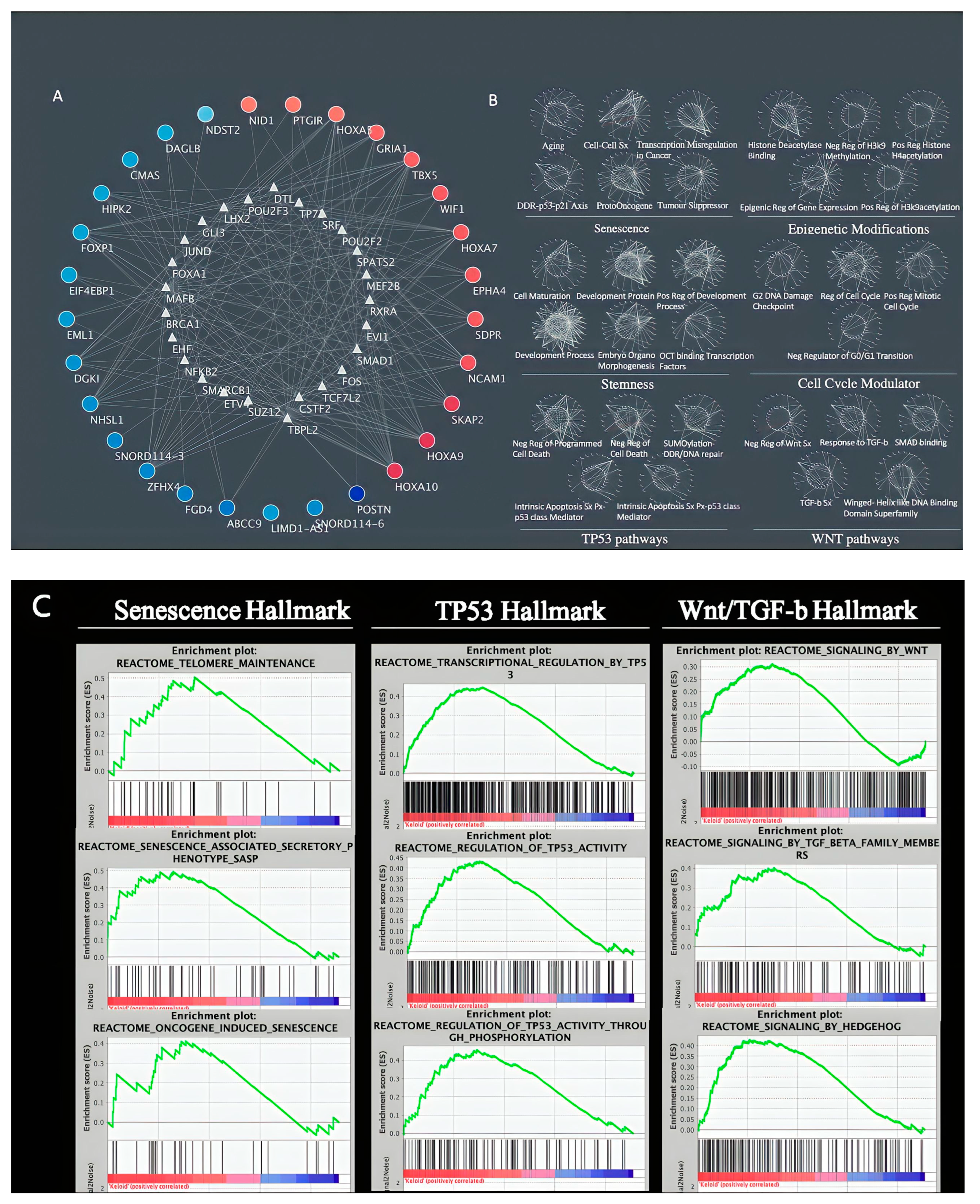
3. Results
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Milanovic, M.; Fan, D.N.Y.; Belenki, D.; Däbritz, J.H.M.; Zhao, Z.; Yu, Y.; Dörr, J.R.; Dimitrova, L.; Lenze, D.; Monteiro Barbosa, I.A.; et al. Senescence-associated reprogramming promotes cancer stemness. Nature 2018, 553, 96–100. [Google Scholar] [CrossRef] [PubMed]
- Asano, N.; Takeuchi, A.; Imatani, A.; Saito, M.; Jin, X.; Hatta, W.; Uno, K.; Koike, T.; Masamune, A. Wnt Signaling and Aging of the Gastrointestinal Tract. Int. J. Mol. Sci. 2022, 23, 12210. [Google Scholar] [CrossRef] [PubMed]
- Domen, A.; Deben, C.; Verswyvel, J.; Flieswasser, T.; Prenen, H.; Peeters, M.; Lardon, F.; Wouters, A. Cellular senescence in cancer: Clinical detection and prognostic implications. J. Exp. Clin. Cancer Res. 2022, 41, 360. [Google Scholar] [CrossRef] [PubMed]
- Piskorz, W.M.; Cechowska-Pasko, M. Senescence of Tumor Cells in Anticancer Therapy—Beneficial and Detrimental Effects. Int. J. Mol. Sci. 2022, 23, 11082. [Google Scholar] [CrossRef] [PubMed]
- Wen, G.-M.; Xu, X.-Y.; Xia, P. Metabolism in Cancer Stem Cells: Targets for Clinical Treatment. Cells 2022, 11, 3790. [Google Scholar] [CrossRef] [PubMed]
- Cruickshanks, H.A.; McBryan, T.; Nelson, D.M.; VanderKraats, N.D.; Shah, P.P.; Van Tuyn, J.; Rai, T.S.; Brock, C.; Donahue, G.; Dunican, D.S.; et al. Senescent cells harbour features of the cancer epigenome. Nat. Cell Biol. 2013, 15, 1495–1506. [Google Scholar] [CrossRef] [PubMed]
- Cassidy, L.D.; Narita, M. Autophagy at the intersection of aging, senescence, and cancer. Mol. Oncol. 2022, 16, 3259–3275. [Google Scholar] [CrossRef]
- Crouch, J.; Shvedova, M.; Thanapaul, R.J.R.S.; Botchkarev, V.; Roh, D. Epigenetic Regulation of Cellular Senescence. Cells 2022, 11, 672. [Google Scholar] [CrossRef]
- Prasanna, P.G.; Citrin, D.E.; Hildesheim, J.; Ahmed, M.M.; Venkatachalam, S.; Riscuta, G.; Xi, D.; Zheng, G.; van Deursen, J.; Goronzy, J.; et al. Therapy-Induced Senescence: Opportunities to Improve Anticancer Therapy. J. Natl. Cancer Inst. 2021, 113, 1285–1298. [Google Scholar] [CrossRef]
- Roger, L.; Tomas, F.; Gire, V. Mechanisms and Regulation of Cellular Senescence. Int. J. Mol. Sci. 2021, 22, 13173. [Google Scholar] [CrossRef]
- Hoare, M.; Das, T.; Alexander, G. Ageing, telomeres, senescence, and liver injury. J. Hepatol. 2010, 53, 950–961. [Google Scholar] [CrossRef] [PubMed]
- Faget, D.V.; Ren, Q.; Stewart, S.A. Unmasking senescence: Context-dependent effects of SASP in cancer. Nat. Rev. Cancer 2019, 19, 439–453. [Google Scholar] [CrossRef] [PubMed]
- Shang, S.; Hua, F.; Hu, Z.W. The regulation of beta-catenin activity and function in cancer: Therapeutic opportunities. Oncotarget 2017, 8, 33972–33989. [Google Scholar] [CrossRef] [PubMed]
- Martin-Orozco, E.; Sanchez-Fernandez, A.; Ortiz-Parra, I.; Nicolas, M.A.-S. WNT Signaling in Tumors: The Way to Evade Drugs and Immunity. Front. Immunol. 2019, 10, 2854. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.; Chen, L.; Li, L.; Qu, S.; Yu, B.; Sun, Y.; Wan, F.; Chen, X.; Liang, R.; Zhu, X. A positive feedback loop between Wnt/beta-catenin signaling and hTERT regulates the cancer stem cell-like traits in radioresistant nasopharyngeal carcinoma cells. J. Cell Biochem. 2020, 121, 4612–4622. [Google Scholar] [CrossRef]
- Wang, L.; Lankhorst, L.; Bernards, R. Exploiting senescence for the treatment of cancer. Nat. Rev. Cancer 2022, 22, 340–355. [Google Scholar] [CrossRef]
- Saleh, T.; Carpenter, V.J.; Tyutyunyk-Massey, L.; Murray, G.; Leverson, J.; Souers, A.; Alotaibi, M.; Faber, A.; Reed, J.; Harada, H.; et al. Clearance of therapy-induced senescent tumor cells by the senolytic ABT-263 via interference with BCL-X(L)-BAX interaction. Mol. Oncol. 2020, 14, 2504–2519. [Google Scholar]
- Zhu, Y.; Tchkonia, T.; Fuhrmann-Stroissnigg, H.; Dai, H.; Ling, Y.; Stout, M.; Pirtskhalava, T.; Giorgadze, N.; Johnson, K.; Giles, C.; et al. Identification of a novel senolytic agent, navitoclax, targeting the Bcl-2 family of anti-apoptotic factors. Aging Cell 2016, 15, 428–435. [Google Scholar] [CrossRef]
- Yamamoto, M.; Sanomachi, T.; Suzuki, S.; Togashi, K.; Sugai, A.; Seino, S.; Sato, A.; Okada, M.; Kitanaka, C. Gemcitabine radiosensitization primes irradiated malignant meningioma cells for senolytic elimination by navitoclax. Neurooncol. Adv. 2021, 3, vdab148. [Google Scholar] [CrossRef]
- Ahmadinejad, F.; Bos, T.; Hu, B.; Britt, E.; Koblinski, J.; Souers, A.J.; Leverson, J.D.; Faber, A.C.; Gewirtz, D.A.; Harada, H. Senolytic-Mediated Elimination of Head and Neck Tumor Cells Induced Into Senescence by Cisplatin. Mol. Pharmacol. 2022, 101, 168–180. [Google Scholar] [CrossRef]
- Carpenter, V.; Saleh, T.; Lee, S.M.; Murray, G.; Reed, J.; Souers, A.; Faber, A.C.; Harada, H.; Gewirtz, D.A. Androgen-deprivation induced senescence in prostate cancer cells is permissive for the development of castration-resistance but susceptible to senolytic therapy. Biochem. Pharmacol. 2021, 193, 114765. [Google Scholar] [CrossRef] [PubMed]
- Justice, J.N.; Nambiar, A.M.; Tchkonia, T.; LeBrasseur, N.K.; Pascual, R.; Hashmi, S.K.; Prata, L.; Masternak, M.M.; Kritchevsky, S.B.; Musi, N.; et al. Senolytics in idiopathic pulmonary fibrosis: Results from a first-in-human, open-label, pilot study. EBioMedicine 2019, 40, 554–563. [Google Scholar] [CrossRef] [PubMed]
- Jia, Q.; Cao, H.; Shen, D.; Li, S.; Yan, L.; Chen, C.; Xing, S.; Dou, F. Quercetin protects against atherosclerosis by regulating the expression of PCSK9, CD36, PPARgamma, LXRalpha and ABCA1. Int. J. Mol. Med. 2019, 44, 893–902. [Google Scholar] [CrossRef]
- Wong, S.; Chin, K.-Y.; Ima-Nirwana, S. Quercetin as an Agent for Protecting the Bone: A Review of the Current Evidence. Int. J. Mol. Sci. 2020, 21, 6448. [Google Scholar] [CrossRef] [PubMed]
- He, K.; Barsoumian, H.; Yang, L.; Sezen, D.; Wasley, M.; Masrorpour, F.; Cortez, M.; Welsh, J. Senolytic Cocktail Dasatinib Plus Quercetin Enhances the Antitumor Effect of Senescence-Inducing Radiotherapy in a Preclinical Model of Melanoma. Int. J. Radiat. Oncol. Biol. Phys. 2021, 111, S57. [Google Scholar] [CrossRef]
- Kovacovicova, K.; Skolnaja, M.; Heinmaa, M.; Mistrik, M.; Pata, P.; Pata, I.; Bartek, J.; Vinciguerra, M. Senolytic Cocktail Dasatinib+Quercetin (D+Q) Does Not Enhance the Efficacy of Senescence-Inducing Chemotherapy in Liver Cancer. Front. Oncol. 2018, 8, 459. [Google Scholar] [CrossRef]
- Tan, S.; Khumalo, N.P.; Bayat, A. Understanding Keloid Pathobiology From a Quasi-Neoplastic Perspective: Less of a Scar and More of a Chronic Inflammatory Disease With Cancer-Like Tendencies. Front. Immunol. 2019, 10, 1810. [Google Scholar] [CrossRef]
- De Felice, B.; Ciarmiello, L.; Mondola, P.; Damiano, S.; Seru, R.; Argenziano, C.; Nacca, M.; Santoriello, M.; Garbi, C. Differential p63 and p53 expression in human keloid fibroblasts and hypertrophic scar fibroblasts. DNA Cell Biol. 2007, 26, 541–547. [Google Scholar] [CrossRef]
- Saed, G.M.; Ladin, D.; Olson, J.; Han, X.; Hou, Z.; Fivenson, D. Analysis of p53 gene mutations in keloids using polymerase chain reaction-based single-strand conformational polymorphism and DNA sequencing. Arch. Dermatol. 1998, 134, 963–967. [Google Scholar] [CrossRef]
- Ladin, D.A.; Hou, Z.; Patel, D.; McPhail, M.; Olson, J.C.; Saed, G.M.; Fivenson, D.P. p53 and apoptosis alterations in keloids and keloid fibroblasts. Wound Repair Regen. 1998, 6, 28–37. [Google Scholar] [CrossRef]
- Chen, Z.Y.; Yu, X.F.; Huang, J.Q.; Li, D.L. The mechanisms of beta-catenin on keloid fibroblast cells proliferation and apoptosis. Eur. Rev. Med. Pharmacol. Sci. 2018, 22, 888–895. [Google Scholar]
- Cai, Y.; Zhu, S.; Yang, W.; Pan, M.; Wang, C.; Wu, W. Downregulation of beta-catenin blocks fibrosis via Wnt2 signaling in human keloid fibroblasts. Tumour Biol. 2017, 39, 1010428317707423. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.-S.; Liang, Y.-C.; Wu, P.; Kulber, D.A.; Tanabe, K.; Chuong, C.-M.; Widelitz, R.; Tuan, T.-L. STAT3 signalling pathway is implicated in keloid pathogenesis by preliminary transcriptome and open chromatin analyses. Exp. Dermatol. 2019, 28, 480–484. [Google Scholar] [CrossRef] [PubMed]
- Yu, D.; Shang, Y.; Yuan, J.; Ding, S.; Luo, S.; Hao, L. Wnt/beta-Catenin Signaling Exacerbates Keloid Cell Proliferation by Regulating Telomerase. Cell Physiol. Biochem. 2016, 39, 2001–2013. [Google Scholar] [CrossRef] [PubMed]
- Grant, C.; Chudakova, D.A.; Itinteang, T.; Chibnall, A.M.; Brasch, H.D.; Davis, P.F.; Tan, S.T. Expression of embryonic stem cell markers in keloid-associated lymphoid tissue. J. Clin. Pathol. 2016, 69, 643–646. [Google Scholar] [CrossRef]
- Varmeh, S.; Egia, A.; McGrouther, D.; Tahan, S.R.; Bayat, A.; Pandolfi, P.P. Cellular senescence as a possible mechanism for halting progression of keloid lesions. Genes Cancer 2011, 2, 1061–1066. [Google Scholar] [CrossRef] [PubMed]
- Barrett, T.; Wilhite, S.E.; Ledoux, P.; Evangelista, C.; Kim, I.F.; Tomashevsky, M.; Marshall, K.A.; Phillippy, K.H.; Sherman, P.M.; Holko, M.; et al. NCBI GEO: Archive for functional genomics data sets—update. Nucleic Acids Res. 2012, 41, D991–D995. [Google Scholar] [CrossRef] [PubMed]
- Hahn, J.M.; Glaser, K.; McFarland, K.L.; Aronow, B.J.; Boyce, S.T.; Supp, D.M. Keloid-derived keratinocytes exhibit an abnormal gene expression profile consistent with a distinct causal role in keloid pathology. Wound Repair Regen. 2013, 21, 530–544. [Google Scholar] [CrossRef] [PubMed]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Merico, D.; Isserlin, R.; Stueker, O.; Emili, A.; Bader, G.D. Enrichment Map: A Network-Based Method for Gene-Set Enrichment Visualization and Interpretation. PLoS ONE 2010, 5, e13984. [Google Scholar] [CrossRef]
- Janky, R.; Verfaillie, A.; Imrichova, H.; Van de Sande, B.; Standaert, L.; Christiaens, V.; Hulselmans, G.; Herten, K.; Sanchez, M.N.; Potier, D.; et al. iRegulon: From a gene list to a gene regulatory network using large motif and track collections. PLOS Comput. Biol. 2014, 10, e1003731. [Google Scholar] [CrossRef]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [PubMed]
- Liberzon, A.; Subramanian, A.; Pinchback, R.; Thorvaldsdóttir, H.; Tamayo, P.; Mesirov, J.P. Molecular signatures database (MSigDB) 3.0. Bioinformatics 2011, 27, 1739–1740. [Google Scholar] [CrossRef] [PubMed]
- Huang, D.W.; Sherman, B.T.; Lempicki, R.A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 2009, 4, 44–57. [Google Scholar] [CrossRef]
- Huang, D.W.; Sherman, B.T.; Lempicki, R.A. Bioinformatics enrichment tools: Paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 2009, 37, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Di Micco, R.; Krizhanovsky, V.; Baker, D.; d’Adda di Fagagna, F. Cellular senescence in ageing: From mechanisms to therapeutic opportunities. Nat. Rev. Mol. Cell Biol. 2021, 22, 75–95. [Google Scholar] [CrossRef]
- Raman, V.; Martensen, S.A.; Reisman, D.; Evron, E.; Odenwald, W.F.; Jaffee, E.; Marks, J.; Sukumar, S. Compromised HOXA5 function can limit p53 expression in human breast tumours. Nature 2000, 405, 974–978. [Google Scholar] [CrossRef]
- Svingen, T.; Tonissen, K. Hox transcription factors and their elusive mammalian gene targets. Heredity 2006, 97, 88–96. [Google Scholar] [CrossRef]
- Chang, C.-J.; Chen, Y.-L.; Hsieh, C.-H.; Liu, Y.-J.; Yu, S.-L.; Chen, J.J.; Wang, C.-C. HOXA5 and p53 cooperate to suppress lung cancer cell invasion and serve as good prognostic factors in non-small cell lung cancer. J. Cancer 2017, 8, 1071–1081. [Google Scholar] [CrossRef]
- Akdemir, K.C.; Jain, A.K.; Allton, K.; Aronow, B.; Xu, X.; Cooney, A.J.; Li, W.; Barton, M.C. Genome-wide profiling reveals stimulus-specific functions of p53 during differentiation and DNA damage of human embryonic stem cells. Nucleic Acids Res. 2013, 42, 205–223. [Google Scholar] [CrossRef]
- Sturmlechner, I.; Zhang, C.; Sine, C.C.; van Deursen, E.-J.; Jeganathan, K.B.; Hamada, N.; Grasic, J.; Friedman, D.; Stutchman, J.T.; Can, I.; et al. p21 produces a bioactive secretome that places stressed cells under immunosurveillance. Science 2021, 374, eabb3420. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.; Silveira, D.A.; Mombach, J.C.M. ATM/miR-34a-5p axis regulates a p21-dependent senescence-apoptosis switch in non-small cell lung cancer: A Boolean model of G1/S checkpoint regulation. FEBS Lett. 2020, 594, 227–239. [Google Scholar] [CrossRef] [PubMed]
- Limandjaja, G.C.; Niessen, F.B.; Scheper, R.J.; Gibbs, S. The Keloid Disorder: Heterogeneity, Histopathology, Mechanisms and Models. Front. Cell Dev. Biol. 2020, 8, 360. [Google Scholar] [CrossRef] [PubMed]
- Lu, F.; Gao, J.; Ogawa, R.; Hyakusoku, H.; Ou, C. Biological differences between fibroblasts derived from peripheral and central areas of keloid tissues. Plast. Reconstr. Surg. 2007, 120, 625–630. [Google Scholar] [CrossRef] [PubMed]
- Appleton, I.; Brown, N.J.; Willoughby, D.A. Apoptosis, necrosis, and proliferation: Possible implications in the etiology of keloids. Am. J. Pathol. 1996, 149, 1441–1447. [Google Scholar]
- Akasaka, Y.; Fujita, K.; Ishikawa, Y.; Asuwa, N.; Inuzuka, K.; Ishihara, M.; Ito, M.; Masuda, T.; Akishima, Y.; Zhang, L.; et al. Detection of apoptosis in keloids and a comparative study on apoptosis between keloids, hypertrophic scars, normal healed flat scars, and dermatofibroma. Wound Repair Regen. 2001, 9, 501–506. [Google Scholar] [CrossRef]
- Huang, C.; Akaishi, S.; Hyakusoku, H.; Ogawa, R. Are keloid and hypertrophic scar different forms of the same disorder? A fibroproliferative skin disorder hypothesis based on keloid findings. Int. Wound J. 2014, 11, 517–522. [Google Scholar] [CrossRef]
- Le, A.D.; Zhang, Q.; Wu, Y.; Messadi, D.V.; Akhondzadeh, A.; Nguyen, A.L.; Aghaloo, T.L.; Kelly, A.P.; Bertolami, C.N. Elevated vascular endothelial growth factor in keloids: Relevance to tissue fibrosis. Cells Tissues Organs 2004, 176, 87–94. [Google Scholar] [CrossRef]
- Touchi, R.; Ueda, K.; Kurokawa, N.; Tsuji, M. Central regions of keloids are severely ischaemic. J. Plast. Reconstr. Aesthe.t Surg. 2016, 69, e35–e41. [Google Scholar] [CrossRef]
- Louw, L.; Van Der Westhuizen, J.P.; De Wit, L.D.; Edwards, G. Keloids: Peripheral and central differences in cell morphology and fatty acid compositions of lipids. Adv. Exp. Med. Biol. 1997, 407, 515–520. [Google Scholar]
- Lim, I.J.; Phan, T.-T.; Bay, B.-H.; Qi, R.; Huynh, H.; Tan, W.T.-L.; Lee, S.-T.; Longaker, M.T. Fibroblasts cocultured with keloid keratinocytes: Normal fibroblasts secrete collagen in a keloidlike manner. Am. J. Physiol. Cell Physiol. 2002, 283, C212–C222. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.; Chen, J.; Xu, B.; Long, X.; Qin, H.; Zhao, R.C.; Wang, X. Keloid-derived keratinocytes acquire a fibroblast-like appearance and an enhanced invasive capacity in a hypoxic microenvironment in vitro. Int. J. Mol. Med. 2015, 35, 1246–1256. [Google Scholar] [CrossRef] [PubMed]
- Funayama, E.; Chodon, T.; Oyama, A.; Sugihara, T. Keratinocytes promote proliferation and inhibit apoptosis of the underlying fibroblasts: An important role in the pathogenesis of keloid. J. Invest. Dermatol. 2003, 121, 1326–1331. [Google Scholar] [CrossRef] [PubMed]








Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, C.-Y.; Wu, C.-W.; Lin, T.-Y. Role of Senescence-Resumed Proliferation in Keloid Pathogenesis. Future Pharmacol. 2023, 3, 198-212. https://doi.org/10.3390/futurepharmacol3010014
Wang C-Y, Wu C-W, Lin T-Y. Role of Senescence-Resumed Proliferation in Keloid Pathogenesis. Future Pharmacology. 2023; 3(1):198-212. https://doi.org/10.3390/futurepharmacol3010014
Chicago/Turabian StyleWang, Ching-Yun, Chieh-Wen Wu, and Ting-Yi Lin. 2023. "Role of Senescence-Resumed Proliferation in Keloid Pathogenesis" Future Pharmacology 3, no. 1: 198-212. https://doi.org/10.3390/futurepharmacol3010014