
Mechanisms of Mitochondrial Oxidative Stress in Brain Injury: From Pathophysiology to Therapeutics

{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
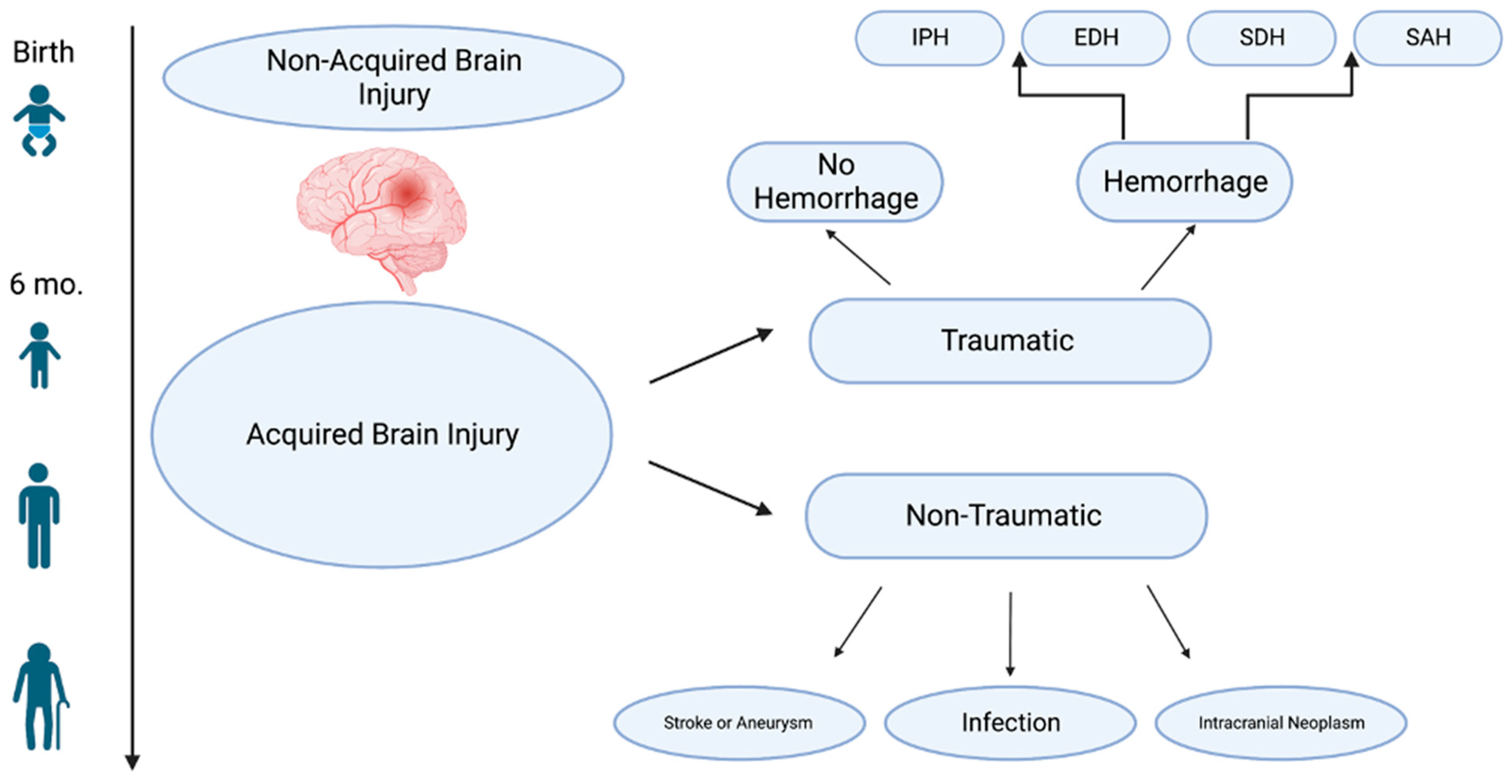
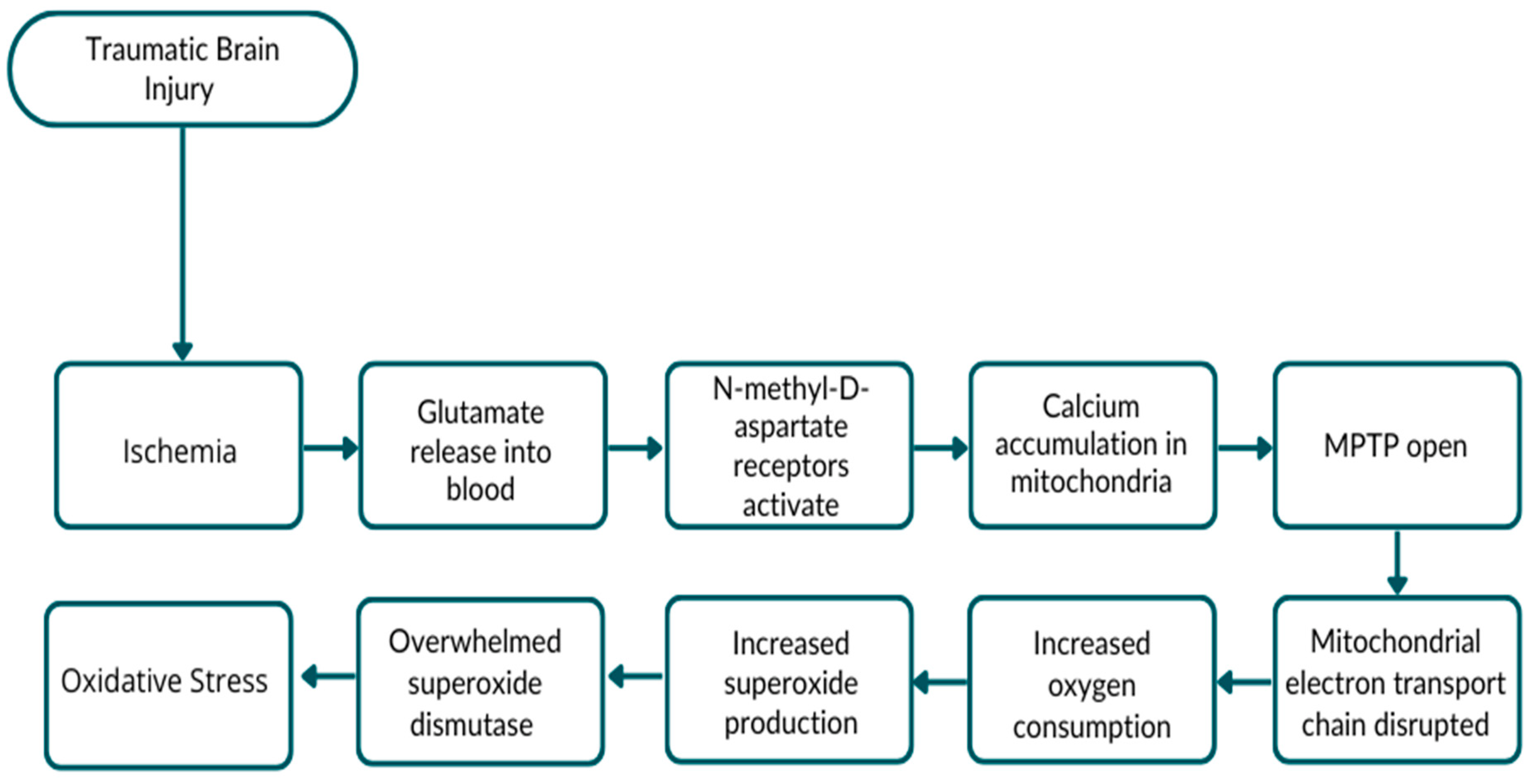
2. Traumatic Brain Injury
3. Traumatic Intracranial Hemorrhage
4. Ischemic Stroke
5. Hemorrhagic Stroke
6. Aneurysm
7. Brain Tumor
8. Cranial Infection
9. Conclusions
Funding
Conflicts of Interest
Abbreviations
References
- Wolff, C.B. Oxygen delivery: The principal role of the circulation. Adv. Exp. Med. Biol. 2013, 789, 37–42. [Google Scholar] [CrossRef]
- Rodriguez-Rodriguez, A.; Egea-Guerrero, J.J.; Murillo-Cabezas, F.; Carrillo-Vico, A. Oxidative stress in traumatic brain injury. Curr. Med. Chem. 2014, 21, 1201–1211. [Google Scholar] [CrossRef]
- Hiebert, J.B.; Shen, Q.; Thimmesch, A.R.; Pierce, J.D. Traumatic brain injury and mitochondrial dysfunction. Chem. J. Med. Sci. 2015, 350, 132–138. [Google Scholar] [CrossRef]
- Cornelius, C.; Crupi, R.; Calabrese, V.; Graziano, A.; Milone, P.; Pennisi, G.; Radak, Z.; Calabrese, E.J.; Cuzzocrea, S. Traumatic brain injury: Oxidative stress and neuroprotection. Antioxid. Redox Signal. 2013, 19, 836–853. [Google Scholar] [CrossRef]
- Ng, S.Y.; Lee, A.Y.W. Traumatic Brain Injuries: Pathophysiology and Potential Therapeutic Targets. Front. Cell. Neurosci. 2019, 13, 528. [Google Scholar] [CrossRef] [PubMed]
- Goldman, L.; Siddiqui, E.M.; Khan, A.; Jahan, S.; Rehman, M.U.; Mehan, S.; Sharma, R.; Budkin, S.; Kumar, S.N.; Sahu, A.; et al. Understanding Acquired Brain Injury: A Review. Biomedicines 2022, 10, 2167. [Google Scholar] [CrossRef] [PubMed]
- Berger, R.; Garnier, Y. Perinatal brain injury. J. Perinat. Med. 2000, 28, 261–285. [Google Scholar] [CrossRef] [PubMed]
- Dumpa, V.; Kamity, R. Birth Trauma. In StatPearls; StatPearls: Treasure Island, FL, USA, 2022. [Google Scholar]
- Gilmore, N.; Katz, D.I.; Kiran, S. Acquired Brain Injury in Adults: A Review of Pathophysiology, Recovery, and Rehabilitation. Perspect. ASHA Spec. Interest. Groups 2021, 6, 714–727. [Google Scholar] [CrossRef] [PubMed]
- Georges, A.; Das, J.M. Traumatic Brain Injury. In StatPearls; StatPearls: Treasure Island, FL, USA, 2022. [Google Scholar]
- Kaur, P.; Sharma, S. Recent Advances in Pathophysiology of Traumatic Brain Injury. Curr. Neuropharmacol. 2018, 16, 1224–1238. [Google Scholar] [CrossRef]
- Capizzi, A.; Woo, J.; Verduzco-Gutierrez, M. Traumatic Brain Injury: An Overview of Epidemiology, Pathophysiology, and Medical Management. Med. Clin. N. Am. 2020, 104, 213–238. [Google Scholar] [CrossRef]
- Ribas, V.; Garcia-Ruiz, C.; Fernandez-Checa, J.C. Glutathione and mitochondria. Front. Pharmacol. 2014, 5, 151. [Google Scholar] [CrossRef] [Green Version]
- Purves, D.; Augustine, G.; Fitzpatrick, D.; Katz, L.; LaMantia, A.; McNamara, J.; Williams, S. Neuroscience, 2nd ed.; Sinauer Associates: Sunderland, MA, USA, 2001. [Google Scholar]
- Thapa, K.; Khan, H.; Singh, T.G.; Kaur, A. Traumatic Brain Injury: Mechanistic Insight on Pathophysiology and Potential Therapeutic Targets. J. Mol. Neurosci. 2021, 71, 1725–1742. [Google Scholar] [CrossRef] [PubMed]
- Parkin, G.M.; Udawela, M.; Gibbons, A.; Dean, B. Glutamate transporters, EAAT1 and EAAT2, are potentially important in the pathophysiology and treatment of schizophrenia and affective disorders. World J. Psychiatry 2018, 8, 51–63. [Google Scholar] [CrossRef] [PubMed]
- Sivasubramanian, M.K.; Monteiro, R.; Balasubramanian, P.; Subramanian, M. Oxidative Stress-Induced Senescence Alters Glutamate Transporter Expression in Human Brainstem Astrocytes. FASEB J. 2020, 34, 1. [Google Scholar] [CrossRef]
- Miralles, V.J.; Martinez-Lopez, I.; Zaragoza, R.; Borras, E.; Garcia, C.; Pallardo, F.V.; Vina, J.R. Na+ dependent glutamate transporters (EAAT1, EAAT2, and EAAT3) in primary astrocyte cultures: Effect of oxidative stress. Brain Res. 2001, 922, 21–29. [Google Scholar] [CrossRef]
- Davis, C.K.; Vemuganti, R. Antioxidant therapies in traumatic brain injury. Neurochem. Int. 2022, 152, 105255. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.H.; Cha, M.; Lee, B.H. Neuroprotective Effect of Antioxidants in the Brain. Int. J. Mol. Sci. 2020, 21, 7152. [Google Scholar] [CrossRef]
- Zhou, Z.; Chen, H.; Zhang, K.; Yang, H.; Liu, J.; Huang, Q. Protective effect of nerve growth factor on neurons after traumatic brain injury. J. Basic Clin. Physiol. Pharmacol. 2003, 14, 217–224. [Google Scholar] [CrossRef]
- Genrikhs, E.E.; Voronkov, D.N.; Kapkaeva, M.R.; Gudasheva, T.A.; Glibka, Y.A.; Isaev, N.K.; Stelmashook, E.V. The delayed protective effect of GK-2, a dipeptide mimetic of Nerve Growth Factor, in a model of rat traumatic brain injury. Brain Res. Bull. 2018, 140, 148–153. [Google Scholar] [CrossRef]
- Chiaretti, A.; Conti, G.; Falsini, B.; Buonsenso, D.; Crasti, M.; Manni, L.; Soligo, M.; Fantacci, C.; Genovese, O.; Calcagni, M.L.; et al. Intranasal Nerve Growth Factor administration improves cerebral functions in a child with severe traumatic brain injury: A case report. Brain Inj. 2017, 31, 1538–1547. [Google Scholar] [CrossRef] [Green Version]
- Garbarino, V.R.; Orr, M.E.; Rodriguez, K.A.; Buffenstein, R. Mechanisms of oxidative stress resistance in the brain: Lessons learned from hypoxia tolerant extremophilic vertebrates. Arch. Biochem. Biophys. 2015, 576, 8–16. [Google Scholar] [CrossRef] [Green Version]
- Eshhar, N.; Striem, S.; Biegon, A. HU-211, a non-psychotropic cannabinoid, rescues cortical neurones from excitatory amino acid toxicity in culture. Neuroreport 1993, 5, 237–240. [Google Scholar] [CrossRef]
- Knoller, N.; Levi, L.; Shoshan, I.; Reichenthal, E.; Razon, N.; Rappaport, Z.H.; Biegon, A. Dexanabinol (HU-211) in the treatment of severe closed head injury: A randomized, placebo-controlled, phase II clinical trial. Crit. Care Med. 2002, 30, 548–554. [Google Scholar] [CrossRef] [PubMed]
- Petronilli, V.; Nicolli, A.; Costantini, P.; Colonna, R.; Bernardi, P. Regulation of the permeability transition pore, a voltage-dependent mitochondrial channel inhibited by cyclosporin A. Biochim. Biophys. Acta 1994, 1187, 255–259. [Google Scholar] [CrossRef] [PubMed]
- Clarke, S.J.; McStay, G.P.; Halestrap, A.P. Sanglifehrin A acts as a potent inhibitor of the mitochondrial permeability transition and reperfusion injury of the heart by binding to cyclophilin-D at a different site from cyclosporin A. J. Biol. Chem. 2002, 277, 34793–34799. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hall, E.D. Beneficial effects of acute intravenous ibuprofen on neurologic recovery of head-injured mice: Comparison of cyclooxygenase inhibition with inhibition of thromboxane A2 synthetase or 5-lipoxygenase. Cent. Nerv. Syst. Trauma 1985, 2, 75–83. [Google Scholar] [CrossRef] [PubMed]
- Caceres, J.A.; Goldstein, J.N. Intracranial hemorrhage. Emerg. Med. Clin. N. Am. 2012, 30, 771–794. [Google Scholar] [CrossRef] [Green Version]
- Tenny, S.; Thorell, W. Intracranial Hemorrhage. In StatPearls; StatPearls: Treasure Island, FL, USA, 2022. [Google Scholar]
- Griswold, D.P.; Fernandez, L.; Rubiano, A.M. Traumatic Subarachnoid Hemorrhage: A Scoping Review. J. Neurotrauma 2022, 39, 35–48. [Google Scholar] [CrossRef]
- Topkoru, B.; Egemen, E.; Solaroglu, I.; Zhang, J.H. Early Brain Injury or Vasospasm? An Overview of Common Mechanisms. Curr. Drug Targets 2017, 18, 1424–1429. [Google Scholar] [CrossRef]
- Ayer, R.E.; Zhang, J.H. The clinical significance of acute brain injury in subarachnoid hemorrhage and opportunity for intervention. Cereb. Hemorrhage 2008, 105, 179–184. [Google Scholar] [CrossRef]
- Jelinek, M.; Jurajda, M.; Duris, K. The Role of Oxidative Stress in Early Brain Injury after Subarachnoid Hemorrhage. Oxidative Med. Cell. Longev. 2020, 2020, 8877116. [Google Scholar] [CrossRef]
- Youle, R.J.; van der Bliek, A.M. Mitochondrial fission, fusion, and stress. Science 2012, 337, 1062–1065. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gaetani, P.; Pasqualin, A.; Rodriguez y Baena, R.; Borasio, E.; Marzatico, F. Oxidative stress in the human brain after subarachnoid hemorrhage. J. Neurosurg. 1998, 89, 748–754. [Google Scholar] [CrossRef] [Green Version]
- Marzatico, F.; Gaetani, P.; Cafe, C.; Spanu, G.; Rodriguez y Baena, R. Antioxidant enzymatic activities after experimental subarachnoid hemorrhage in rats. Acta Neurol. Scand. 1993, 87, 62–66. [Google Scholar] [CrossRef] [PubMed]
- Shao, L.; Chen, S.; Ma, L. Secondary Brain Injury by Oxidative Stress after Cerebral Hemorrhage: Recent Advances. Front. Cell. Neurosci. 2022, 16, 853589. [Google Scholar] [CrossRef] [PubMed]
- Moro, M.A.; Almeida, A.; Bolanos, J.P.; Lizasoain, I. Mitochondrial respiratory chain and free radical generation in stroke. Free Radic. Biol. Med. 2005, 39, 1291–1304. [Google Scholar] [CrossRef]
- Won, S.J.; Kim, D.Y.; Gwag, B.J. Cellular and molecular pathways of ischemic neuronal death. J. Biochem. Mol. Biol. 2002, 35, 67–86. [Google Scholar] [CrossRef] [Green Version]
- Murphy, A.N.; Fiskum, G.; Beal, M.F. Mitochondria in neurodegeneration: Bioenergetic function in cell life and death. J. Cereb. Blood Flow Metab. 1999, 19, 231–245. [Google Scholar] [CrossRef] [Green Version]
- Ismail, H.; Shakkour, Z.; Tabet, M.; Abdelhady, S.; Kobaisi, A.; Abedi, R.; Nasrallah, L.; Pintus, G.; Al-Dhaheri, Y.; Mondello, S.; et al. Traumatic Brain Injury: Oxidative Stress and Novel Anti-Oxidants Such as Mitoquinone and Edaravone. Antioxidants 2020, 9, 943. [Google Scholar] [CrossRef]
- Lewen, A.; Matz, P.; Chan, P.H. Free radical pathways in CNS injury. J. Neurotrauma 2000, 17, 871–890. [Google Scholar] [CrossRef]
- Rodriguez y Baena, R.; Gaetani, P.; Silvani, V.; Spanu, G.; Marzatico, F. Effect of nimodipine on mitochondrial respiration in different rat brain areas after subarachnoid haemorrhage. In Acta Neurochirurgica Supplement, Proceedings of the 8 th European Congress of Neurosurgery, Barcelona, September 6–11, 1987: Volume 2 Spinal Cord and Spine Pathologies Basic Research in Neurosurgery; Springer: Vienna, Austria, 1988; Volume 43, pp. 177–181. [Google Scholar] [CrossRef]
- Kowalczyk, P.; Sulejczak, D.; Kleczkowska, P.; Bukowska-Osko, I.; Kucia, M.; Popiel, M.; Wietrak, E.; Kramkowski, K.; Wrzosek, K.; Kaczynska, K. Mitochondrial Oxidative Stress-A Causative Factor and Therapeutic Target in Many Diseases. Int. J. Mol. Sci. 2021, 22, 13384. [Google Scholar] [CrossRef] [PubMed]
- Duan, J.; Yuan, W.; Jiang, J.; Wang, J.; Yan, X.; Liu, F.; Liu, A. ASK1 inhibitor NQDI-1 decreases oxidative stress and neuroapoptosis via the ASK1/p38 and JNK signaling pathway in early brain injury after subarachnoid hemorrhage in rats. Mol. Med. Rep. 2023, 27, 47. [Google Scholar] [CrossRef]
- Wu, Q.; Gao, C.; Wang, H.; Zhang, X.; Li, Q.; Gu, Z.; Shi, X.; Cui, Y.; Wang, T.; Chen, X.; et al. Mdivi-1 alleviates blood-brain barrier disruption and cell death in experimental traumatic brain injury by mitigating autophagy dysfunction and mitophagy activation. Int. J. Biochem. Cell Biol. 2018, 94, 44–55. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Yu, J.; Ma, D.; Weng, X. Edaravone Improves the Post-traumatic Brain Injury Dysfunction in Learning and Memory by Modulating Nrf2/ARE Signal Pathway. Clinics 2021, 76, e3131. [Google Scholar] [CrossRef]
- Sullivan, P.G.; Thompson, M.B.; Scheff, S.W. Cyclosporin A attenuates acute mitochondrial dysfunction following traumatic brain injury. Exp. Neurol. 1999, 160, 226–234. [Google Scholar] [CrossRef]
- Cheng, G.; Kong, R.H.; Zhang, L.M.; Zhang, J.N. Mitochondria in traumatic brain injury and mitochondrial-targeted multipotential therapeutic strategies. Br. J. Pharmacol. 2012, 167, 699–719. [Google Scholar] [CrossRef] [Green Version]
- Schiavone, S.; Neri, M.; Trabace, L.; Turillazzi, E. The NADPH oxidase NOX2 mediates loss of parvalbumin interneurons in traumatic brain injury: Human autoptic immunohistochemical evidence. Sci. Rep. 2017, 7, 8752. [Google Scholar] [CrossRef] [Green Version]
- Di Pietro, V.; Yakoub, K.M.; Caruso, G.; Lazzarino, G.; Signoretti, S.; Barbey, A.K.; Tavazzi, B.; Lazzarino, G.; Belli, A.; Amorini, A.M. Antioxidant Therapies in Traumatic Brain Injury. Antioxidants 2020, 9, 260. [Google Scholar] [CrossRef] [Green Version]
- Granger, M.; Eck, P. Dietary Vitamin C in Human Health. Adv. Food Nutr Res. 2018, 83, 281–310. [Google Scholar] [CrossRef] [PubMed]
- Tavazzi, B.; Signoretti, S.; Lazzarino, G.; Amorini, A.M.; Delfini, R.; Cimatti, M.; Marmarou, A.; Vagnozzi, R. Cerebral oxidative stress and depression of energy metabolism correlate with severity of diffuse brain injury in rats. Neurosurgery 2005, 56, 582–589; discussion 582–589. [Google Scholar] [CrossRef]
- Razmkon, A.; Sadidi, A.; Sherafat-Kazemzadeh, E.; Mehrafshan, A.; Jamali, M.; Malekpour, B.; Saghafinia, M. Administration of vitamin C and vitamin E in severe head injury: A randomized double-blind controlled trial. Clin. Neurosurg. 2011, 58, 133–137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koza, L.; Linseman, D.A. Glutathione precursors shield the brain from trauma. Neural Regen. Res. 2019, 14, 1701–1702. [Google Scholar] [CrossRef]
- Lin, P.H.; Kuo, L.T.; Luh, H.T. The Roles of Neurotrophins in Traumatic Brain Injury. Life 2022, 12, 26. [Google Scholar] [CrossRef]
- Lin, Y.; Ma, H.Y.; Wang, Y.; He, J.; Liu, H.J. Identification of Potential Core Genes for the Rupture of Intracranial Aneurysms by a Bioinformatics Analysis. Front. Genet. 2022, 13, 875007. [Google Scholar] [CrossRef] [PubMed]
- Hui, C.; Tadi, P.; Patti, L. Ischemic Stroke. In StatPearls; StatPearls: Treasure Island, FL, USA, 2022. [Google Scholar]
- Campbell, B.C.V.; De Silva, D.A.; Macleod, M.R.; Coutts, S.B.; Schwamm, L.H.; Davis, S.M.; Donnan, G.A. Ischaemic stroke. Nat. Rev. Dis. Prim. 2019, 5, 70. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Lu, J.; Manaenko, A.; Tang, J.; Hu, Q. Mitochondria in Ischemic Stroke: New Insight and Implications. Aging Dis. 2018, 9, 924–937. [Google Scholar] [CrossRef] [Green Version]
- Tian, H.; Chen, X.; Liao, J.; Yang, T.; Cheng, S.; Mei, Z.; Ge, J. Mitochondrial quality control in stroke: From the mechanisms to therapeutic potentials. J. Cell. Mol. Med. 2022, 26, 1000–1012. [Google Scholar] [CrossRef]
- Su, Z.; Ye, Y.; Shen, C.; Qiu, S.; Sun, Y.; Hu, S.; Xiong, X.; Li, Y.; Li, L.; Wang, H. Pathophysiology of Ischemic Stroke: Noncoding RNA Role in Oxidative Stress. Oxid. Med. Cell. Longev. 2022, 2022, 5815843. [Google Scholar] [CrossRef]
- Wen, B.; Xu, K.; Huang, R.; Jiang, T.; Wang, J.; Chen, J.; Chen, J.; He, B. Preserving mitochondrial function by inhibiting GRP75 ameliorates neuron injury under ischemic stroke. Mol. Med. Rep. 2022, 25, 165. [Google Scholar] [CrossRef]
- Yang, K.; Chen, Z.; Gao, J.; Shi, W.; Li, L.; Jiang, S.; Hu, H.; Liu, Z.; Xu, D.; Wu, L. The Key Roles of GSK-3beta in Regulating Mitochondrial Activity. Cell. Physiol. Biochem. 2017, 44, 1445–1459. [Google Scholar] [CrossRef]
- Panickar, K.S.; Anderson, R.A. Effect of polyphenols on oxidative stress and mitochondrial dysfunction in neuronal death and brain edema in cerebral ischemia. Int. J. Mol. Sci. 2011, 12, 8181–8207. [Google Scholar] [CrossRef] [Green Version]
- Montano, A.; Hanley, D.F.; Hemphill, J.C., 3rd. Hemorrhagic stroke. Handb. Clin. Neurol. 2021, 176, 229–248. [Google Scholar] [CrossRef] [PubMed]
- Unnithan, A.K.A.; Das, J.M.; Mehta, P. Hemorrhagic Stroke. In StatPearls; StatPearls: Treasure Island, FL, USA, 2022. [Google Scholar]
- Rajashekar, D.; Liang, J.W. Intracerebral Hemorrhage. In StatPearls; StatPearls: Treasure Island, FL, USA, 2022. [Google Scholar]
- Ziu, E.; Khan Suheb, M.Z.; Mesfin, F.B. Subarachnoid Hemorrhage. In StatPearls; StatPearls: Treasure Island, FL, USA, 2022. [Google Scholar]
- Rymer, M.M. Hemorrhagic stroke: Intracerebral hemorrhage. Mo. Med. 2011, 108, 50–54. [Google Scholar]
- Lantigua, H.; Ortega-Gutierrez, S.; Schmidt, J.M.; Lee, K.; Badjatia, N.; Agarwal, S.; Claassen, J.; Connolly, E.S.; Mayer, S.A. Subarachnoid hemorrhage: Who dies, and why? Crit. Care 2015, 19, 309. [Google Scholar] [CrossRef] [Green Version]
- Persson, H.C.; Tornbom, K.; Sunnerhagen, K.S.; Tornbom, M. Consequences and coping strategies six years after a subarachnoid hemorrhage—A qualitative study. PLoS ONE 2017, 12, e0181006. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Visser-Meily, J.M.; Rhebergen, M.L.; Rinkel, G.J.; van Zandvoort, M.J.; Post, M.W. Long-term health-related quality of life after aneurysmal subarachnoid hemorrhage: Relationship with psychological symptoms and personality characteristics. Stroke 2009, 40, 1526–1529. [Google Scholar] [CrossRef] [Green Version]
- Polster, S.P.; Carrion-Penagos, J.; Lyne, S.B.; Gregson, B.A.; Cao, Y.; Thompson, R.E.; Stadnik, A.; Girard, R.; Money, P.L.; Lane, K.; et al. Intracerebral Hemorrhage Volume Reduction and Timing of Intervention Versus Functional Benefit and Survival in the MISTIE III and STICH Trials. Neurosurgery 2021, 88, 961–970. [Google Scholar] [CrossRef]
- Lu, H.; Jiang, M.; Lu, L.; Zheng, G.; Dong, Q. Ultrastructural mitochondria changes in perihematomal brain and neuroprotective effects of Huperzine A after acute intracerebral hemorrhage. Neuropsychiatr. Dis. Treat. 2015, 11, 2649–2657. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Jungsuwadee, P.; Vore, M.; Butterfield, D.A.; St Clair, D.K. Collateral damage in cancer chemotherapy: Oxidative stress in nontargeted tissues. Mol. Interv. 2007, 7, 147–156. [Google Scholar] [CrossRef] [PubMed]
- Zheng, J.; Shi, L.; Liang, F.; Xu, W.; Li, T.; Gao, L.; Sun, Z.; Yu, J.; Zhang, J. Sirt3 Ameliorates Oxidative Stress and Mitochondrial Dysfunction After Intracerebral Hemorrhage in Diabetic Rats. Front. Neurosci. 2018, 12, 414. [Google Scholar] [CrossRef]
- Malisch, T.W.; Guglielmi, G.; Vinuela, F.; Duckwiler, G.; Gobin, Y.P.; Martin, N.A.; Frazee, J.G.; Chmiel, J.S. Unruptured aneurysms presenting with mass effect symptoms: Response to endosaccular treatment with Guglielmi detachable coils. Part I. Symptoms of cranial nerve dysfunction. J. Neurosurg. 1998, 89, 956–961. [Google Scholar] [CrossRef]
- Keedy, A. An overview of intracranial aneurysms. McGill J. Med. 2006, 9, 141–146. [Google Scholar] [CrossRef]
- Jersey, A.M.; Foster, D.M. Cerebral Aneurysm. In StatPearls; StatPearls: Treasure Island, FL, USA, 2022. [Google Scholar]
- Belavadi, R.; Gudigopuram, S.V.R.; Raguthu, C.C.; Gajjela, H.; Kela, I.; Kakarala, C.L.; Hassan, M.; Sange, I. Surgical Clipping Versus Endovascular Coiling in the Management of Intracranial Aneurysms. Cureus 2021, 13, e20478. [Google Scholar] [CrossRef] [PubMed]
- Molyneux, A.J.; Kerr, R.S.; Yu, L.M.; Clarke, M.; Sneade, M.; Yarnold, J.A.; Sandercock, P.; International Subarachnoid Aneurysm Trial (ISAT) Collaborative Group. International subarachnoid aneurysm trial (ISAT) of neurosurgical clipping versus endovascular coiling in 2143 patients with ruptured intracranial aneurysms: A randomised comparison of effects on survival, dependency, seizures, rebleeding, subgroups, and aneurysm occlusion. Lancet 2005, 366, 809–817. [Google Scholar] [CrossRef] [PubMed]
- Starke, R.M.; Chalouhi, N.; Ali, M.S.; Jabbour, P.M.; Tjoumakaris, S.I.; Gonzalez, L.F.; Rosenwasser, R.H.; Koch, W.J.; Dumont, A.S. The role of oxidative stress in cerebral aneurysm formation and rupture. Curr. Neurovasc. Res. 2013, 10, 247–255. [Google Scholar] [CrossRef] [Green Version]
- Zhao, W.; Zhang, H.; Su, J.Y. MicroRNA-29a contributes to intracranial aneurysm by regulating the mitochondrial apoptotic pathway. Mol. Med. Rep. 2018, 18, 2945–2954. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Llorens-Martin, M.; Lopez-Domenech, G.; Soriano, E.; Avila, J. GSK3beta is involved in the relief of mitochondria pausing in a Tau-dependent manner. PLoS ONE 2011, 6, e27686. [Google Scholar] [CrossRef] [Green Version]
- Shi, Y.; Li, S.; Song, Y.; Liu, P.; Yang, Z.; Liu, Y.; Quan, K.; Yu, G.; Fan, Z.; Zhu, W. Nrf-2 signaling inhibits intracranial aneurysm formation and progression by modulating vascular smooth muscle cell phenotype and function. J. Neuroinflamm. 2019, 16, 185. [Google Scholar] [CrossRef]
- Patrushev, M.; Kasymov, V.; Patrusheva, V.; Ushakova, T.; Gogvadze, V.; Gaziev, A. Mitochondrial permeability transition triggers the release of mtDNA fragments. Cell. Mol. Life Sci. 2004, 61, 3100–3103. [Google Scholar] [CrossRef]
- Patrushev, M.; Kasymov, V.; Patrusheva, V.; Ushakova, T.; Gogvadze, V.; Gaziev, A.I. Release of mitochondrial DNA fragments from brain mitochondria of irradiated mice. Mitochondrion 2006, 6, 43–47. [Google Scholar] [CrossRef]
- West, A.P.; Khoury-Hanold, W.; Staron, M.; Tal, M.C.; Pineda, C.M.; Lang, S.M.; Bestwick, M.; Duguay, B.A.; Raimundo, N.; MacDuff, D.A.; et al. Mitochondrial DNA stress primes the antiviral innate immune response. Nature 2015, 520, 553–557. [Google Scholar] [CrossRef] [Green Version]
- Chavda, V.; Chaurasia, B.; Garg, K.; Deora, H.; Umana, G.E.; Palmisciano, P.; Scalia, G.; Lu, B. Molecular mechanisms of oxidative stress in stroke and cancer. Brain Disord. 2022, 5, 100029. [Google Scholar] [CrossRef]
- Shimada, K.; Crother, T.R.; Karlin, J.; Dagvadorj, J.; Chiba, N.; Chen, S.; Ramanujan, V.K.; Wolf, A.J.; Vergnes, L.; Ojcius, D.M.; et al. Oxidized mitochondrial DNA activates the NLRP3 inflammasome during apoptosis. Immunity 2012, 36, 401–414. [Google Scholar] [CrossRef] [Green Version]
- Patterson, J.C.; Joughin, B.A.; van de Kooij, B.; Lim, D.C.; Lauffenburger, D.A.; Yaffe, M.B. ROS and Oxidative Stress Are Elevated in Mitosis during Asynchronous Cell Cycle Progression and Are Exacerbated by Mitotic Arrest. Cell Syst. 2019, 8, 163–167 e162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guntuku, L.; Naidu, V.G.; Yerra, V.G. Mitochondrial Dysfunction in Gliomas: Pharmacotherapeutic Potential of Natural Compounds. Curr. Neuropharmacol. 2016, 14, 567–583. [Google Scholar] [CrossRef] [Green Version]
- White, M.J.; McArthur, K.; Metcalf, D.; Lane, R.M.; Cambier, J.C.; Herold, M.J.; van Delft, M.F.; Bedoui, S.; Lessene, G.; Ritchie, M.E.; et al. Apoptotic caspases suppress mtDNA-induced STING-mediated type I IFN production. Cell 2014, 159, 1549–1562. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ushio-Fukai, M.; Nakamura, Y. Reactive oxygen species and angiogenesis: NADPH oxidase as target for cancer therapy. Cancer Lett. 2008, 266, 37–52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sabatino, M.E.; Grondona, E.; Sosa, L.D.V.; Mongi Bragato, B.; Carreno, L.; Juarez, V.; da Silva, R.A.; Remor, A.; de Bortoli, L.; de Paula Martins, R.; et al. Oxidative stress and mitochondrial adaptive shift during pituitary tumoral growth. Free Radic. Biol. Med. 2018, 120, 41–55. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Finkel, T. From sulfenylation to sulfhydration: What a thiolate needs to tolerate. Sci. Signal 2012, 5, pe10. [Google Scholar] [CrossRef]
- Ren, X.; Keeney, J.T.R.; Miriyala, S.; Noel, T.; Powell, D.K.; Chaiswing, L.; Bondada, S.; St Clair, D.K.; Butterfield, D.A. The triangle of death of neurons: Oxidative damage, mitochondrial dysfunction, and loss of choline-containing biomolecules in brains of mice treated with doxorubicin. Advanced insights into mechanisms of chemotherapy induced cognitive impairment (“chemobrain”) involving TNF-alpha. Free Radic. Biol. Med. 2019, 134, 1–8. [Google Scholar] [CrossRef]
- Koedel, U.; Pfister, H.W. Oxidative stress in bacterial meningitis. Brain Pathol. 1999, 9, 57–67. [Google Scholar] [CrossRef] [PubMed]
- Wnek, M.; Ressel, L.; Ricci, E.; Rodriguez-Martinez, C.; Guerrero, J.C.; Ismail, Z.; Smith, C.; Kipar, A.; Sodeik, B.; Chinnery, P.F.; et al. Herpes simplex encephalitis is linked with selective mitochondrial damage; a post-mortem and in vitro study. Acta Neuropathol. 2016, 132, 433–451. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Derakhshan, M.; Willcocks, M.M.; Salako, M.A.; Kass, G.E.N.; Carter, M.J. Human herpesvirus 1 protein US3 induces an inhibition of mitochondrial electron transport. J. Gen. Virol. 2006, 87 Pt 8, 2155–2159. [Google Scholar] [CrossRef]
- Latchman, D.S. Effect of herpes simplex virus type 2 infection on mitochondrial gene expression. J. Gen. Virol. 1988, 69 Pt 6, 1405–1410. [Google Scholar] [CrossRef] [PubMed]
- Ledur, P.F.; Karmirian, K.; Pedrosa, C.; Souza, L.R.Q.; Assis-de-Lemos, G.; Martins, T.M.; Ferreira, J.; de Azevedo Reis, G.F.; Silva, E.S.; Silva, D.; et al. Zika virus infection leads to mitochondrial failure, oxidative stress and DNA damage in human iPSC-derived astrocytes. Sci. Rep. 2020, 10, 1218. [Google Scholar] [CrossRef] [Green Version]
- Gu, M.; Mei, X.L.; Zhao, Y.N. Sepsis and Cerebral Dysfunction: BBB Damage, Neuroinflammation, Oxidative Stress, Apoptosis and Autophagy as Key Mediators and the Potential Therapeutic Approaches. Neurotox. Res. 2021, 39, 489–503. [Google Scholar] [CrossRef]
- Barichello, T.; Generoso, J.S.; Simoes, L.R.; Elias, S.G.; Quevedo, J. Role of oxidative stress in the pathophysiology of pneumococcal meningitis. Oxid. Med. Cell. Longev. 2013, 2013, 371465. [Google Scholar] [CrossRef] [Green Version]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nguyen, A.; Patel, A.B.; Kioutchoukova, I.P.; Diaz, M.J.; Lucke-Wold, B. Mechanisms of Mitochondrial Oxidative Stress in Brain Injury: From Pathophysiology to Therapeutics. Oxygen 2023, 3, 163-178. https://doi.org/10.3390/oxygen3020012
Nguyen A, Patel AB, Kioutchoukova IP, Diaz MJ, Lucke-Wold B. Mechanisms of Mitochondrial Oxidative Stress in Brain Injury: From Pathophysiology to Therapeutics. Oxygen. 2023; 3(2):163-178. https://doi.org/10.3390/oxygen3020012
Chicago/Turabian StyleNguyen, Andrew, Anjali B. Patel, Ivelina P. Kioutchoukova, Michael J. Diaz, and Brandon Lucke-Wold. 2023. "Mechanisms of Mitochondrial Oxidative Stress in Brain Injury: From Pathophysiology to Therapeutics" Oxygen 3, no. 2: 163-178. https://doi.org/10.3390/oxygen3020012