
Screening and Druggability Analysis of Marine Active Metabolites against SARS-CoV-2: An Integrative Computational Approach


 , , ,
, , ,  , , and
, , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Software/Servers Used
2.2. Target Protein Preparation
2.3. Ligand Preparation
2.4. Drug-Likeness Calculations
2.5. Prediction of Pharmacokinetic Parameters
2.6. Molecular Docking Studies
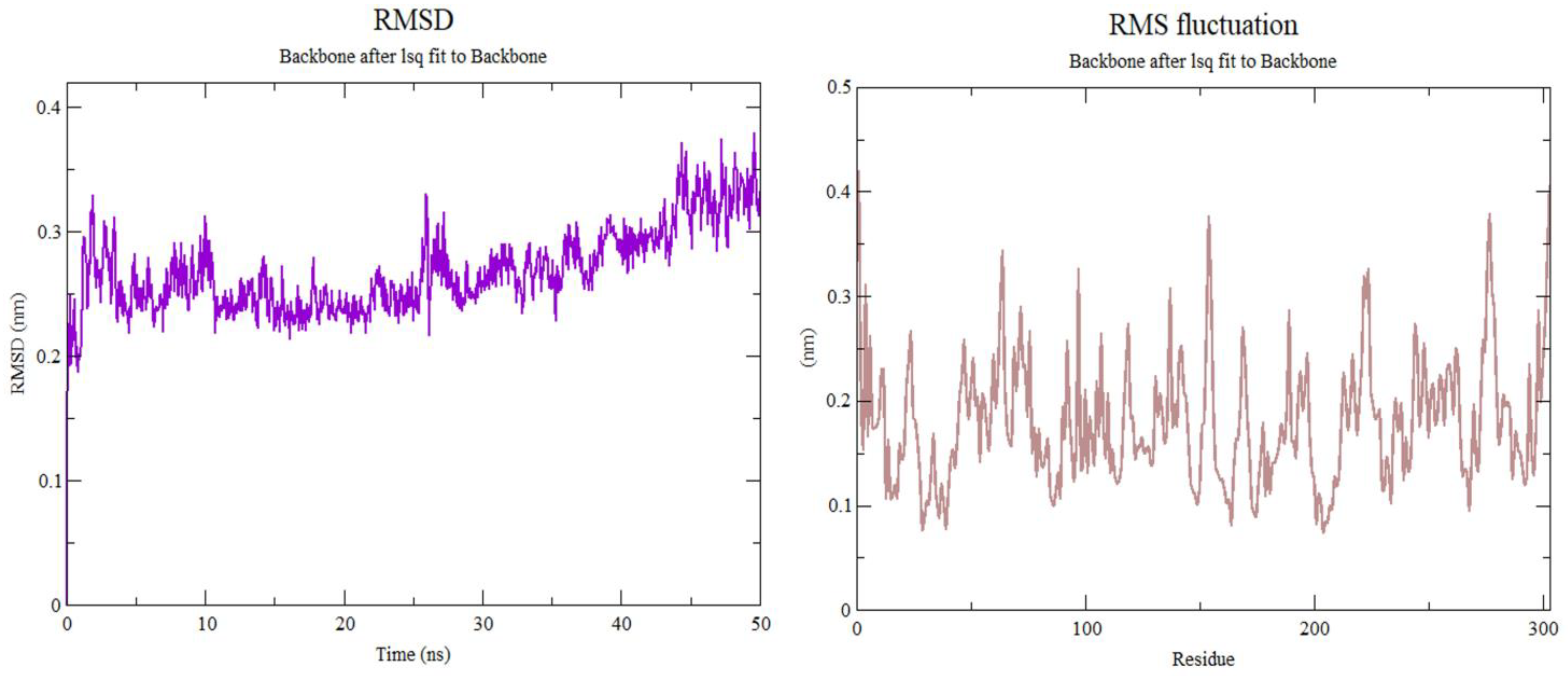
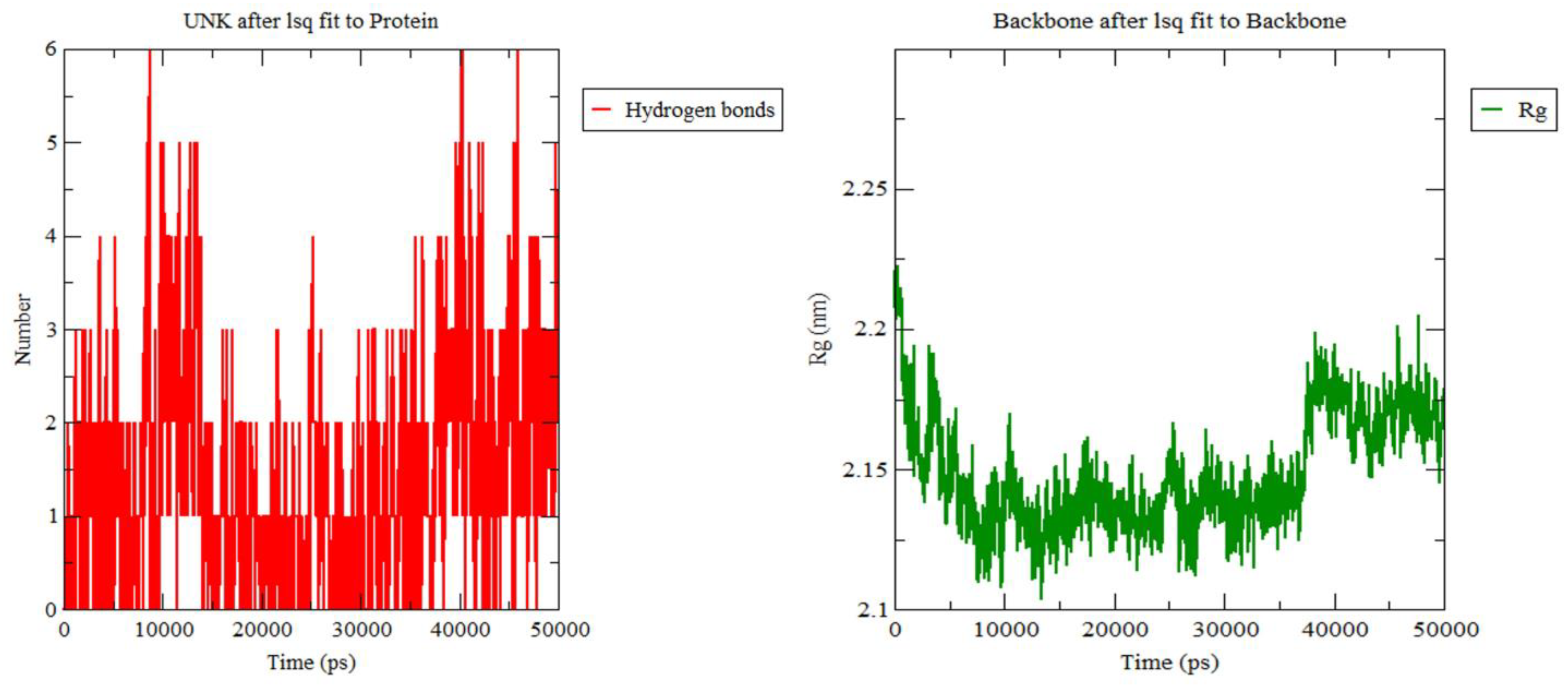
2.7. Molecular Dynamic Simulation
3. Results and Discussion
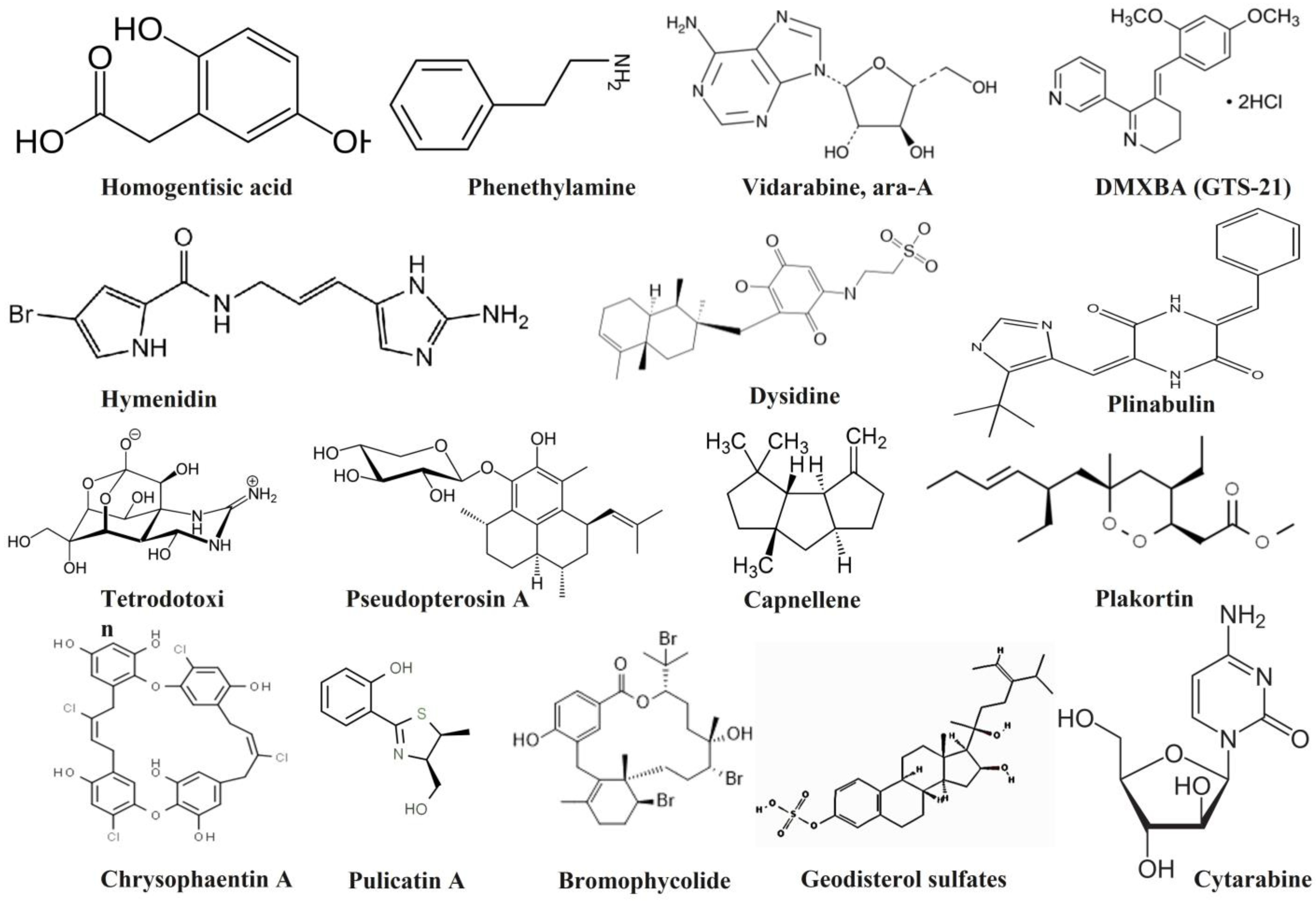
3.1. Drug-Likeness Properties
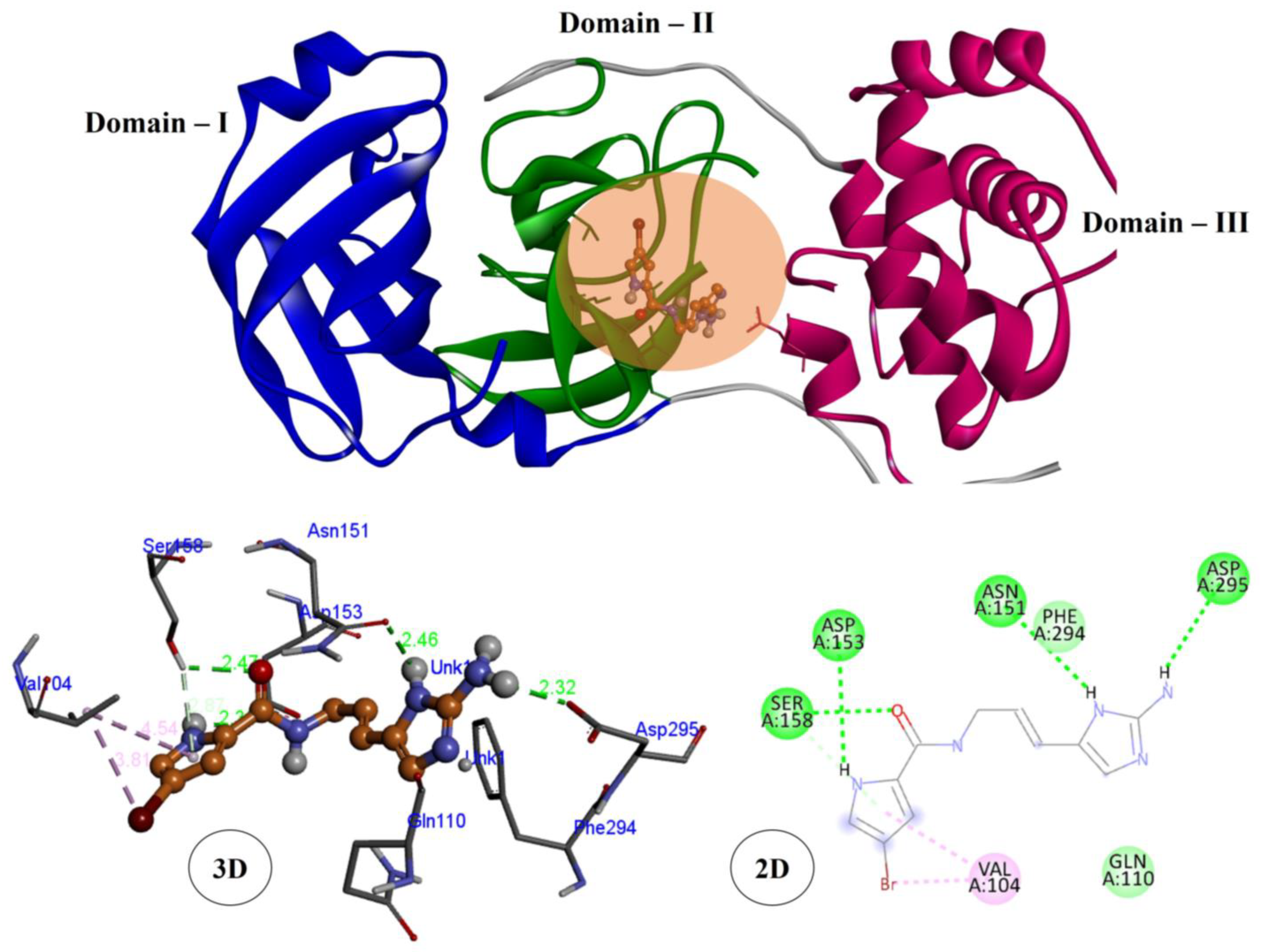
3.2. Molecular Docking Studies
3.3. Molecular Dynamics Simulation
3.4. Pharmacokinetic Properties Analysis
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Abd El-Aziz, T.M.; Stockand, J.D. Recent Progress and Challenges in Drug Development against COVID-19 Coronavirus (SARS-CoV-2)-an Update on the Status. Infect. Genet. Evol. 2020, 83, 104327. [Google Scholar] [CrossRef] [PubMed]
- Idda, M.L.; Soru, D.; Floris, M. Overview of the First 6 Months of Clinical Trials for COVID-19 Pharmacotherapy: The Most Studied Drugs. Front. Public Health 2020, 8, 497. [Google Scholar] [CrossRef] [PubMed]
- Prathiviraj, R.; Saranya, S.; Bharathi, M.; Chellapandi, P. A Hijack Mechanism of Indian SARS-CoV-2 Isolates for Relapsing Contemporary Antiviral Therapeutics. Comput. Biol. Med. 2021, 132, 104315. [Google Scholar] [CrossRef] [PubMed]
- Prathiviraj, R.; Chellapandi, P.; Begum, A.; Kiran, G.S.; Selvin, J. Identification of Genotypic Variants and Its Proteomic Mutations of Brazilian SARS-CoV-2 Isolates. Virus Res. 2022, 307, 198618. [Google Scholar] [CrossRef] [PubMed]
- Singh, M.; de Wit, E. Antiviral Agents for the Treatment of COVID-19: Progress and Challenges. Cell Rep. Med. 2022, 3, 100549. [Google Scholar] [CrossRef]
- Li, H.; Xue, Q.; Xu, X. Involvement of the Nervous System in SARS-CoV-2 Infection. Neurotox. Res. 2020, 38, 1–7. [Google Scholar] [CrossRef]
- Rahman, M.M.; Bibi, S.; Rahaman, M.S.; Rahman, F.; Islam, F.; Khan, M.S.; Hasan, M.M.; Parvez, A.; Hossain, M.A.; Maeesa, S.K.; et al. Natural therapeutics and nutraceuticals for lung diseases: Traditional significance, phytochemistry, and pharmacology. Biomed. Pharmacother. 2022, 150, 113041. [Google Scholar] [CrossRef]
- Varga, Z.; Flammer, A.J.; Steiger, P.; Haberecker, M.; Andermatt, R.; Zinkernagel, A.S.; Mehra, M.R.; Schuepbach, R.A.; Ruschitzka, F.; Moch, H. Endothelial Cell Infection and Endotheliitis in COVID-19. Lancet 2020, 395, 1417–1418. [Google Scholar] [CrossRef]
- Kim, D.; Lee, J.-Y.; Yang, J.-S.; Kim, J.W.; Kim, V.N.; Chang, H. The Architecture of SARS-CoV-2 Transcriptome. Cell 2020, 181, 914–921. [Google Scholar] [CrossRef]
- Pal, M.; Berhanu, G.; Desalegn, C.; Kandi, V. Severe acute respiratory syndrome coronavirus-2 (SARS-CoV-2): An update. Cureus 2020, 12, e7423. [Google Scholar] [CrossRef]
- Yoshimoto, F.K. The Proteins of Severe Acute Respiratory Syndrome Coronavirus-2 (SARS CoV-2 or n-COV19), the Cause of COVID-19. Protein J. 2020, 39, 198–216. [Google Scholar] [CrossRef] [PubMed]
- Bibi, S.; Khan, M.S.; El-Kafrawy, S.A.; Alandijany, T.A.; El-Daly, M.M.; Yousafi, Q.; Fatima, D.; Faizo, A.A.; Bajrai, L.H.; Azhar, E.I. Virtual screening and molecular dynamics simulation analysis of Forsythoside A as a plant-derived inhibitor of SARS-CoV-2 3CLpro. Saudi Pharm. J. 2022, 30, 979–1002. [Google Scholar] [CrossRef] [PubMed]
- Bianchi, M.; Benvenuto, D.; Giovanetti, M.; Angeletti, S.; Ciccozzi, M.; Pascarella, S. Sars-CoV-2 Envelope and Membrane Proteins: Differences from Closely Related Proteins Linked to Cross-Species Transmission. BioMed Res. Int. 2020, 2020, 4389089. [Google Scholar] [CrossRef] [PubMed]
- Snijder, E.J.; Decroly, E.; Ziebuhr, J. Coronaviruses. Adv. Virus Res. 2016, 96, 59–126. [Google Scholar]
- Jackson, C.B.; Farzan, M.; Chen, B.; Choe, H. Mechanisms of SARS-CoV-2 Entry into Cells. Nat. Rev. Mol. Cell Biol. 2022, 23, 3–20. [Google Scholar] [CrossRef]
- Biswas, P.; Hasan, M.M.; Dey, D.; dos Santos Costa, A.C.; Polash, S.A.; Bibi, S.; Ferdous, N.; Kaium, M.; Rahman, M.D.; Jeet, F.K.; et al. Candidate antiviral drugs for COVID-19 and their environmental implications: A comprehensive analysis. Environ. Sci. Pollut. Res. 2021, 28, 59570–59593. [Google Scholar] [CrossRef]
- Alanagreh, L.; Alzoughool, F.; Atoum, M. The Human Coronavirus Disease COVID-19: Its Origin, Characteristics, and Insights into Potential Drugs and Its Mechanisms. Pathogens 2020, 9, 331. [Google Scholar] [CrossRef]
- Wu, C.; Liu, Y.; Yang, Y.; Zhang, P.; Zhong, W.; Wang, Y.; Wang, Q.; Xu, Y.; Li, M.; Li, X. Analysis of Therapeutic Targets for SARS-CoV-2 and Discovery of Potential Drugs by Computational Methods. Acta Pharm. Sin. B 2020, 10, 766–788. [Google Scholar] [CrossRef]
- Salehi, B.; Sharifi-Rad, J.; Seca, A.M.; Pinto, D.C.; Michalak, I.; Trincone, A.; Mishra, A.P.; Nigam, M.; Zam, W.; Martins, N. Current Trends on Seaweeds: Looking at Chemical Composition, Phytopharmacology, and Cosmetic Applications. Molecules 2019, 24, 4182. [Google Scholar] [CrossRef] [Green Version]
- Tanna, B.; Mishra, A. Nutraceutical Potential of Seaweed Polysaccharides: Structure, Bioactivity, Safety, and Toxicity. Compr. Rev. Food Sci. Food Saf. 2019, 18, 817–831. [Google Scholar] [CrossRef] [Green Version]
- Dyshlovoy, S.A.; Honecker, F. Marine Compounds and Cancer: The First Two Decades of XXI Century. Mar. Drugs 2019, 18, 20. [Google Scholar] [CrossRef] [PubMed]
- Kumar, L.D.; Prathiviraj, R.; Selvakumar, M.; Guna, R.; Abbirami, E.; Sivasudha, T. HRLC-ESI-MS Based Identification of Active Small Molecules from Cissus Quadrangularis and Likelihood of Their Action towards the Primary Targets of Osteoarthritis. J. Mol. Struct. 2020, 1199, 127048. [Google Scholar] [CrossRef]
- Murugesan, S.; Srinivasan, V.; Lakshmanan, D.K.; Venkateswaran, M.R.; Jayabal, S.; Nadar, M.M.; Kathiravan, A.; Jhonsi, M.A.; Thilagar, S.; Periyasamy, S. Evaluation of the Anti-Rheumatic Properties of Thymol Using Carbon Dots as Nanocarriers on FCA Induced Arthritic Rats. Food Funct. 2021, 12, 5038–5050. [Google Scholar] [CrossRef] [PubMed]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and Computational Approaches to Estimate Solubility and Permeability in Drug Discovery and Development Settings. Adv. Drug Deliv. Rev. 1997, 23, 3–25. [Google Scholar] [CrossRef]
- Pires, D.E.; Blundell, T.L.; Ascher, D.B. PkCSM: Predicting Small-Molecule Pharmacokinetic and Toxicity Properties Using Graph-Based Signatures. J. Med. Chem. 2015, 58, 4066–4072. [Google Scholar] [CrossRef]
- Selvakumar, M.; Palanichamy, P.; Arumugam, V.; Venkatesan, M.; Aathmanathan, S.; Krishnamoorthy, H.; Pugazhendhi, A. In Silico Potential of Nutraceutical Plant of Pithecellobium Dulce against GRP78 Target Protein for Breast Cancer. Appl. Nanosci. 2021, 1–13. [Google Scholar] [CrossRef]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High Performance Molecular Simulations through Multi-Level Parallelism from Laptops to Supercomputers. SoftwareX 2015, 1, 19–25. [Google Scholar] [CrossRef] [Green Version]
- Tian, C.; Kasavajhala, K.; Belfon, K.A.; Raguette, L.; Huang, H.; Migues, A.N.; Bickel, J.; Wang, Y.; Pincay, J.; Wu, Q. Ff19SB: Amino-Acid-Specific Protein Backbone Parameters Trained against Quantum Mechanics Energy Surfaces in Solution. J. Chem. Theor. Comput. 2019, 16, 528–552. [Google Scholar] [CrossRef]
- Salgarello, M.; Visconti, G.; Barone-Adesi, L. Interlocking Circumareolar Suture with Undyed Polyamide Thread: A Personal Experience. Aesthetic Plast. Surg. 2013, 37, 1061–1062. [Google Scholar] [CrossRef]
- Hammad, S.; Bouaziz-Terrachet, S.; Meghnem, R.; Meziane, D. Pharmacophore Development, Drug-Likeness Analysis, Molecular Docking, and Molecular Dynamics Simulations for Identification of New CK2 Inhibitors. J. Mol. Model. 2020, 26, 160. [Google Scholar] [CrossRef]
- Wang, J.; Wang, W.; Kollman, P.A.; Case, D.A. Automatic Atom Type and Bond Type Perception in Molecular Mechanical Calculations. J. Mol. Graph. Model. 2006, 25, 247–260. [Google Scholar] [CrossRef] [PubMed]
- Yusof, I.; Segall, M.D. Considering the Impact Drug-like Properties Have on the Chance of Success. Drug Discov. Today 2013, 18, 659–666. [Google Scholar] [CrossRef] [PubMed]
- Hu, Q.; Xiong, Y.; Zhu, G.-H.; Zhang, Y.-N.; Zhang, Y.-W.; Huang, P.; Ge, G.-B. The SARS-CoV-2 Main Protease (Mpro): Structure, Function, and Emerging Therapies for COVID-19. MedComm 2022, 3, e151. [Google Scholar] [CrossRef]
- Vishvakarma, V.K.; Singh, M.B.; Jain, P.; Kumari, K.; Singh, P. Hunting the Main Protease of SARS-CoV-2 by Plitidepsin: Molecular Docking and Temperature-Dependent Molecular Dynamics Simulations. Amino Acids 2022, 54, 205–213. [Google Scholar] [CrossRef] [PubMed]
- Bibi, S.; Hasan, M.M.; Wang, Y.B.; Papadakos, S.P.; Yu, H. Cordycepin as a Promising Inhibitor of SARS-CoV-2 RNA dependent RNA polymerase (RdRp). Curr. Med. Chem. 2022, 29, 152–162. [Google Scholar] [CrossRef] [PubMed]
- Rauf, A.; Rashid, U.; Khalil, A.A.; Khan, S.A.; Anwar, S.; Alafnan, A.; Alamri, A.; Rengasamy, K.R. Docking-Based Virtual Screening and Identification of Potential COVID-19 Main Protease Inhibitors from Brown Algae. S. Afr. J. Bot. 2021, 143, 428–434. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, S.; Das, S.; Ahmad, I.; Patel, H. In Silico Validation of Anti-Viral Drugs Obtained from Marine Sources as a Potential Target against SARS-CoV-2 Mpro. J. Indian Chem. Soc. 2021, 98, 100272. [Google Scholar] [CrossRef]
- Ramadhan, D.S.F.; Siharis, F.; Abdurrahman, S.; Isrul, M.; Fakih, T.M. In Silico Analysis of Marine Natural Product from Sponge (Clathria Sp.) for Their Activity as Inhibitor of SARS-CoV-2 Main Protease. J. Biomol. Struct. Dyn. 2021, 40, 11526–11532. [Google Scholar] [CrossRef]
- Swain, S.S.; Singh, S.R.; Sahoo, A.; Panda, P.K.; Hussain, T.; Pati, S. Integrated Bioinformatics–Cheminformatics Approach toward Locating Pseudo-Potential Antiviral Marine Alkaloids against SARS-CoV-2-Mpro. Proteins Struct. Funct. Bioinform. 2022, 90, 1617–1633. [Google Scholar] [CrossRef]
- Baildya, N.; Ghosh, N.N.; Chattopadhyay, A.P. Inhibitory Activity of Hydroxychloroquine on COVID-19 Main Protease: An Insight from MD-Simulation Studies. J. Mol. Struct. 2020, 1219, 128595. [Google Scholar] [CrossRef]
- Krupanidhi, S.; Abraham Peele, K.; Venkateswarulu, T.C.; Ayyagari, V.S.; Nazneen Bobby, M.; John Babu, D.; Venkata Narayana, A.; Aishwarya, G. Screening of Phytochemical Compounds of Tinospora Cordifolia for Their Inhibitory Activity on SARS-CoV-2: An in Silico Study. J. Biomol. Struct. Dyn. 2021, 39, 5799–5803. [Google Scholar] [CrossRef] [PubMed]
- Singh, M.B.; Vishvakarma, V.K.; Lal, A.A.; Chandra, R.; Jain, P.; Singh, P. A Comparative Study of 5-Fluorouracil, Doxorubicin, Methotrexate, Paclitaxel for Their Inhibition Ability for Mpro of NCoV: Molecular Docking and Molecular Dynamics Simulations. J. Indian Chem. Soc. 2022, 99, 100790. [Google Scholar] [CrossRef]
- Chen, D.; Oezguen, N.; Urvil, P.; Ferguson, C.; Dann, S.M.; Savidge, T.C. Regulation of Protein-Ligand Binding Affinity by Hydrogen Bond Pairing. Sci. Adv. 2016, 2, e1501240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aungst, B.J. Optimizing Oral Bioavailability in Drug Discovery: An Overview of Design and Testing Strategies and Formulation Options. J. Pharm. Sci. 2017, 106, 921–929. [Google Scholar] [CrossRef] [PubMed]
- Bergström, C.A. In Silico Predictions of Drug Solubility and Permeability: Two Rate-Limiting Barriers to Oral Drug Absorption. Basic Clin. Pharmacol. Toxicol. 2005, 96, 156–161. [Google Scholar] [CrossRef]
- Igel, S.; Drescher, S.; Mürdter, T.; Hofmann, U.; Heinkele, G.; Tegude, H.; Glaeser, H.; Brenner, S.S.; Somogyi, A.A.; Omari, T. Increased Absorption of Digoxin from the Human Jejunum Due to Inhibition of Intestinal Transporter-Mediated Efflux. Clin. Pharmacokinet. 2007, 46, 777–785. [Google Scholar] [CrossRef]
- König, J.; Müller, F.; Fromm, M.F. Transporters and Drug-Drug Interactions: Important Determinants of Drug Disposition and Effects. Pharmacol. Rev. 2013, 65, 944–966. [Google Scholar] [CrossRef] [Green Version]
- Han, Y.; Zhang, J.; Hu, C.Q.; Zhang, X.; Ma, B.; Zhang, P. In Silico ADME and Toxicity Prediction of Ceftazidime and Its Impurities. Front. Pharmacol. 2019, 10, 434. [Google Scholar] [CrossRef]
- Bibi, Z. Role of Cytochrome P450 in Drug Interactions. Nutr. Metab. 2008, 5, 27. [Google Scholar] [CrossRef] [Green Version]
- Shargel, L.; Wu-Pong, S.; Yu, A.B.C. Chapter 6. Drug Elimination and Clearance. In Applied Biopharmaceutics & Pharmacokinetics, 6th ed.; The McGraw-Hill Companies: New York, NY, USA, 2012; p. 106. [Google Scholar]
- Kirkland, D.; Zeiger, E.; Madia, F.; Gooderham, N.; Kasper, P.; Lynch, A.; Morita, T.; Ouedraogo, G.; Morte, J.M.P.; Pfuhler, S. Can in Vitro Mammalian Cell Genotoxicity Test Results Be Used to Complement Positive Results in the Ames Test and Help Predict Carcinogenic or in Vivo Genotoxic Activity? I. Reports of Individual Databases Presented at an eurl ecvam Workshop. Mutat. Res. Genet. Toxicol. Environ. Mutagen. 2014, 775, 55–68. [Google Scholar] [CrossRef]
- Creanza, T.M.; Delre, P.; Ancona, N.; Lentini, G.; Saviano, M.; Mangiatordi, G.F. Structure-Based Prediction of HERG-Related Cardiotoxicity: A Benchmark Study. J. Chem. Inf. Model. 2021, 61, 4758–4770. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | MW | logP | AlogP | HBA | HBD | TSPA | AMR | nRB | nAtom | RC | n RigidB | nArom Ring | nHB |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Cytarabine, ara-C | 243.09 | −2.193 | −2.942 | 8 | 4 | 128.61 | 52.82 | 2 | 30 | 2 | 16 | 0 | 12 |
| Vidarabine, ara-A | 267.1 | −2.367 | −3.453 | 9 | 4 | 136.26 | 62.91 | 2 | 32 | 3 | 19 | 2 | 13 |
| Tetrodotoxin | 319.1 | −3.581 | −4.239 | 11 | 8 | 190.25 | 61.71 | 1 | 39 | 4 | 24 | 0 | 19 |
| DMXBA | 308.15 | 1.262 | −0.538 | 4 | 0 | 43.18 | 97.19 | 4 | 43 | 3 | 21 | 2 | 4 |
| Plinabulin | 336.16 | 3.008 | 0.565 | 6 | 3 | 82.59 | 102.06 | 3 | 45 | 3 | 24 | 2 | 9 |
| Pseudopterosin A | 432.25 | 4.368 | 0.885 | 6 | 4 | 99.38 | 119.61 | 3 | 67 | 4 | 31 | 1 | 10 |
| Chrysophaentin A | 676.02 | 5.424 | 3.052 | 8 | 6 | 139.84 | 187.65 | 0 | 68 | 5 | 48 | 4 | 14 |
| Phenethylamine | 121.09 | 1.106 | 0.725 | 1 | 1 | 26.02 | 43.12 | 2 | 20 | 1 | 7 | 1 | 2 |
| Geodisterol sulfates | 506.27 | 5.203 | 1.597 | 6 | 3 | 112.44 | 139.82 | 7 | 77 | 4 | 31 | 1 | 9 |
| Bromophycolides | 662.02 | 6.273 | 4.125 | 4 | 2 | 66.76 | 145.79 | 1 | 71 | 3 | 35 | 1 | 6 |
| Plakortin | 312.23 | 5.484 | 0.253 | 4 | 0 | 44.76 | 80.06 | 9 | 54 | 1 | 13 | 0 | 4 |
| Homogentisic acid | 168.04 | 0.036 | −0.123 | 4 | 3 | 77.76 | 44.72 | 2 | 20 | 1 | 10 | 1 | 7 |
| Hymenidin | 309.02 | 0.93 | −1.68 | 6 | 4 | 91.54 | 72.63 | 5 | 30 | 2 | 14 | 2 | 10 |
| Dysidine | 451.2 | 6.237 | 0.807 | 7 | 3 | 129.15 | 120.8 | 6 | 64 | 3 | 27 | 0 | 10 |
| Capnellene | 220.18 | 3.585 | 1.41 | 1 | 1 | 20.23 | 65.87 | 0 | 40 | 3 | 18 | 0 | 2 |
| Pulicatin A | 223.07 | 0.881 | 0.66 | 3 | 2 | 78.12 | 65.21 | 2 | 28 | 2 | 14 | 1 | 5 |
| S. No | Compound Name | PubChem ID | Chemical Class | Main Protease (6LU7) |
|---|---|---|---|---|
| 1 | Homogentisic acid | 780 | Phenolics | −5.6 |
| 2 | Phenethylamine | 1001 | Alkaloid | −4.8 |
| 3 | Cytarabine, ara-C | 6253 | Nucleoside | −6.2 |
| 4 | Vidarabine, ara-A | 21704 | Nucleoside | −6.1 |
| 5 | DMXBA (GTS-21) | 5310985 | Alkaloid | −5.5 |
| 6 | Hymenidin | 6439099 | Alkaloid | −6.4 |
| 7 | Plinabulin | 9949641 | Alkaloid | −6.4 |
| 8 | Dysidine | 10321583 | Terpene | −5.9 |
| 9 | Tetrodotoxin | 11174599 | Alkaloid | −6.3 |
| 10 | Pseudopterosin A | 11732783 | Glycoside | −6.3 |
| 11 | Capnellene | 14060593 | Terpene | −6.0 |
| 12 | Bromophycolides | 21778345 | Terpene | −6.0 |
| 13 | Geodisterol sulfates | 44254699 | Steroid | −6.6 |
| 14 | Plakortin | 44417613 | Polyketide | −5.4 |
| 15 | Chrysophaentin A | 46872004 | Shikimate | −6.6 |
| 16 | Pulicatin A | 136020617 | Alkaloid | −5.5 |
| 17 | Paracetamol | 1983 | Standard drug | −6.2 |
| 18 | HCQ | 3652 | Standard drug | −6.6 |
| Compound Name | Main Protease (6LU7) |
|---|---|
| Geodisterol sulfates | VAL104, PHE294 |
| Chrysophaentin A | GLU290, LYS137, LYS5, TYR126 |
| Hymenidin | SER158, ASP153, ASN151, ASP295, VAL104 |
| Plinabulin (NPI-2358) | GLN110, THR111, PHE294 |
| Tetrodotoxin | ASN151, ASP153, PHE 294 |
| Paracetamol | THR111, ASN151, ASP295 |
| HCQ | GLN110, THR111, ILE106, VAL101, VAL104 |
| Property | Name | Tetrodotoxin | Chrysophaentin A | Geodisterol Sulfates | Hymenidin | Plinabulin | Unit |
|---|---|---|---|---|---|---|---|
| Absorption | Water solubility | −2.244 | −2.898 | −3.231 | −2.893 | −2.894 | log mol/L |
| Caco2 permeability | 0.557 | −0.859 | 0.551 | −0.336 | −0.128 | log Papp in 10- cm/s | |
| Intestinal absorption (human) | 36.93 | 100 | 49.98 | 71.261 | 65.663 | % Absorbed | |
| Skin permeability | −2.735 | −2.735 | −2.735 | −2.735 | −2.735 | log Kp | |
| P-glycoprotein substrate | Yes | Yes | Yes | Yes | Yes | Yes/No | |
| P-glycoprotein I inhibitor | No | Yes | No | No | No | Yes/No | |
| P-glycoprotein II inhibitor | No | Yes | Yes | No | No | Yes/No | |
| Distribution | VDss (human) | −1.053 | −1.24 | −1.205 | −0.367 | 0.325 | log L/kg |
| Fraction unbound (human) | 0.8 | 0.143 | 0.08 | 0.458 | 0.101 | Numeric (Fu) | |
| BBB permeability | −1.149 | −2 | −0.893 | −1.288 | −0.285 | log BB | |
| CNS permeability | −5.174 | −2.487 | −2.763 | −4.592 | −2.619 | log PS | |
| Metabolism | CYP2D6 substrate | No | No | No | No | No | Yes/No |
| CYP3A4 substrate | No | Yes | No | No | No | Yes/No | |
| CYP1A2 inhibitor | No | No | No | No | Yes | Yes/No | |
| CYP2C19 inhibitor | No | No | No | No | No | Yes/No | |
| CYP2C9 inhibitor | No | No | No | No | No | Yes/No | |
| CYP2D6 inhibitor | No | No | No | No | Yes | Yes/No | |
| CYP3A4 inhibitor | No | No | No | No | No | Yes/No | |
| Excretion | Total clearance score | 0.663 | −0.211 | 0.27 | 1.027 | 0.457 | log mL/min/kg |
| Renal OCT2 substrate | No | No | No | No | No | Yes/No | |
| Toxicity | AMES toxicity | No | No | No | No | Yes | Yes/No |
| Max. tolerated dose (human) | 0.44 | 0.432 | −0.098 | 0.551 | 0.424 | log mg/kg/day | |
| hERG I inhibitor | No | No | No | No | No | Yes/No | |
| hERG II inhibitor | No | Yes | No | No | Yes | Yes/No | |
| Oral rat acute toxicity (LD50) | 2.061 | 2.512 | 2.686 | 2.507 | 2.669 | mol/kg | |
| Oral at chronic toxicity | 5.252 | 2.535 | 2.247 | 2.19 | 1.662 | log mg/kg bw/day | |
| Hepatotoxicity | No | No | No | Yes | No | Yes/No | |
| Skin sensitization | No | No | No | No | No | Yes/No | |
| T.Pyriformis toxicity | 0.285 | 0.285 | 0.285 | 0.285 | 0.285 | log ug/L | |
| Minnow toxicity | 7.311 | −0.711 | −0.305 | 2.477 | 4.67 | log mM |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Murugesan, S.; Ragavendran, C.; Ali, A.; Arumugam, V.; Lakshmanan, D.K.; Palanichamy, P.; Venkatesan, M.; Kamaraj, C.; Luna-Arias, J.P.; Fabián, F.-L.; et al. Screening and Druggability Analysis of Marine Active Metabolites against SARS-CoV-2: An Integrative Computational Approach. Int. J. Transl. Med. 2023, 3, 27-41. https://doi.org/10.3390/ijtm3010003
Murugesan S, Ragavendran C, Ali A, Arumugam V, Lakshmanan DK, Palanichamy P, Venkatesan M, Kamaraj C, Luna-Arias JP, Fabián F-L, et al. Screening and Druggability Analysis of Marine Active Metabolites against SARS-CoV-2: An Integrative Computational Approach. International Journal of Translational Medicine. 2023; 3(1):27-41. https://doi.org/10.3390/ijtm3010003
Chicago/Turabian StyleMurugesan, Selvakumar, Chinnasamy Ragavendran, Amir Ali, Velusamy Arumugam, Dinesh Kumar Lakshmanan, Palanikumar Palanichamy, Manigandan Venkatesan, Chinnaperumal Kamaraj, Juan Pedro Luna-Arias, Fernández-Luqueño Fabián, and et al. 2023. "Screening and Druggability Analysis of Marine Active Metabolites against SARS-CoV-2: An Integrative Computational Approach" International Journal of Translational Medicine 3, no. 1: 27-41. https://doi.org/10.3390/ijtm3010003