
Radiation and Diabetic Retinopathy: A Dark Synergy

Abstract
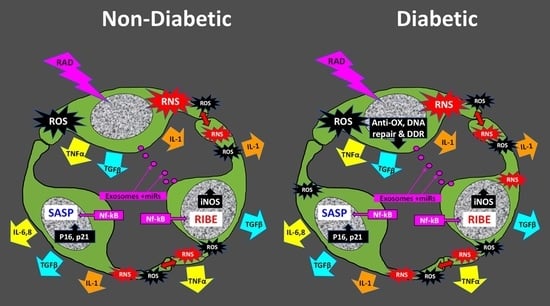
:
1. Introduction
2. Vascular Histopathology in Radiation and Diabetic Retinopathies: Similar Yet Distinctive
3. Telangiectasia as a Distinctive Vascular Change in Radiation Retinopathy
3.1. Telangiectasia: Endothelial Phenotype and Paracrine Signaling from VEGF to Norrin
3.2. VEGF: Mitogen and Morphogen in Vascular Development
3.3. Vascular Stability and the Barrier Phenotype: Angiopoietin/Tie-2 and Norrin
3.4. Norrin: A Role in Telangiectasia?
3.5. The Vascular Wall in Telangiectasia: TGFβ in Control of the Extracellular Matrix
4. Reactive Oxygen and Reactive Nitrogen Species in RR
4.1. Reactive Oxygen Species: Initiators and Source of Chronic Cell Injury in RR
4.2. Reactive Nitrogen Species: Products and Effectors of Oxidative Stress
4.3. NOS Uncoupling: Source of ROS and RNS
4.4. S-Glutathionylation of eNOS
5. The Radiation-Induced Bystander Effect
Role of Cellular Senescence in the RIBE
6. ROS and Impaired Antioxidant Defense in the Pathogenesis of DR
6.1. NADPH Oxidase as a Source of ROS in DR
6.2. Role of iNOS in Vascular Dysfunction in DR and RR
6.3. Nitrositive Stress in Neuro-Glial Pathology in DR and RR
7. Chromatin Structure: A Determinant of Radiation Sensitivity
7.1. DSB Repair in Heterochromatin
7.2. Heterochromatin: Constitutive or Facultative

7.3. DNA Damage: Chromatin Sensitivity vs. Cellular Sensitivity
8. Telangiectasia in ATM: DNA Damage and/or Oxidative Stress
9. DNA Damage and Repair in Diabetes: Roles for RAGE and miR200
Oxidant-Induced Single-Strand Breaks and Poly-(ADP-Ribose) Polymerase in DR
10. Vascular Repair in DR and RR: Role of Endothelial Progenitor Cells
Micro-RNAs in Vascular Repair
11. Addition of Radiation to DR Provides an Animal Model with Unique Features
12. Possible Therapeutic Interventions
13. Conclusions: Combined Vascular Insult of RR on a Background of DR
Funding
Conflicts of Interest
Abbreviations
| AG | aminoguanidine |
| AT | ataxia telangiectasia mutated |
| BH4 | tetrahydrobiopterin |
| BL | basal lamina |
| BM | basement membrane |
| cHC | constitutive heterochromatin |
| DR | diabetic retinopathy |
| DSBs | double-strand DNA breaks |
| EC | endothelial cell |
| eNOS | endothelial nitric oxide synthase |
| EPC | endothelial progenitor cell |
| ETC | electron transport chain |
| FAD | flavin adenine dinucleotide |
| fHC | facultative heterochromatin |
| γH2AX | phospho-histone H2AX |
| GSH | reduced glutathione |
| GSSH | oxidised glutathione |
| GSX | glutathione peroxide |
| H2O2 | hydrogen peroxide |
| HbA1C | hemoglobin-A1C |
| HC | heterochromatin |
| HLArg | hydroxy-L-arginine |
| IL-1 | interleukin-1 |
| IL-6 | interleukin-6 |
| IL-8 | interleukin-8 |
| iNOS | inducible nitric oxide synthase |
| IR | ionizing radiation |
| IRMA | intraretinal microvascular abnormalities |
| LADs | lamina-associated domains |
| miR | microRNA |
| miR | microRNA |
| MnSOD | manganese superoxide dismutase |
| MRN | MRE11-Rad50-Nbs1 complex |
| MSCs | mesenchymal stem cells |
| NAD+ | oxidised nicotinamide adenine dinucleotide |
| NADPH | reduced nicotinamide adenine dinucleotide phosphate |
| NF-kB | nuclear factor kappa-B |
| NHEJ | Non-homologous end joining |
| nNOS | neuronal nitric oxide synthase |
| NO | nitric oxide |
| NOX2- | NADPH-oxidase-2 |
| NVU | neurovascular unit |
| P21 | cyclin-dependent kinase inhibitor-1 |
| P53 | tumor suppressor protein-53 |
| PAR | poly-ADP-ribose |
| PARP | poly-ADP-ribose polymerase |
| PDR | proliferative diabetic retinopathy |
| RAGE | receptor for advanced glycation end-products |
| RIBE | radiation-induced bystander effect |
| RIF | radiation-induced foci |
| RNS | reactive nitrogen species |
| ROS | reactive oxygen species |
| RR | radiation retinopathy |
| SASP- | senescence-associated secretory phenotype |
| SSBs | single-strand DNA breaks |
| STZ | streptozotocin |
| TBI | total body irradiation |
| TGFβ | transforming growth factor beta |
| TNFα | tumor necrosis factor alpha, |
| VEGF | vascular endothelial growth factor |
| 8-OHdG | 8-hydroxydeoxy-guanosine |
References
- Dhir, S.P.; Joshi, A.V.; Banerjee, A.K. Radiation Retinopathy in Diabetes Mellitus Report of a Case. Acta Radiol. Oncol. 1982, 21, 111–113. [Google Scholar] [CrossRef]
- Brown, G.C.; Shields, J.A.; Sanborn, G.; Augsburger, J.J.; Savino, P.J.; Schatz, N.J. Radiation Retinopathy. Ophthalmology 1982, 89, 1494–1501. [Google Scholar] [CrossRef] [PubMed]
- Amoaku, W.M.K.; Archer, D.B. Cephalic radiation and retinal vasculopathy. Eye 1990, 4, 195–203. [Google Scholar] [CrossRef] [PubMed]
- Viebahn, M.; Barricks, M.E.; Osterloh, M.D. Synergism between diabetic and radiation retinopathy: Case report and review. Br. J. Ophthalmol. 1991, 75, 629–632. [Google Scholar] [CrossRef] [PubMed]
- Archer, D.B.; Amoaku, W.M.K.; Gardiner, T.A. Radiation retinopathy—Clinical, histopathological, ultrastructural and experimental correlations. Eye 1991, 5, 239–251. [Google Scholar] [CrossRef]
- Archer, D.B. Responses of retinal and choroidal vessels to ionising radiation. Eye 1993, 7, 1–13. [Google Scholar] [CrossRef]
- Gardiner, T.A.; Amoaku, W.M.K.; Archer, D.B. The Combined Effect of Diabetes and Ionising Radiation on the Retinal Vasculature of the Rate. Curr. Eye Res. 1993, 12, 1009–1014. [Google Scholar] [CrossRef]
- Stitt, A.W.; Anderson, H.R.; Gardiner, T.A.; McIntyre, I.; Archer, D.B. The combined effects of diabetes and ionising radiation on the rat retina: An ultrastructural study. Curr. Eye Res. 1994, 13, 79–86. [Google Scholar] [CrossRef]
- García-O’Farrill, N.; Pugazhendhi, S.; Karth, P.A.; Hunter, A.A. Radiation retinopathy intricacies and advances in management. Semin. Ophthalmol. 2022, 37, 417–435. [Google Scholar] [CrossRef]
- Sahoo, N.K.; Ranjan, R.; Tyagi, M.; Agrawal, H.; Reddy, S. Radiation Retinopathy: Detection and Management Strategies. Clin. Ophthalmol. 2021, 15, 3797–3809. [Google Scholar] [CrossRef]
- Yu, H.J.; Schefler, A.C.; Houston, H.R.C.O. Radiation Retinopathy—A Review of Past and Current Treatment Strategies. US Ophthalmic Rev. 2020, 13, 34–39. [Google Scholar] [CrossRef]
- Souto, E.B.; Campos, J.R.; Da Ana, R.; Fangueiro, J.F.; Martins-Gomes, C.; Durazzo, A.; Lucarini, M.; López, E.S.; Espina, M.; García, M.L.; et al. Diabetic Retinopathy and Ocular Melanoma: How Far We Are? Appl. Sci. 2020, 10, 2777. [Google Scholar] [CrossRef]
- Powell, B.E.; Chin, K.J.; Finger, P.T. Early anti-VEGF treatment for radiation maculopathy and optic neuropathy: Lessons learned. Eye 2022, 36, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Stitt, A.W.; Curtis, T.M.; Chen, M.; Medina, R.J.; McKay, G.J.; Jenkins, A.; Gardiner, T.A.; Lyons, T.J.; Hammes, H.-P.; Simó, R.; et al. The progress in understanding and treatment of diabetic retinopathy. Prog. Retin. Eye Res. 2016, 51, 156–186. [Google Scholar] [CrossRef]
- Stitt, A.W.; O’Neill, C.L.; O’Doherty, M.T.; Archer, D.B.; Gardiner, T.A.; Medina, R.J. Vascular stem cells and ischaemic retinopathies. Prog. Retin. Eye Res. 2011, 30, 149–166. [Google Scholar] [CrossRef]
- Garner, A.; Ashton, N.; Tripathi, R.; Kohner, E.M.; Bulpitt, C.J.; Dollery, C.T. Pathogenesis of hypertensive retinopathy. An experimental study in the monkey. Br. J. Ophthalmol. 1975, 59, 3–44. [Google Scholar] [CrossRef] [Green Version]
- Amoaku, W.M.K.; Archer, D.B. Fluorescein angiographic features, natural course and treatment of radiation retinopathy. Eye 1990, 4, 657–667. [Google Scholar] [CrossRef]
- Irvine, A.R.; Alvarado, J.A.; Wara, W.M.; Morris, B.W.; Wood, I.S. Radiation retinopathy: An experimental model for the ischemic--proliferative retinopathies. Trans. Am. Ophthalmol. Soc. 1981, 79, 103–122. [Google Scholar]
- Curtis, T.M.; Gardiner, T.A.; Stitt, A.W. Microvascular lesions of diabetic retinopathy: Clues towards understanding pathogenesis? Eye 2009, 23, 1496–1508. [Google Scholar] [CrossRef] [Green Version]
- Webster, A.R.; Anderson, J.R.; Richards, E.M.; Moore, A.T. Ischaemic retinopathy occurring in patients receiving bone marrow allografts and campath-1G: A clinicopathological study. Br. J. Ophthalmol. 1995, 79, 687–691. [Google Scholar] [CrossRef] [Green Version]
- Archerz, D.B.; Gardiner, T.A. Ionizing radiation and the retina. Curr. Opin. Ophthalmol. 1994, 5, 59–65. [Google Scholar] [CrossRef] [PubMed]
- Sigler, E.J.; Randolph, J.C.; Calzada, J.I.; Wilson, M.W.; Haik, B.G. Current management of Coats disease. Surv. Ophthalmol. 2014, 59, 30–46. [Google Scholar] [CrossRef]
- Shields, J.A.; Shields, C.L.; Honavar, S.; Demirci, H. Clinical variations and complications of Coats disease in 150 cases: The 2000 Sanford Gifford Memorial Lecture. Am. J. Ophthalmol. 2001, 131, 561–571. [Google Scholar] [CrossRef] [PubMed]
- McGettrick, P.M.; Loeffler, K.U. Bilateral Coats’ disease in an infant (a clinical, angiographic, light and electron microscopic study). Eye 1987, 1, 136–145. [Google Scholar] [CrossRef] [Green Version]
- Tripathi, R.; Ashton, N. Electron microscopical study of Coat’s disease. Br. J. Ophthalmol. 1971, 55, 289–301. [Google Scholar] [CrossRef] [Green Version]
- Ishibashi, T.; Inomata, H. Ultrastructure of retinal vessels in diabetic patients. Br. J. Ophthalmol. 1993, 77, 574–578. [Google Scholar] [CrossRef] [Green Version]
- Darche, M.; Verschueren, A.; Farias, D.C.; Borella, Y.; Paques, M. Confocal microscopy of telangiectatic capillaries (Tel Caps) and other features of microvascular remodeling following branch retinal vein occlusion. J. Anat. 2022. [Google Scholar] [CrossRef]
- Gardiner, T.A.; Stitt, A.W. Pericyte and Vascular Smooth Muscle Death in Diabetic Retinopathy Involves Autophagy. Int. J. Transl. Med. 2022, 2, 26–40. [Google Scholar] [CrossRef]
- Ashton, N. Vascular basement membrane changes in diabetic retinopathy. Montgomery lecture, 1973. Br. J. Ophthalmol. 1974, 58, 344–366. [Google Scholar] [CrossRef] [Green Version]
- Ishikawa, T. Fine structure of retinal vessels in man and the macaque monkey. Investig. Ophthalmol. 1963, 2, 1–15. [Google Scholar]
- Hogan, M.J.; Feeney, L. The ultrastructure of the retinal vessels: II. The small vessels. J. Ultrastruct. Res. 1963, 9, 29–46. [Google Scholar] [CrossRef] [PubMed]
- McConnell, H.L.; Kersch, C.N.; Woltjer, R.L.; Neuwelt, E.A. The Translational Significance of the Neurovascular Unit. J. Biol. Chem. 2017, 292, 762–770. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Metea, M.R.; Newman, E.A. Signalling within the neurovascular unit in the mammalian retina. Exp. Physiol. 2007, 92, 635–640. [Google Scholar] [CrossRef]
- Ward, N.L.; Lamanna, J.C. The neurovascular unit and its growth factors: Coordinated response in the vascular and nervous systems. Neurol. Res. 2004, 26, 870–883. [Google Scholar] [CrossRef] [PubMed]
- Janzer, R.C.; Raff, M.C. Astrocytes induce blood–brain barrier properties in endothelial cells. Nature 1987, 325, 253–257. [Google Scholar] [CrossRef]
- Benjamin, L.E.; Hemo, I.; Keshet, E. A plasticity window for blood vessel remodelling is defined by pericyte coverage of the preformed endothelial network and is regulated by PDGF-B and VEGF. Development 1998, 125, 1591–1598. [Google Scholar] [CrossRef]
- Uemura, A.; Ogawa, M.; Hirashima, M.; Fujiwara, T.; Koyama, S.; Takagi, H.; Honda, Y.; Wiegand, S.J.; Yancopoulos, G.D.; Nishikawa, S.-I. Recombinant angiopoietin-1 restores higher-order architecture of growing blood vessels in mice in the absence of mural cells. J. Clin. Investig. 2002, 110, 1619–1628. [Google Scholar] [CrossRef]
- Daneman, R.; Zhou, L.; Kebede, A.A.; Barres, B.A. Pericytes are required for blood–brain barrier integrity during embryogenesis. Nature 2010, 468, 562–566. [Google Scholar] [CrossRef] [Green Version]
- Armulik, A.; Genové, G.; Mäe, M.; Nisancioglu, M.H.; Wallgard, E.; Niaudet, C.; He, L.; Norlin, J.; Lindblom, P.; Strittmatter, K.; et al. Pericytes regulate the blood & ndash; brain barrier. Nature 2010, 468, 557–561. [Google Scholar] [CrossRef] [Green Version]
- Leung, D.W.; Cachianes, G.; Kuang, W.-J.; Goeddel, D.V.; Ferrara, N. Vascular Endothelial Growth Factor Is a Secreted Angiogenic Mitogen. Science 1989, 246, 1306–1309. [Google Scholar] [CrossRef]
- Senger, D.R.; Galli, S.J.; Dvorak, A.M.; Perruzzi, C.A.; Harvey, V.S.; Dvorak, H.F. Tumor Cells Secrete a Vascular Permeability Factor That Promotes Accumulation of Ascites Fluid. Science 1983, 219, 983–985. [Google Scholar] [CrossRef] [PubMed]
- Ferrara, N.; Davis-Smyth, T. The Biology of Vascular Endothelial Growth Factor. Endocr. Rev. 1997, 18, 4–25. [Google Scholar] [CrossRef] [PubMed]
- Carmeliet, P.; Storkebaum, E. Vascular and neuronal effects of VEGF in the nervous system: Implications for neurological disorders. Semin. Cell Dev. Biol. 2002, 13, 39–53. [Google Scholar] [CrossRef] [PubMed]
- Lange, C.; Storkebaum, E.; de Almodovar, C.R.; Dewerchin, M.; Carmeliet, P. Vascular endothelial growth factor: A neurovascular target in neurological diseases. Nat. Rev. Neurol. 2016, 12, 439–454. [Google Scholar] [CrossRef]
- Selvam, S.; Kumar, T.; Fruttiger, M. Retinal vasculature development in health and disease. Prog. Retin. Eye Res. 2018, 63, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Gerhardt, H.; Golding, M.; Fruttiger, M.; Ruhrberg, C.; Lundkvist, A.; Abramsson, A.; Jeltsch, M.; Mitchell, C.; Alitalo, K.; Shima, D.; et al. VEGF guides angiogenic sprouting utilizing endothelial tip cell filopodia. J. Cell Biol. 2003, 161, 1163–1177. [Google Scholar] [CrossRef] [PubMed]
- Holderfield, M.T.; Hughes, C.C. Crosstalk Between Vascular Endothelial Growth Factor, Notch, and Transforming Growth Factor-β in Vascular Morphogenesis. Circ. Res. 2008, 102, 637–652. [Google Scholar] [CrossRef] [Green Version]
- Marlier, A.; Schmidt-Ott, K.M.; Gallagher, A.-R.; Barasch, J.; Karihaloo, A. Vegf as an epithelial cell morphogen modulates branching morphogenesis of embryonic kidney by directly acting on the ureteric bud. Mech. Dev. 2009, 126, 91–98. [Google Scholar] [CrossRef]
- Shinkai, M.; Shinkai, T.; Montedonico, S.; Puri, P. Effect of VEGF on the branching morphogenesis of normal and nitrofen-induced hypoplastic fetal rat lung explants. J. Pediatr. Surg. 2006, 41, 781–786. [Google Scholar] [CrossRef]
- Roberts, W.; Palade, G. Increased microvascular permeability and endothelial fenestration induced by vascular endothelial growth factor. J. Cell Sci. 1995, 108, 2369–2379. [Google Scholar] [CrossRef]
- Esser, S.; Wolburg, K.; Wolburg, H.; Breier, G.; Kurzchalia, T.; Risau, W. Vascular Endothelial Growth Factor Induces Endothelial Fenestrations In Vitro. J. Cell Biol. 1998, 140, 947–959. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blaauwgeers, H.G.; Holtkamp, G.M.; Rutten, H.; Witmer, A.N.; Koolwijk, P.; Partanen, T.A.; Alitalo, K.; Kroon, M.E.; Kijlstra, A.; van Hinsbergh, V.W.; et al. Polarized Vascular Endothelial Growth Factor Secretion by Human Retinal Pigment Epithelium and Localization of Vascular Endothelial Growth Factor Receptors on the Inner Choriocapillaris: Evidence for a Trophic Paracrine Relation. Am. J. Pathol. 1999, 155, 421–428. [Google Scholar] [CrossRef] [PubMed]
- Sonnen, K.F.; Janda, C.Y. Signalling dynamics in embryonic development. Biochem. J. 2021, 478, 4045–4070. [Google Scholar] [CrossRef] [PubMed]
- Perrimon, N.; Pitsouli, C.; Shilo, B.-Z. Signaling Mechanisms Controlling Cell Fate and Embryonic Patterning. Cold Spring Harb. Perspect. Biol. 2012, 4, a005975. [Google Scholar] [CrossRef] [Green Version]
- Saharinen, P.; Eklund, L.; Alitalo, K. Therapeutic targeting of the angiopoietin–TIE pathway. Nat. Rev. Drug Discov. 2017, 16, 635–661. [Google Scholar] [CrossRef]
- Daneman, R.; Agalliu, D.; Zhou, L.; Kuhnert, F.; Kuo, C.J.; Barres, B.A. Wnt/β-catenin signaling is required for CNS, but not non-CNS, angiogenesis. Proc. Natl. Acad. Sci. USA 2009, 106, 641–646. [Google Scholar] [CrossRef] [Green Version]
- Daneman, R.; Prat, A. The Blood–Brain Barrier. Cold Spring Harb. Perspect. Biol. 2015, 7, a020412. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Y.; Wang, Y.; Tischfield, M.; Williams, J.; Smallwood, P.M.; Rattner, A.; Taketo, M.M.; Nathans, J. Canonical WNT signaling components in vascular development and barrier formation. J. Clin. Investig. 2014, 124, 3825–3846. [Google Scholar] [CrossRef] [Green Version]
- Xu, Q.; Wang, Y.; Dabdoub, A.; Smallwood, P.M.; Williams, J.; Woods, C.; Kelley, M.W.; Jiang, L.; Tasman, W.; Zhang, K.; et al. Vascular Development in the Retina and Inner Ear: Control by Norrin and Frizzled-4, a High-Affinity Ligand-Receptor Pair. Cell 2004, 116, 883–895. [Google Scholar] [CrossRef] [Green Version]
- Ye, X.; Wang, Y.; Nathans, J. The Norrin/Frizzled4 signaling pathway in retinal vascular development and disease. Trends Mol. Med. 2010, 16, 417–425. [Google Scholar] [CrossRef] [Green Version]
- Black, G.; Redmond, R.M. The molecular biology of Norrie’s disease. Eye 1994, 8, 491–496. [Google Scholar] [CrossRef] [PubMed]
- Perez-Vilar, J.; Hill, R.L. Norrie Disease Protein (Norrin) Forms Disulfide-linked Oligomers Associated with the Extracellular Matrix. J. Biol. Chem. 1997, 272, 33410–33415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ye, X.; Smallwood, P.; Nathans, J. Expression of the Norrie disease gene (Ndp) in developing and adult mouse eye, ear, and brain. Gene Expr. Patterns 2011, 11, 151–155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Rattner, A.; Zhou, Y.; Williams, J.; Smallwood, P.M.; Nathans, J. Norrin/Frizzled4 Signaling in Retinal Vascular Development and Blood Brain Barrier Plasticity. Cell 2012, 151, 1332–1344. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Black, G.C.M.; Perveen, R.; Bonshek, R.; Cahill, M.; Clayton-Smith, J.; Lloyd, I.C.; McLeod, D. Coats’ disease of the retina (unilateral retinal telangiectasis) caused by somatic mutation in the NDP gene: A role for norrin in retinal angiogenesis. Hum. Mol. Genet. 1999, 8, 2031–2035. [Google Scholar] [CrossRef] [Green Version]
- Richter, M.; Gottanka, J.; May, C.A.; Welge-Lüssen, U.; Berger, W.; Lütjen-Drecoll, E. Retinal vasculature changes in Norrie disease mice. Investig. Opthalmology Vis. Sci. 1998, 39, 2450–2457. [Google Scholar]
- Luhmann, U.F.O.; Lin, J.; Acar, N.; Lammel, S.; Feil, S.; Grimm, C.; Seeliger, M.W.; Hammes, H.-P.; Berger, W. Role of the Norrie Disease Pseudoglioma Gene in Sprouting Angiogenesis during Development of the Retinal Vasculature. Investig. Opthalmology Vis. Sci. 2005, 46, 3372–3382. [Google Scholar] [CrossRef]
- Beck, S.C.; Feng, Y.; Sothilingam, V.; Garrido, M.G.; Tanimoto, N.; Acar, N.; Shan, S.; Seebauer, B.; Berger, W.; Hammes, H.-P.; et al. Long-term consequences of developmental vascular defects on retinal vessel homeostasis and function in a mouse model of Norrie disease. PLoS ONE 2017, 12, e0178753. [Google Scholar] [CrossRef] [Green Version]
- Zuercher, J.; Fritzsche, M.; Feil, S.; Mohn, L.; Berger, W. Norrin stimulates cell proliferation in the superficial retinal vascular plexus and is pivotal for the recruitment of mural cells. Hum. Mol. Genet. 2012, 21, 2619–2630. [Google Scholar] [CrossRef] [Green Version]
- Stitt, A.W.; Gardiner, T.A.; Archer, D.B. Histological and ultrastructural investigation of retinal microaneurysm development in diabetic patients. Br. J. Ophthalmol. 1995, 79, 362–367. [Google Scholar] [CrossRef] [Green Version]
- Gardiner, T.A.; Archer, D.B.; Curtis, T.M.; Stitt, A.W. Arteriolar Involvement in the Microvascular Lesions of Diabetic Retinopathy: Implications for Pathogenesis. Microcirculation 2007, 14, 25–38. [Google Scholar] [CrossRef] [PubMed]
- Orlidge, A.; D’Amore, P.A. Inhibition of capillary endothelial cell growth by pericytes and smooth muscle cells. J. Cell Biol. 1987, 105, 1455–1462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, W.; Fruttiger, M.; Zhu, L.; Chung, S.H.; Barnett, N.L.; Kirk, J.K.; Lee, S.R.; Coorey, N.J.; Killingsworth, M.C.; Sherman, L.S.; et al. Conditional Muller Cell Ablation Causes Independent Neuronal and Vascular Pathologies in a Novel Transgenic Model. J. Neurosci. 2012, 32, 15715–15727. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barcellos-Hoff, M.H.; Park, C.; Wright, E.G. Radiation and the microenvironment—Tumorigenesis and therapy. Nat. Rev. Cancer 2005, 5, 867–875. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barcellos-Hoff, M.H.; Dix, T.A. Redox-mediated activation of latent transforming growth factor-beta 1. Mol. Endocrinol. 1996, 10, 1077–1083. [Google Scholar] [CrossRef] [Green Version]
- Prud’Homme, G.J. Pathobiology of transforming growth factor β in cancer, fibrosis and immunologic disease, and therapeutic considerations. Lab. Investig. 2007, 87, 1077–1091. [Google Scholar] [CrossRef] [Green Version]
- Straub, J.M.; New, J.; Hamilton, C.D.; Lominska, C.; Shnayder, Y.; Thomas, S.M. Radiation-induced fibrosis: Mechanisms and implications for therapy. J. Cancer Res. Clin. Oncol. 2015, 141, 1985–1994. [Google Scholar] [CrossRef] [Green Version]
- Bringmann, A.; Iandiev, I.; Pannicke, T.; Wurm, A.; Hollborn, M.; Wiedemann, P.; Osborne, N.N.; Reichenbach, A. Cellular signaling and factors involved in Müller cell gliosis: Neuroprotective and detrimental effects. Prog. Retin. Eye Res. 2009, 28, 423–451. [Google Scholar] [CrossRef]
- Ponsioen, T.L.; Van Luyn, M.J.; Van Der Worp, R.J.; Pas, H.H.; Hooymans, J.M.; Los, L.I. Human retinal Müller cells synthesize collagens of the vitreous and vitreoretinal interface in vitro. Mol. Vis. 2008, 14, 652–660. [Google Scholar]
- Conedera, F.M.; Pousa, A.M.Q.; Presby, D.M.; Mercader, N.; Enzmann, V.; Tschopp, M. Diverse Signaling by TGFβ Isoforms in Response to Focal Injury is Associated with Either Retinal Regeneration or Reactive Gliosis. Cell Mol. Neurobiol. 2020, 41, 43–62. [Google Scholar] [CrossRef] [Green Version]
- Görlach, A.; Berchner-Pfannschmidt, U.; Wotzlaw, C.; Cool, R.H.; Fandrey, J.; Acker, H.; Jungermann, K.; Kietzmann, T. Reactive oxygen species modulate HIF-1 mediated PAI-1 expression: Involvement of the GTPase Rac1. Thromb. Haemost. 2003, 89, 926–935. [Google Scholar] [CrossRef] [PubMed]
- Tominaga, K.; Suzuki, H.I. TGF-β Signaling in Cellular Senescence and Aging-Related Pathology. Int. J. Mol. Sci. 2019, 20, 5002. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shao, C.; Folkard, M.; Prise, K.M. Role of TGF-β1 and nitric oxide in the bystander response of irradiated glioma cells. Oncogene 2008, 27, 434–440. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Díaz-Coránguez, M.; Lin, C.-M.; Liebner, S.; Antonetti, D.A. Norrin restores blood-retinal barrier properties after vascular endothelial growth factor–induced permeability. J. Biol. Chem. 2020, 295, 4647–4660. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chidiac, R.; Abedin; Macleod, G.; Yang, A.; Thibeault, P.E.; Blazer, L.L.; Adams, J.J.; Zhang, L.; Roehrich, H.; Jo, H.; et al. A Norrin/Wnt surrogate antibody stimulates endothelial cell barrier function and rescues retinopathy. EMBO Mol. Med. 2021, 13, e13977. [Google Scholar] [CrossRef]
- Desouky, O.; Ding, N.; Zhou, G. Targeted and non-targeted effects of ionizing radiation. J. Radiat. Res. Appl. Sci. 2015, 8, 247–254. [Google Scholar] [CrossRef] [Green Version]
- Förstermann, U.; Sessa, W.C. Nitric oxide synthases: Regulation and function. Eur. Heart J. 2012, 33, 829–837. [Google Scholar] [CrossRef] [Green Version]
- Pacher, P.; Beckman, J.S.; Liaudet, L. Nitric Oxide and Peroxynitrite in Health and Disease. Physiol. Rev. 2007, 87, 315–424. [Google Scholar] [CrossRef] [Green Version]
- Beckman, J.S.; Beckman, T.W.; Chen, J.; Marshall, P.A.; Freeman, B.A. Apparent hydroxyl radical production by peroxynitrite: Implications for endothelial injury from nitric oxide and superoxide. Proc. Natl. Acad. Sci. USA 1990, 87, 1620–1624. [Google Scholar] [CrossRef] [Green Version]
- Indo, H.P.; Yen, H.-C.; Nakanishi, I.; Matsumoto, K.-I.; Tamura, M.; Nagano, Y.; Matsui, H.; Gusev, O.; Cornette, R.; Okuda, T.; et al. A mitochondrial superoxide theory for oxidative stress diseases and aging. J. Clin. Biochem. Nutr. 2015, 56, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Lundberg, J.O.; Weitzberg, E. Nitric oxide signaling in health and disease. Cell 2022, 185, 2853–2878. [Google Scholar] [CrossRef] [PubMed]
- Radi, R. Oxygen radicals, nitric oxide, and peroxynitrite: Redox pathways in molecular medicine. Proc. Natl. Acad. Sci. USA 2018, 115, 5839–5848. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stuehr, D.; Pou, S.; Rosen, G.M. Oxygen Reduction by Nitric-oxide Synthases. J. Biol. Chem. 2001, 276, 14533–14536. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuzkaya, N.; Weissmann, N.; Harrison, D.G.; Dikalov, S. Interactions of Peroxynitrite, Tetrahydrobiopterin, Ascorbic Acid, and Thiols: Implications for Uncoupling Endothelial Nitric-Oxide Synthase. J. Biol. Chem. 2003, 278, 22546–22554. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, W.; Druhan, L.J.; Chen, C.-A.; Hemann, C.; Chen, Y.-R.; Berka, V.; Tsai, A.-L.; Zweier, J.L. Peroxynitrite Induces Destruction of the Tetrahydrobiopterin and Heme in Endothelial Nitric Oxide Synthase: Transition from Reversible to Irreversible Enzyme Inhibition. Biochemistry 2010, 49, 3129–3137. [Google Scholar] [CrossRef] [Green Version]
- Joussen, A.M.; Poulaki, V.; Mitsiades, N.; Kirchhof, B.; Koizumi, K.; Döhmen, S.; Adamis, A.P. Nonsteroidal anti-inflammatory drugs prevent early diabetic retinopathy via TNF-α suppression. FASEB J. 2002, 16, 438–440. [Google Scholar] [CrossRef]
- Adamis, A.P.; Berman, A.J. Immunological mechanisms in the pathogenesis of diabetic retinopathy. Semin. Immunopathol. 2008, 30, 65–84. [Google Scholar] [CrossRef]
- Yakovlev, V.A. Role of nitric oxide in the radiation-induced bystander effect. Redox Biol. 2015, 6, 396–400. [Google Scholar] [CrossRef] [Green Version]
- Gunnett, C.; Lund, D.; McDowell, A.; Faraci, F.; Heistad, D. Mechanisms of Inducible Nitric Oxide Synthase–Mediated Vascular Dysfunction. Arter. Thromb. Vasc. Biol. 2005, 25, 1617–1622. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.-A.; Wang, T.-Y.; Varadharaj, S.; Reyes, L.A.; Hemann, C.; Hassan Talukder, M.A.; Chen, Y.-R.; Druhan, L.J.; Zweier, J.L. S-glutathionylation uncouples eNOS and regulates its cellular and vascular function. Nature 2010, 468, 1115–1118. [Google Scholar] [CrossRef] [Green Version]
- Yoshida, T.; Goto, S.; Kawakatsu, M.; Urata, Y.; Li, T.-S. Mitochondrial dysfunction, a probable cause of persistent oxidative stress after exposure to ionizing radiation. Free Radic. Res. 2012, 46, 147–153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ricci, C.; Pastukh, V.; Leonard, J.; Turrens, J.; Wilson, G.; Schaffer, D.; Schaffer, S.W. Mitochondrial DNA damage triggers mitochondrial-superoxide generation and apoptosis. Am. J. Physiol. Cell Physiol. 2008, 294, C413–C422. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Azzam, E.I.; Jay-Gerin, J.-P.; Pain, D. Ionizing radiation-induced metabolic oxidative stress and prolonged cell injury. Cancer Lett. 2012, 327, 48–60. [Google Scholar] [CrossRef] [Green Version]
- Havaki, S.; Kotsinas, A.; Chronopoulos, E.; Kletsas, D.; Georgakilas, A.; Gorgoulis, V.G. The role of oxidative DNA damage in radiation induced bystander effect. Cancer Lett. 2015, 356, 43–51. [Google Scholar] [CrossRef]
- Prise, K.M.; O’Sullivan, J.M. Radiation-induced bystander signalling in cancer therapy. Nat. Rev. Cancer 2009, 9, 351–360. [Google Scholar] [CrossRef] [PubMed]
- Mothersill, C.; Seymour, C. Radiation-Induced Bystander Effects: Past History and Future Directions. Radiat. Res. 2001, 155, 759–767. [Google Scholar] [CrossRef] [PubMed]
- Hei, T.K.; Zhou, H.; Ivanov, V.; Hong, M.; Lieberman, H.B.; Brenner, D.J.; Amundson, S.A.; Geard, C.R. Mechanism of radiation-induced bystander effects: A unifying model. J. Pharm. Pharmacol. 2008, 60, 943–950. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, H.; Ivanov, V.N.; Lien, Y.-C.; Davidson, M.; Hei, T.K. Mitochondrial Function and Nuclear Factor-κB–Mediated Signaling in Radiation-Induced Bystander Effects. Cancer Res. 2008, 68, 2233–2240. [Google Scholar] [CrossRef] [Green Version]
- Hong, C.-W.; Kim, Y.-M.; Pyo, H.; Lee, J.-H.; Kim, S.; Lee, S.; Noh, J.M. Involvement of inducible nitric oxide synthase in radiation-induced vascular endothelial damage. J. Radiat. Res. 2013, 54, 1036–1042. [Google Scholar] [CrossRef] [Green Version]
- Koturbash, I.; Zemp, F.J.; Kutanzi, K.; Luzhna, L.; Loree, J.; Kolb, B.; Kovalchuk, O. Sex-Specific microRNAome deregulation in the shielded bystander spleen of cranially exposed mice. Cell Cycle 2008, 7, 1658–1667. [Google Scholar] [CrossRef] [Green Version]
- Xu, S.; Ding, N.; Pei, H.; Hu, W.; Wei, W.; Zhang, X.; Zhou, G.; Wang, J. MiR-21 is involved in radiation-induced bystander effects. RNA Biol. 2014, 11, 1161–1170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haimovich, G.; Ecker, C.M.; Dunagin, M.C.; Eggan, E.; Raj, A.; Gerst, J.E.; Singer, R.H. Intercellular mRNA trafficking via membrane nanotube-like extensions in mammalian cells. Proc. Natl. Acad. Sci. USA 2017, 114, E9873–E9882. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, S.; Wang, J.; Ding, N.; Hu, W.; Zhang, X.; Wang, B.; Hua, J.; Wei, W.; Zhu, Q. Exosome-mediated microRNA transfer plays a role in radiation-induced bystander effect. RNA Biol. 2015, 12, 1355–1363. [Google Scholar] [CrossRef] [Green Version]
- Nawaz, M.; Fatima, F. Extracellular Vesicles, Tunneling Nanotubes, and Cellular Interplay: Synergies and Missing Links. Front. Mol. Biosci. 2017, 4, 50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raposo, G.; Stoorvogel, W. Extracellular vesicles: Exosomes, microvesicles, and friends. J. Cell Biol. 2013, 200, 373–383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shu, Z.; Tan, J.; Miao, Y.; Zhang, Q. The role of microvesicles containing microRNAs in vascular endothelial dysfunction. J. Cell Mol. Med. 2019, 23, 7933–7945. [Google Scholar] [CrossRef]
- Li, M.; You, L.; Xue, J.; Lu, Y. Ionizing Radiation-Induced Cellular Senescence in Normal, Non-transformed Cells and the Involved DNA Damage Response: A Mini Review. Front. Pharmacol. 2018, 9, 522. [Google Scholar] [CrossRef] [PubMed]
- Childs, B.G.; Durik, M.; Baker, D.J.; Van Deursen, J.M. Cellular senescence in aging and age-related disease: From mechanisms to therapy. Nat. Med. 2015, 21, 1424–1435. [Google Scholar] [CrossRef] [Green Version]
- Kumari, R.; Jat, P. Mechanisms of Cellular Senescence: Cell Cycle Arrest and Senescence Associated Secretory Phenotype. Front. Cell Dev. Biol. 2021, 9, 645593. [Google Scholar] [CrossRef]
- Huang, T.T.; Wuerzberger-Davis, S.M.; Wu, Z.-H.; Miyamoto, S. Sequential Modification of NEMO/IKKγ by SUMO-1 and Ubiquitin Mediates NF-κB Activation by Genotoxic Stress. Cell 2003, 115, 565–576. [Google Scholar] [CrossRef] [Green Version]
- Childs, B.G.; Baker, D.J.; Kirkland, J.L.; Campisi, J.; Van Deursen, J.M. Senescence and apoptosis: Dueling or complementary cell fates? EMBO Rep. 2014, 15, 1139–1153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hagelstrom, R.T.; Askin, K.F.; Williams, A.J.; Ramaiah, L.; Desaintes, C.; Goodwin, E.H.; Ullrich, R.L.; Bailey, S.M. DNA-PKcs and ATM influence generation of ionizing radiation-induced bystander signals. Oncogene 2008, 27, 6761–6769. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Campisi, J.; d’Adda di Fagagna, F. Cellular senescence: When bad things happen to good cells. Nat. Rev. Mol. Cell Biol. 2007, 8, 729–740. [Google Scholar] [CrossRef]
- Nelson, G.; Wordsworth, J.; Wang, C.; Jurk, D.; Lawless, C.; Martin-Ruiz, C.; von Zglinicki, T. A senescent cell bystander effect: Senescence-induced senescence. Aging Cell 2012, 11, 345–349. [Google Scholar] [CrossRef] [Green Version]
- Bertelli, P.M.; Pedrini, E.; Hughes, D.; McDonnell, S.; Pathak, V.; Peixoto, E.; Guduric-Fuchs, J.; Stitt, A.W.; Medina, R.J. Long term high glucose exposure induces premature senescence in retinal endothelial cells. Front. Physiol. 2022, 13, 929118. [Google Scholar] [CrossRef]
- Maeda, M.; Hayashi, T.; Mizuno, N.; Hattori, Y.; Kuzuya, M. Intermittent High Glucose Implements Stress-Induced Senescence in Human Vascular Endothelial Cells: Role of Superoxide Production by NADPH Oxidase. PLoS ONE 2015, 10, e0123169. [Google Scholar] [CrossRef] [PubMed]
- Guerrero, F.; Carmona, A.; Obrero, T.; Jiménez, M.J.; Soriano, S.; Moreno, J.A.; Martín-Malo, A.; Aljama, P. Role of endothelial microvesicles released by p-cresol on endothelial dysfunction. Sci. Rep. 2020, 10, 10657. [Google Scholar] [CrossRef]
- Terlecki-Zaniewicz, L.; Lämmermann, I.; Latreille, J.; Bobbili, M.R.; Pils, V.; Schosserer, M.; Weinmüllner, R.; Dellago, H.; Skalicky, S.; Pum, D.; et al. Small extracellular vesicles and their miRNA cargo are anti-apoptotic members of the senescence-associated secretory phenotype. Aging 2018, 10, 1103–1132. [Google Scholar] [CrossRef]
- Colpani, O.; Spinetti, G. MicroRNAs orchestrating senescence of endothelial and vascular smooth muscle cells. Vasc. Biol. 2019, 1, H75–H81. [Google Scholar] [CrossRef] [Green Version]
- Kale, A.; Sharma, A.; Stolzing, A.; Desprez, P.-Y.; Campisi, J. Role of immune cells in the removal of deleterious senescent cells. Immun. Ageing 2020, 17, 16. [Google Scholar] [CrossRef]
- Acosta, J.C.; Banito, A.; Wuestefeld, T.; Georgilis, A.; Janich, P.; Morton, J.P.; Athineos, D.; Kang, T.-W.; Lasitschka, F.; Andrulis, M.; et al. A complex secretory program orchestrated by the inflammasome controls paracrine senescence. Nature 2013, 15, 978–990. [Google Scholar] [CrossRef] [PubMed]
- Gasek, N.S.; Kuchel, G.A.; Kirkland, J.L.; Xu, M. Strategies for targeting senescent cells in human disease. Nat. Aging 2021, 1, 870–879. [Google Scholar] [CrossRef]
- Takeda, A.; Shigematsu, N.; Suzuki, S.; Fujii, M.; Kawata, T.; Kawaguchi, O.; Uno, T.; Takano, H.; Kubo, A.; Ito, H. Late retinal complications of radiation therapy for nasal and paranasal malignancies: Relationship between irradiated-dose area and severity. Int. J. Radiat. Oncol. 1999, 44, 599–605. [Google Scholar] [CrossRef] [PubMed]
- Baynes, J.W. Role of Oxidative Stress in Development of Complications in Diabetes. Diabetes 1991, 40, 405–412. [Google Scholar] [CrossRef] [PubMed]
- Nishikawa, T.; Edelstein, D.; Du, X.L.; Yamagishi, S.-I.; Matsumura, T.; Kaneda, Y.; Yorek, M.A.; Beebe, D.J.; Oates, P.J.; Hammes, H.-P.; et al. Normalizing mitochondrial superoxide production blocks three pathways of hyperglycaemic damage. Nature 2000, 404, 787–790. [Google Scholar] [CrossRef]
- Brownlee, M. The Pathobiology of Diabetic Complications: A Unifying Mechanism. Diabetes 2005, 54, 1615–1625. [Google Scholar] [CrossRef] [Green Version]
- Kowluru, R.A.; Abbas, S.N. Diabetes-Induced Mitochondrial Dysfunction in the Retina. Investig. Opthalmology Vis. Sci. 2003, 44, 5327–5334. [Google Scholar] [CrossRef] [Green Version]
- Kowluru, R.A. Diabetic Retinopathy: Mitochondrial Dysfunction and Retinal Capillary Cell Death. Antioxid. Redox Signal. 2005, 7, 1581–1587. [Google Scholar] [CrossRef]
- Kowluru, R.A.; Kowluru, V.; Xiong, Y.; Ho, Y.-S. Overexpression of mitochondrial superoxide dismutase in mice protects the retina from diabetes-induced oxidative stress. Free Radic. Biol. Med. 2006, 41, 1191–1196. [Google Scholar] [CrossRef]
- Giacco, F.; Brownlee, M. Oxidative Stress and Diabetic Complications. Circ. Res. 2010, 107, 1058–1070. [Google Scholar] [CrossRef] [Green Version]
- Handy, D.; Loscalzo, J. Redox Regulation of Mitochondrial Function. Antioxid. Redox Signal. 2012, 16, 1323–1367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mailloux, R.J. Mitochondrial Antioxidants and the Maintenance of Cellular Hydrogen Peroxide Levels. Oxidative Med. Cell Longev. 2018, 2018, 7857251. [Google Scholar] [CrossRef] [PubMed]
- Ježek, P.; Hlavatá, L. Mitochondria in homeostasis of reactive oxygen species in cell, tissues, and organism. Int. J. Biochem. Cell Biol. 2005, 37, 2478–2503. [Google Scholar] [CrossRef] [PubMed]
- Starkov, A.A.; Andreyev, A.Y.; Zhang, S.F.; Starkova, N.N.; Korneeva, M.; Syromyatnikov, M.; Popov, V.N. Scavenging of H2O2 by mouse brain mitochondria. J. Bioenerg. Biomembr. 2014, 46, 471–477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gaucher, C.; Boudier, A.; Bonetti, J.; Clarot, I.; Leroy, P.; Parent, M. Glutathione: Antioxidant Properties Dedicated to Nanotechnologies. Antioxidants 2018, 7, 62. [Google Scholar] [CrossRef] [Green Version]
- Griffith, O.W. Biologic and pharmacologic regulation of mammalian glutathione synthesis. Free Radic. Biol. Med. 1999, 27, 922–935. [Google Scholar] [CrossRef]
- Handy, D.E.; Loscalzo, J. The role of glutathione peroxidase-1 in health and disease. Free Radic. Biol. Med. 2022, 188, 146–161. [Google Scholar] [CrossRef]
- Flohé, L.; Toppo, S.; Orian, L. The glutathione peroxidase family: Discoveries and mechanism. Free Radic. Biol. Med. 2022, 187, 113–122. [Google Scholar] [CrossRef]
- Mikkelsen, R.B.; Wardman, P. Biological chemistry of reactive oxygen and nitrogen and radiation-induced signal transduction mechanisms. Oncogene 2003, 22, 5734–5754. [Google Scholar] [CrossRef] [Green Version]
- Agardh, C.-D.; Agardh, E.; Qian, Y.; Hultberg, B. Glutathione levels are reduced in diabetic rat retina but are not influenced by ischemia followed by recirculation. Metabolism 1998, 47, 269–272. [Google Scholar] [CrossRef]
- Sekhar, R.V.; McKay, S.V.; Patel, S.G.; Guthikonda, A.P.; Reddy, V.T.; Balasubramanyam, A.; Jahoor, F. Glutathione Synthesis Is Diminished in Patients with Uncontrolled Diabetes and Restored by Dietary Supplementation With Cysteine and Glycine. Diabetes Care 2011, 34, 162–167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lutchmansingh, F.K.; Hsu, J.W.; Bennett, F.I.; Badaloo, A.; McFarlane-Anderson, N.; Gordon-Strachan, G.M.; Wright-Pascoe, R.A.; Jahoor, F.; Boyne, M.S. Glutathione metabolism in type 2 diabetes and its relationship with microvascular complications and glycemia. PLoS ONE 2018, 13, e0198626. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Padmaja, S.; Squadrito, G.L.; Pryor, W.A. Inactivation of Glutathione Peroxidase by Peroxynitrite. Arch. Biochem. Biophys. 1998, 349, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Kowluru, R. Diabetic Retinopathy and NADPH Oxidase-2: A Sweet Slippery Road. Antioxidants 2021, 10, 783. [Google Scholar] [CrossRef]
- Al-Shabrawey, M.; Rojas, M.; Sanders, T.; Behzadian, A.; El-Remessy, A.; Bartoli, M.; Parpia, A.K.; Liou, G.; Caldwell, R. Role of NADPH Oxidase in Retinal Vascular Inflammation. Investig. Opthalmology Vis. Sci. 2008, 49, 3239–3244. [Google Scholar] [CrossRef]
- Al-Shabrawey, M.; Bartoli, M.; El-Remessy, A.B.; Ma, G.; Matragoon, S.; Lemtalsi, T.; Caldwell, R.W. Role of NADPH Oxidase and Stat3 in Statin-Mediated Protection against Diabetic Retinopathy. Investig. Opthalmology Vis. Sci. 2008, 49, 3231–3238. [Google Scholar] [CrossRef] [Green Version]
- Fortuño, A.; José, G.S.; Moreno, M.U.; Díez, J.; Zalba, G. Oxidative stress and vascular remodelling. Exp. Physiol. 2005, 90, 457–462. [Google Scholar] [CrossRef]
- Inoguchi, T.; Sonta, T.; Tsubouchi, H.; Etoh, T.; Kakimoto, M.; Sonoda, N.; Sato, N.; Sekiguchi, N.; Kobayashi, K.; Sumimoto, H.; et al. Protein Kinase C–Dependent Increase in Reactive Oxygen Species (ROS) Production in Vascular Tissues of Diabetes. J. Am. Soc. Nephrol. 2003, 14, S227–S232. [Google Scholar] [CrossRef] [Green Version]
- Drummond, G.R.; Sobey, C.G. Endothelial NADPH oxidases: Which NOX to target in vascular disease? Trends Endocrinol. Metab. 2014, 25, 452–463. [Google Scholar] [CrossRef]
- Bubb, K.J.; Birgisdottir, A.B.; Tang, O.; Hansen, T.; Figtree, G.A. Redox modification of caveolar proteins in the cardiovascular system—Role in cellular signalling and disease. Free Radic. Biol. Med. 2017, 109, 61–74. [Google Scholar] [CrossRef]
- Caldwell, R.B.; Zhang, W.; Romero, M.J. Vascular dysfunction in retinopathy—An emerging role for arginase. Brain Res. Bull. 2010, 81, 303–309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kowluru, R.A.; Kowluru, A.; Veluthakal, R.; Mohammad, G.; Syed, I.; Santos, J.M.; Mishra, M. Tiam1-Rac1 signalling axis-mediated activation of NADPH oxidase-2 initiates mitochondrial damage in the development of diabetic retinopathy. Diabetologia 2014, 57, 1047–1056. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Liu, X.; Yanoff, M.; Cohen, S.; Ye, X. Cultured retinal capillary pericytes die by apoptosis after an abrupt fluctuation from high to low glucose levels: A comparative study with retinal capillary endothelial cells. Diabetologia 1996, 39, 537–547. [Google Scholar] [CrossRef] [PubMed]
- Zheng, L.; Du, Y.; Miller, C.; Gubitosi-Klug, R.A.; Kern, T.S.; Ball, S.; Berkowitz, B.A. Critical role of inducible nitric oxide synthase in degeneration of retinal capillaries in mice with streptozotocin-induced diabetes. Diabetologia 2007, 50, 1987–1996. [Google Scholar] [CrossRef] [Green Version]
- Antonetti, D.A.; Silva, P.S.; Stitt, A.W. Current understanding of the molecular and cellular pathology of diabetic retinopathy. Nat. Rev. Endocrinol. 2021, 17, 195–206. [Google Scholar] [CrossRef]
- Simó, R.; Stitt, A.W.; Gardner, T.W. Neurodegeneration in diabetic retinopathy: Does it really matter? Diabetologia 2018, 61, 1902–1912. [Google Scholar] [CrossRef] [Green Version]
- Vielma, A.H.; Retamal, M.A.; Schmachtenberg, O. Nitric oxide signaling in the retina: What have we learned in two decades? Brain Res. 2012, 1430, 112–125. [Google Scholar] [CrossRef]
- Eggers, E.D.; Carreon, T.A. The effects of early diabetes on inner retinal neurons. Vis. Neurosci. 2020, 37, E006. [Google Scholar] [CrossRef]
- Roufail, E.; Soulis, T.; Boel, E.; Cooper, M.E.; Rees, S. Depletion of nitric oxide synthase-containing neurons in the diabetic retina: Reversal by aminoguanidine. Diabetologia 1998, 41, 1419–1425. [Google Scholar] [CrossRef] [Green Version]
- Corbett, J.A.; McDaniel, M.L. Selective inhibition of inducible nitric oxide synthase by aminoguanidine. Methods Enzymol. 1996, 268, 398–408. [Google Scholar] [CrossRef]
- Nejad, P.; Farhad, A.; Razavi, S. The use of aminoguanidine, a selective inducible nitric oxide synthase inhibitor, to evaluate the role of nitric oxide on periapical healing. Dent. Res. J. 2011, 8, 197–202. [Google Scholar] [CrossRef]
- Amoaku, W.M.K.; Frew, L.; Mahon, G.J.; Gardiner, T.A.; Archer, D.B. Early ultrastructural changes after low-dose X-irradiation in the retina of the rat. Eye 1989, 3, 638–646. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amoakul, W.M.K.; Mahon, G.J.; Gardiner, T.A.; Frew, L.; Archer, D.B. Late ultrastructural changes in the retina of the rat following low-dose X-irradiation. Graefe’s Arch. Clin. Exp. Ophthalmol. 1992, 230, 569–574. [Google Scholar] [CrossRef] [PubMed]
- McLeod, D.; Marshall, J.; Kohner, E.M.; Bird, A.C. The role of axoplasmic transport in the pathogenesis of retinal cotton-wool spots. Br. J. Ophthalmol. 1977, 61, 177–191. [Google Scholar] [CrossRef] [Green Version]
- Carter-Dawson, L.D.; Lavail, M.M. Rods and cones in the mouse retina. I. Structural analysis using light and electron microscopy. J. Comp. Neurol. 1979, 188, 245–262. [Google Scholar] [CrossRef] [PubMed]
- Solovei, I.; Kreysing, M.; Lanctôt, C.; Kösem, S.; Peichl, L.; Cremer, T.; Guck, J.; Joffe, B. Nuclear Architecture of Rod Photoreceptor Cells Adapts to Vision in Mammalian Evolution. Cell 2009, 137, 356–368. [Google Scholar] [CrossRef] [Green Version]
- Wheeler, K.T.; Wierowski, J.V. DNA Accessibility: A Determinant of Mammalian Cell Differentiation? Radiat. Res. 1983, 93, 312–318. [Google Scholar] [CrossRef] [PubMed]
- Caldecott, K.W. Single-strand break repair and genetic disease. Nat. Rev. Genet. 2008, 9, 619–631. [Google Scholar] [CrossRef] [PubMed]
- Lieber, M.R. The Mechanism of Double-Strand DNA Break Repair by the Nonhomologous DNA End-Joining Pathway. Annu. Rev. Biochem. 2010, 79, 181–211. [Google Scholar] [CrossRef] [Green Version]
- Iyama, T.; Wilson, D.M., III. DNA repair mechanisms in dividing and non-dividing cells. DNA Repair 2013, 12, 620–636. [Google Scholar] [CrossRef] [Green Version]
- Frock, R.L.; Sadeghi, C.; Meng, J.; Wang, J.L. DNA End Joining: G0-ing to the Core. Biomolecules 2021, 11, 1487. [Google Scholar] [CrossRef]
- Ciccia, A.; Elledge, S.J. The DNA Damage Response: Making It Safe to Play with Knives. Mol. Cell 2010, 40, 179–204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harper, J.W.; Elledge, S.J. The DNA Damage Response: Ten Years after. Mol. Cell 2007, 28, 739–745. [Google Scholar] [CrossRef] [PubMed]
- Lam, F.C. The DNA damage response—From cell biology to human disease. J. Transl. Genet. Genom. 2022, 6, 204–222. [Google Scholar] [CrossRef]
- Taylor, A.M.R.; Harnden, D.G.; Arlett, C.F.; Harcourt, S.A.; Lehmann, A.R.; Stevens, S.; Bridges, B.A. Ataxia telangiectasia: A human mutation with abnormal radiation sensitivity. Nature 1975, 258, 427–429. [Google Scholar] [CrossRef] [PubMed]
- Rothblum-Oviatt, C.; Wright, J.; Lefton-Greif, M.A.; McGrath-Morrow, S.A.; Crawford, T.O.; Lederman, H.M. Ataxia telangiectasia: A review. Orphanet J. Rare Dis. 2016, 11, 159. [Google Scholar] [CrossRef] [Green Version]
- McKinnon, P.J. ATM and ataxia telangiectasia. EMBO Rep. 2004, 5, 772–776. [Google Scholar] [CrossRef] [Green Version]
- Riballo, E.; Kühne, M.; Rief, N.; Doherty, A.; Smith, G.C.; Recio, M.-J.; Reis, C.; Dahm, K.; Fricke, A.; Krempler, A.; et al. A Pathway of Double-Strand Break Rejoining Dependent upon ATM, Artemis, and Proteins Locating to γ-H2AX Foci. Mol. Cell 2004, 16, 715–724. [Google Scholar] [CrossRef]
- Savitsky, K.; Bar-Shira, A.; Gilad, S.; Rotman, G.; Ziv, Y.; Vanagaite, L.; Tagle, D.A.; Smith, S.; Uziel, T.; Sfez, S.; et al. A Single Ataxia Telangiectasia Gene with a Product Similar to PI-3 Kinase. Science 1995, 268, 1749–1753. [Google Scholar] [CrossRef]
- Shiloh, Y. ATM and related protein kinases: Safeguarding genome integrity. Nat. Rev. Cancer 2003, 3, 155–168. [Google Scholar] [CrossRef]
- Helt, C.E.; Cliby, W.A.; Keng, P.C.; Bambara, R.A.; O’Reilly, M.A. Ataxia Telangiectasia Mutated (ATM) and ATM and Rad3-related Protein Exhibit Selective Target Specificities in Response to Different Forms of DNA Damage. J. Biol. Chem. 2005, 280, 1186–1192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bakkenist, C.J.; Kastan, M.B. DNA damage activates ATM through intermolecular autophosphorylation and dimer dissociation. Nature 2003, 421, 499–506. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.-H.; Paull, T.T. ATM Activation by DNA Double-Strand Breaks Through the Mre11-Rad50-Nbs1 Complex. Science 2005, 308, 551–554. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.-H.; Paull, T.T. Activation and regulation of ATM kinase activity in response to DNA double-strand breaks. Oncogene 2007, 26, 7741–7748. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stinson, B.M.; Loparo, J.J. Repair of DNA Double-Strand Breaks by the Nonhomologous End Joining Pathway. Annu. Rev. Biochem. 2021, 90, 137–164. [Google Scholar] [CrossRef] [PubMed]
- Sonoda, E.; Hochegger, H.; Saberi, A.; Taniguchi, Y.; Takeda, S. Differential usage of non-homologous end-joining and homologous recombination in double strand break repair. DNA Repair 2006, 5, 1021–1029. [Google Scholar] [CrossRef]
- Noon, A.T.; Shibata, A.; Rief, N.; Löbrich, M.; Stewart, G.; Jeggo, P.A.; Goodarzi, A.A. 53BP1-dependent robust localized KAP-1 phosphorylation is essential for heterochromatic DNA double-strand break repair. Nature 2010, 12, 177–184. [Google Scholar] [CrossRef]
- Goodarzi, A.A.; Noon, A.T.; Deckbar, D.; Ziv, Y.; Shiloh, Y.; Löbrich, M.; Jeggo, P.A. ATM Signaling Facilitates Repair of DNA Double-Strand Breaks Associated with Heterochromatin. Mol. Cell 2008, 31, 167–177. [Google Scholar] [CrossRef]
- Goodarzi, A.A.; Jeggo, P.; Lobrich, M. The influence of heterochromatin on DNA double strand break repair: Getting the strong, silent type to relax. DNA Repair 2010, 9, 1273–1282. [Google Scholar] [CrossRef]
- Goodarzi, A.A.; Jeggo, P.A. The Heterochromatic Barrier to DNA Double Strand Break Repair: How to Get the Entry Visa. Int. J. Mol. Sci. 2012, 13, 11844–11860. [Google Scholar] [CrossRef] [Green Version]
- Falk, M.; Lukasova, E.; Kozubek, S. Higher-order chromatin structure in DSB induction, repair and misrepair. Mutat. Res. Mol. Mech. Mutagen. 2010, 704, 88–100. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.-A.; Kruhlak, M.; Dotiwala, F.; Nussenzweig, A.; Haber, J.E. Heterochromatin is refractory to γ-H2AX modification in yeast and mammals. J. Cell Biol. 2007, 178, 209–218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cowell, I.G.; Sunter, N.J.; Singh, P.; Austin, C.; Durkacz, B.W.; Tilby, M.J. γH2AX Foci Form Preferentially in Euchromatin after Ionising-Radiation. PLoS ONE 2007, 2, e1057. [Google Scholar] [CrossRef] [PubMed]
- Costes, S.V.; Ponomarev, A.; Chen, J.L.; Nguyen, D.; Cucinotta, F.A.; Barcellos-Hoff, M.H. Image-Based Modeling Reveals Dynamic Redistribution of DNA Damage into Nuclear Sub-Domains. PLoS Comput. Biol. 2007, 3, e155. [Google Scholar] [CrossRef] [Green Version]
- Lorat, Y.; Schanz, S.; Schüler, N.; Wennemuth, G.; Rübe, C.; Rübe, C.E. Beyond Repair Foci: DNA Double-Strand Break Repair in Euchromatic and Heterochromatic Compartments Analyzed by Transmission Electron Microscopy. PLoS ONE 2012, 7, e38165. [Google Scholar] [CrossRef]
- Rogakou, E.P.; Pilch, D.R.; Orr, A.H.; Ivanova, V.S.; Bonner, W.M. DNA Double-stranded Breaks Induce Histone H2AX Phosphorylation on Serine. J. Biol. Chem. 1998, 273, 5858–5868. [Google Scholar] [CrossRef] [Green Version]
- Chiolo, I.; Tang, J.; Georgescu, W.; Costes, S.V. Nuclear dynamics of radiation-induced foci in euchromatin and heterochromatin. Mutat. Res. Mol. Mech. Mutagen. 2013, 750, 56–66. [Google Scholar] [CrossRef] [Green Version]
- Rogakou, E.P.; Boon, C.; Redon, C.; Bonner, W.M. Megabase Chromatin Domains Involved in DNA Double-Strand Breaks In Vivo. J. Cell Biol. 1999, 146, 905–916. [Google Scholar] [CrossRef] [Green Version]
- Stiff, T.; O’Driscoll, M.; Rief, N.; Iwabuchi, K.; Löbrich, M.; Jeggo, P.A. ATM and DNA-PK Function Redundantly to Phosphorylate H2AX after Exposure to Ionizing Radiation. Cancer Res. 2004, 64, 2390–2396. [Google Scholar] [CrossRef] [Green Version]
- Misteli, T.; Soutoglou, E. The emerging role of nuclear architecture in DNA repair and genome maintenance. Nat. Rev. Mol. Cell Biol. 2009, 10, 243–254. [Google Scholar] [CrossRef]
- Trojer, P.; Reinberg, D. Facultative Heterochromatin: Is There a Distinctive Molecular Signature? Mol. Cell 2007, 28, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Beisel, C.; Paro, R. Silencing chromatin: Comparing modes and mechanisms. Nat. Rev. Genet. 2011, 12, 123–135. [Google Scholar] [CrossRef] [PubMed]
- Colmenares, S.U.; Karpen, G.H. Heterochromatin: Guardian of the Genome. Annu. Rev. Cell Dev. Biol. 2018, 34, 265–288. [Google Scholar] [CrossRef] [Green Version]
- Grigoryev, S.A.; Bulynko, Y.A.; Popova, E.Y. The end adjusts the means: Heterochromatin remodelling during terminal cell differentiation. Chromosom. Res. 2006, 14, 53–69. [Google Scholar] [CrossRef]
- van Steensel, B.; Belmont, A.S. Lamina-Associated Domains: Links with Chromosome Architecture, Heterochromatin, and Gene Repression. Cell 2017, 169, 780–791. [Google Scholar] [CrossRef] [Green Version]
- Gitlin, J.D.; D’Amore, P.A. Culture of retinal capillary cells using selective growth media. Microvasc. Res. 1983, 26, 74–80. [Google Scholar] [CrossRef]
- Cervelli, T.; Panetta, D.; Navarra, T.; Andreassi, M.G.; Basta, G.; Galli, A.; Salvadori, P.A.; Picano, E.; Del Turco, S. Effects of single and fractionated low-dose irradiation on vascular endothelial cells. Atherosclerosis 2014, 235, 510–518. [Google Scholar] [CrossRef]
- Guelen, L.; Pagie, L.; Brasset, E.; Meuleman, W.; Faza, M.B.; Talhout, W.; Eussen, B.H.; de Klein, A.; Wessels, L.; de Laat, W.; et al. Domain organization of human chromosomes revealed by mapping of nuclear lamina interactions. Nature 2008, 453, 948–951. [Google Scholar] [CrossRef]
- Nazer, E. To be or not be (in the LAD): Emerging roles of lamin proteins in transcriptional regulation. Biochem. Soc. Trans. 2022, 50, 1035–1044. [Google Scholar] [CrossRef]
- Peric-Hupkes, D.; Meuleman, W.; Pagie, L.; Bruggeman, S.W.; Solovei, I.; Brugman, W.; Gräf, S.; Flicek, P.; Kerkhoven, R.M.; van Lohuizen, M.; et al. Molecular Maps of the Reorganization of Genome-Nuclear Lamina Interactions during Differentiation. Mol. Cell 2010, 38, 603–613. [Google Scholar] [CrossRef]
- Meuleman, W.; Peric-Hupkes, D.; Kind, J.; Beaudry, J.-B.; Pagie, L.; Kellis, M.; Reinders, M.; Wessels, L.; van Steensel, B. Constitutive nuclear lamina–genome interactions are highly conserved and associated with A/T-rich sequence. Genome Res. 2013, 23, 270–280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poleshko, A.; Shah, P.P.; Gupta, M.; Babu, A.; Morley, M.P.; Manderfield, L.J.; Ifkovits, J.L.; Calderon, D.; Aghajanian, H.; Sierra-Pagán, J.E.; et al. Genome-Nuclear Lamina Interactions Regulate Cardiac Stem Cell Lineage Restriction. Cell 2017, 171, 573–587.e14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, N.; Gardiner, T.; Archer, D. A Morphologic and Autoradiographic Study of Cell Death and Regeneration in the Retinal Microvasculature of Normal and Diabetic Rats. Am. J. Ophthalmol. 1985, 100, 51–60. [Google Scholar] [CrossRef] [PubMed]
- Stitt, A.W.; McGoldrick, C.; Rice-McCaldin, A.; McCance, D.R.; Glenn, J.V.; Hsu, D.K.; Liu, F.-T.; Thorpe, S.R.; Gardiner, T.A. Impaired Retinal Angiogenesis in Diabetes: Role of Advanced Glycation End Products and Galectin-3. Diabetes 2005, 54, 785–794. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Falk, M.; Lukášová, E.; Kozubek, S. Chromatin structure influences the sensitivity of DNA to γ-radiation. Biochim. Biophys. Acta BBA Mol. Cell Res. 2008, 1783, 2398–2414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, Z.; Deshpande, R.; Paull, T.T. ATM activation in the presence of oxidative stress. Cell Cycle 2010, 9, 4805–4811. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herrup, K.; Li, J.; Chen, J. The role of ATM and DNA damage in neurons: Upstream and downstream connections. DNA Repair 2013, 12, 600–604. [Google Scholar] [CrossRef] [Green Version]
- Guo, Z.; Kozlov, S.; Lavin, M.F.; Person, M.D.; Paull, T.T. ATM Activation by Oxidative Stress. Science 2010, 330, 517–521. [Google Scholar] [CrossRef] [Green Version]
- Cosentino, C.; Grieco, D.; Costanzo, V. ATM activates the pentose phosphate pathway promoting anti-oxidant defence and DNA repair. EMBO J. 2011, 30, 546–555. [Google Scholar] [CrossRef] [Green Version]
- Archer, D.B.; Gardiner, T.A.; Stitt, A.W. Functional Anatomy, Fine Structure and Basic Pathology of the Retinal Vasculature. In Retinal Vascular Disease; Joussen, A.M., Gardner, T.W., Kirchhof, B., Ryan, S.J., Eds.; Springer: Berlin/Heidelberg, Germany, 2007; pp. 3–23. [Google Scholar]
- Barzilai, A.; Yamamoto, K.-I. DNA damage responses to oxidative stress. DNA Repair 2004, 3, 1109–1115. [Google Scholar] [CrossRef]
- Gilad, S.; Chessa, L.; Khosravi, R.; Russell, P.; Galanty, Y.; Piane, M.; Gatti, R.A.; Jorgensen, T.J.; Shiloh, Y.; Bar-Shira, A. Genotype-Phenotype Relationships in Ataxia-Telangiectasia and Variants. Am. J. Hum. Genet. 1998, 62, 551–561. [Google Scholar] [CrossRef] [Green Version]
- Gatti, R.A.; Becker-Catania, S.; Chun, H.H.; Sun, X.; Mitui, M.; Lai, C.-H.; Khanlou, N.; Babaei, M.; Cheng, R.; Clark, C.; et al. The Pathogenesis of Ataxia-Telangiectasia. Learning from a Rosetta Stone. Clin. Rev. Allergy Immunol. 2001, 20, 87–108. [Google Scholar] [CrossRef] [PubMed]
- Mauget-Fay¨sse, M.; Vuillaume, M.; Quaranta, M.; Moullan, N.; Ange’le, S.; Friesen, M.D.; Hall, J. Idiopathic and Radiation-Induced Ocular Telangiectasia: The Involvement of the ATM Gene. Investig. Opthalmology Vis. Sci. 2003, 44, 3257–3262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barbazetto, I.A.; Room, M.; Yannuzzi, N.A.; Barile, G.R.; Merriam, J.E.; Bardal, A.M.C.; Freund, K.B.; Yannuzzi, L.A.; Allikmets, R. ATMGene Variants in Patients with Idiopathic Perifoveal Telangiectasia. Investig. Opthalmology Vis. Sci. 2008, 49, 3806–3811. [Google Scholar] [CrossRef] [PubMed]
- Donath, H.; Hess, U.; Kieslich, M.; Theis, M.; Ohlenschläger, U.; Schubert, R.; Woelke, S.; Zielen, S. Diabetes in Patients with Ataxia Telangiectasia: A National Cohort Study. Front. Pediatr. 2020, 8, 317. [Google Scholar] [CrossRef] [PubMed]
- Nissenkorn, A.; Levy-Shraga, Y.; Banet-Levi, Y.; Lahad, A.; Sarouk, I.; Modan-Moses, D. Endocrine abnormalities in ataxia telangiectasia: Findings from a national cohort. Pediatr. Res. 2016, 79, 889–894. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raz-Prag, D.; Galron, R.; Segev-Amzaleg, N.; Solomon, A.S.; Shiloh, Y.; Barzilai, A.; Frenkel, D. A Role for Vascular Deficiency in Retinal Pathology in a Mouse Model of Ataxia-Telangiectasia. Am. J. Pathol. 2011, 179, 1533–1541. [Google Scholar] [CrossRef]
- Bhatwadekar, A.D.; Duan, Y.; Chakravarthy, H.; Korah, M.; Caballero, S.; Busik, J.V.; Grant, M.B. Ataxia Telangiectasia Mutated Dysregulation Results in Diabetic Retinopathy. Stem Cells 2016, 34, 405–417. [Google Scholar] [CrossRef] [Green Version]
- Ito, K.; Hirao, A.; Arai, F.; Matsuoka, S.; Takubo, K.; Hamaguchi, I.; Nomiyama, K.; Hosokawa, K.; Sakurada, K.; Nakagata, N.; et al. Regulation of oxidative stress by ATM is required for self-renewal of haematopoietic stem cells. Nature 2004, 431, 997–1002. [Google Scholar] [CrossRef]
- Fuks, Z.; Kolesnick, R. Engaging the vascular component of the tumor response. Cancer Cell 2005, 8, 89–91. [Google Scholar] [CrossRef] [Green Version]
- Lorenzi, M.; Montisano, D.F.; Toledo, S.; Barrieux, A. High glucose induces DNA damage in cultured human endothelial cells. J. Clin. Investig. 1986, 77, 322–325. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dandona, P.; Thusu, K.; Cook, S.; Snyder, B.; Makowski, J.; Armstrong, D.; Nicotera, T. Oxidative damage to DNA in diabetes mellitus. Lancet 1996, 347, 444–445. [Google Scholar] [CrossRef] [PubMed]
- Leinonen, J.; Lehtimäki, T.; Toyokuni, S.; Okada, K.; Tanaka, T.; Hiai, H.; Ochi, H.; Laippala, P.; Rantalaiho, V.; Wirta, O.; et al. New biomarker evidence of oxidative DNA damage in patients with non-insulin-dependent diabetes mellitus. FEBS Lett. 1997, 417, 150–152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hinokio, Y.; Suzuki, S.; Hirai, M.; Chiba, M.; Hirai, A.; Toyota, T. Oxidative DNA damage in diabetes mellitus: Its association with diabetic complications. Diabetologia 1999, 42, 995–998. [Google Scholar] [CrossRef] [Green Version]
- Blasiak, J.; Arabski, M.; Krupa, R.; Woźniak, K.; Zadrozny, M.; Kasznicki, J.; Zurawska, M.; Drzewoski, J. DNA damage and repair in type 2 diabetes mellitus. Mutat. Res. Mol. Mech. Mutagen. 2004, 554, 297–304. [Google Scholar] [CrossRef]
- Giovannini, C.; Piaggi, S.; Federico, G.; Scarpato, R. High levels of γ-H2AX foci and cell membrane oxidation in adolescents with type 1 diabetes. Mutat. Res. Mol. Mech. Mutagen. 2014, 770, 128–135. [Google Scholar] [CrossRef]
- Nishikawa, T.; Sasahara, T.; Kiritoshi, S.; Sonoda, K.; Senokuchi, T.; Matsuo, T.; Kukidome, D.; Wake, N.; Matsumura, T.; Miyamura, N.; et al. Evaluation of Urinary 8-Hydroxydeoxy-Guanosine as a Novel Biomarker of Macrovascular Complications in Type 2 Diabetes. Diabetes Care 2003, 26, 1507–1512. [Google Scholar] [CrossRef] [Green Version]
- Simone, S.; Gorin, Y.; Velagapudi, C.; Abboud, H.E.; Habib, S.L. Mechanism of Oxidative DNA Damage in Diabetes: Tuberin Inactivation and Downregulation of DNA Repair Enzyme 8-Oxo-7,8-Dihydro-2’-Deoxyguanosine-DNA Glycosylase. Diabetes 2008, 57, 2626–2636. [Google Scholar] [CrossRef] [Green Version]
- Ciminera, A.K.; Shuck, S.C.; Termini, J. Elevated glucose increases genomic instability by inhibiting nucleotide excision repair. Life Sci. Alliance 2021, 4, e202101159. [Google Scholar] [CrossRef]
- Zhong, A.; Chang, M.; Yu, T.; Gau, R.; Riley, D.J.; Chen, Y.; Chen, P.-L. Aberrant DNA Damage Response and DNA Repair Pathway in High Glucose Conditions. J. Cancer Res. Updates 2018, 7, 64–74. [Google Scholar] [CrossRef] [Green Version]
- Kumar, V.; Fleming, T.; Terjung, S.; Gorzelanny, C.; Gebhardt, C.; Agrawal, R.; Mall, M.A.; Ranzinger, J.; Zeier, M.; Madhusudhan, T.; et al. Homeostatic nuclear RAGE–ATM interaction is essential for efficient DNA repair. Nucleic Acids Res. 2017, 45, 10595–10613. [Google Scholar] [CrossRef] [PubMed]
- Kumar, V.; Agrawal, R.; Pandey, A.; Kopf, S.; Hoeffgen, M.; Kaymak, S.; Bandapalli, O.R.; Gorbunova, V.; Seluanov, A.; Mall, M.A.; et al. Compromised DNA repair is responsible for diabetes-associated fibrosis. EMBO J. 2020, 39, e103477. [Google Scholar] [CrossRef] [PubMed]
- Tsai, K.; Tullis, B.; Breithaupt, K.; Fowers, R.; Jones, N.; Grajeda, S.; Reynolds, P.; Arroyo, J. A Role for RAGE in DNA Double Strand Breaks (DSBs) Detected in Pathological Placentas and Trophoblast Cells. Cells 2021, 10, 857. [Google Scholar] [CrossRef]
- Gorroño, L.E.; López-Díez, R.; Yepuri, G.; Ramirez, L.S.; Reverdatto, S.; Gugger, P.; Shekhtman, A.; Ramasamy, R.; Schmidt, A.M. Receptor for Advanced Glycation End Products (RAGE) and Mechanisms and Therapeutic Opportunities in Diabetes and Cardiovascular Disease: Insights from Human Subjects and Animal Models. Front. Cardiovasc. Med. 2020, 7, 37. [Google Scholar] [CrossRef] [PubMed]
- Glenn, J.V.; Stitt, A.W. The role of advanced glycation end products in retinal ageing and disease. Biochim. Biophys. Acta BBA Gen. Subj. 2009, 1790, 1109–1116. [Google Scholar] [CrossRef] [PubMed]
- McVicar, C.M.; Ward, M.; Colhoun, L.M.; Guduric-Fuchs, J.; Bierhaus, A.; Fleming, T.; Schlotterer, A.; Kolibabka, M.; Hammes, H.-P.; Chen, M.; et al. Role of the receptor for advanced glycation endproducts (RAGE) in retinal vasodegenerative pathology during diabetes in mice. Diabetologia 2015, 58, 1129–1137. [Google Scholar] [CrossRef]
- Qiu, S.; Huang, J. MRN complex is an essential effector of DNA damage repair. J. Zhejiang Univ. B 2021, 22, 31–37. [Google Scholar] [CrossRef]
- Daffu, G.; Del Pozo, C.H.; O’Shea, K.M.; Ananthakrishnan, R.; Ramasamy, R.; Schmidt, A.M. Radical Roles for RAGE in the Pathogenesis of Oxidative Stress in Cardiovascular Diseases and Beyond. Int. J. Mol. Sci. 2013, 14, 19891–19910. [Google Scholar] [CrossRef] [Green Version]
- Bhatt, S.; Gupta, M.K.; Khamaisi, M.; Martinez, R.; Gritsenko, M.A.; Wagner, B.K.; Guye, P.; Busskamp, V.; Shirakawa, J.; Wu, G.; et al. Preserved DNA Damage Checkpoint Pathway Protects against Complications in Long-Standing Type 1 Diabetes. Cell Metab. 2015, 22, 239–252. [Google Scholar] [CrossRef] [Green Version]
- Magenta, A.; Ciarapica, R.; Capogrossi, M.C. The Emerging Role of miR-200 Family in Cardiovascular Diseases. Circ. Res. 2017, 120, 1399–1402. [Google Scholar] [CrossRef]
- Magenta, A.; Cencioni, C.; Fasanaro, P.; Zaccagnini, G.; Greco, S.; Sarra-Ferraris, G.; Antonini, A.; Martelli, F.; Capogrossi, M.C. miR-200c is upregulated by oxidative stress and induces endothelial cell apoptosis and senescence via ZEB1 inhibition. Cell Death Differ. 2011, 18, 1628–1639. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Charron, M.J.; Bonner-Weir, S. Implicating PARP and NAD+ depletion in type I diabetes. Nat. Med. 1999, 5, 269–270. [Google Scholar] [CrossRef] [PubMed]
- Burkart, V.; Wang, Z.-Q.; Radons, J.; Heller, B.; Herceg, Z.; Stingl, L.; Wagner, E.F.; Kolb, H. Mice lacking the poly(ADP-ribose) polymerase gene are resistant to pancreatic beta-cell destruction and diabetes development induced by streptozocin. Nat. Med. 1999, 5, 314–319. [Google Scholar] [CrossRef]
- Chaudhuri, A.R.; Nussenzweig, A. The multifaceted roles of PARP1 in DNA repair and chromatin remodelling. Nat. Rev. Mol. Cell Biol. 2017, 18, 610–621. [Google Scholar] [CrossRef]
- Heeres, J.T.; Hergenrother, P.J. Poly(ADP-ribose) makes a date with death. Curr. Opin. Chem. Biol. 2007, 11, 644–653. [Google Scholar] [CrossRef]
- Zheng, L.; Szabó, C.; Kern, T.S. Poly(ADP-Ribose) Polymerase Is Involved in the Development of Diabetic Retinopathy via Regulation of Nuclear Factor-κB. Diabetes 2004, 53, 2960–2967. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soriano, F.; Virág, L.; Szabó, C. Diabetic endothelial dysfunction: Role of reactive oxygen and nitrogen species production and poly(ADP-ribose) polymerase activation. J. Mol. Med. 2001, 79, 437–448. [Google Scholar] [CrossRef]
- Soriano, F.G.; Virág, L.; Jagtap, P.; Szabó, E.; Mabley, J.G.; Liaudet, L.; Marton, A.; Hoyt, D.G.; Murthy, K.G.K.; Salzman, A.L.; et al. Diabetic endothelial dysfunction: The role of poly(ADP-ribose) polymerase activation. Nat. Med. 2001, 7, 108–113. [Google Scholar] [CrossRef]
- Szabó, C.; Ohshima, H. DNA Damage Induced by Peroxynitrite: Subsequent Biological Effects. Nitric Oxide 1997, 1, 373–385. [Google Scholar] [CrossRef]
- Islam, B.U.; Habib, S.; Ahmad, P.; Allarakha, S.; Moinuddin; Ali, A. Pathophysiological Role of Peroxynitrite Induced DNA Damage in Human Diseases: A Special Focus on Poly(ADP-ribose) Polymerase (PARP). Indian J. Clin. Biochem. 2015, 30, 368–385. [Google Scholar] [CrossRef] [Green Version]
- de la Lastra, J.M.P.; Juan, C.A.; Plou, F.J.; Pérez-Lebeña, E. The Nitration of Proteins, Lipids and DNA by Peroxynitrite Derivatives-Chemistry Involved and Biological Relevance. Stresses 2022, 2, 5. [Google Scholar] [CrossRef]
- Tamir, S.; Burney, S.; Tannenbaum, S.R. DNA Damage by Nitric Oxide. Chem. Res. Toxicol. 1996, 9, 821–827. [Google Scholar] [CrossRef] [PubMed]
- McVicar, C.M.; Hamilton, R.; Colhoun, L.M.; Gardiner, T.A.; Brines, M.; Cerami, A.; Stitt, A.W. Intervention With an Erythropoietin-Derived Peptide Protects Against Neuroglial and Vascular Degeneration During Diabetic Retinopathy. Diabetes 2011, 60, 2995–3005. [Google Scholar] [CrossRef] [Green Version]
- Bailey, D.M.; Lundby, C.; Berg, R.M.G.; Taudorf, S.; Rahmouni, H.; Gutowski, M.; Mulholland, C.W.; Sullivan, J.L.; Swenson, E.R.; McEneny, J.; et al. On the antioxidant properties of erythropoietin and its association with the oxidative-nitrosative stress response to hypoxia in humans. Acta Physiol. 2014, 212, 175–187. [Google Scholar] [CrossRef] [PubMed]
- Katavetin, P.; Tungsanga, K.; Eiam-Ong, S.; Nangaku, M. Antioxidative effects of erythropoietin. Kidney Int. 2007, 72, S10–S15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Browning, A.C.; Alibhai, A.; McIntosh, R.S.; Rotchford, A.P.; Bhan, A.; Amoaku, W.M. Effect of diabetes mellitus and hyperglycemia on the proliferation of human Tenon’s capsule fibroblasts: Implications for wound healing after glaucoma drainage surgery. Wound Repair Regen. 2005, 13, 295–302. [Google Scholar] [CrossRef] [PubMed]
- Waltenberger, J. Impaired collateral vessel development in diabetes: Potential cellular mechanisms and therapeutic implications. Cardiovasc. Res. 2001, 49, 554–560. [Google Scholar] [CrossRef] [Green Version]
- Mizutani, M.; Kern, T.S.; Lorenzi, M. Accelerated death of retinal microvascular cells in human and experimental diabetic retinopathy. J. Clin. Investig. 1996, 97, 2883–2890. [Google Scholar] [CrossRef]
- Loomans, C.J.; de Koning, E.J.; Staal, F.J.; Rookmaaker, M.B.; Verseyden, C.; de Boer, H.C.; Verhaar, M.C.; Braam, B.; Rabelink, T.J.; van Zonneveld, A.-J. Endothelial Progenitor Cell Dysfunction: A Novel Concept in the Pathogenesis of Vascular Complications of Type 1 Diabetes. Diabetes 2004, 53, 195–199. [Google Scholar] [CrossRef] [Green Version]
- Stefánsson, E. Ocular Oxygenation and the Treatment of Diabetic Retinopathy. Surv. Ophthalmol. 2006, 51, 364–380. [Google Scholar] [CrossRef]
- Stefánsson, E.; Hatchell, D.L.; Fisher, B.L.; Sutherland, F.S.; Machemer, R. Panretinal Photocoagulation and Retinal Oxygenation in Normal and Diabetic Cats. Am. J. Ophthalmol. 1986, 101, 657–664. [Google Scholar] [CrossRef] [PubMed]
- Stefansson, E.; Landers, M.B., 3rd; Wolbarsht, M.L. Increased Retinal Oxygen Supply Following Pan-Retinal Photocoagulation and Vitrectomy and Lensectomy. Trans. Am. Ophthalmol. Soc. 1981, 79, 307–334. [Google Scholar] [PubMed]
- Evans, J.R.; Michelessi, M.; Virgili, G. Laser photocoagulation for proliferative diabetic retinopathy. Cochrane Database Syst. Rev. 2014, 11, CD011234. [Google Scholar] [CrossRef]
- Gross, J.G.; Glassman, A.R.; Liu, D.; Sun, J.K.; Antoszyk, A.N.; Baker, C.W.; Bressler, N.M.; Elman, M.J.; Ferris, F.L.; Gardner, T.W.; et al. Five-Year Outcomes of Panretinal Photocoagulation vs. Intravitreous Ranibizumab for Proliferative Diabetic Retinopathy: A Randomized Clinical Trial. JAMA Ophthalmol. 2018, 136, 1138–1148. [Google Scholar] [CrossRef] [Green Version]
- Gage, P.J.; Rhoades, W.; Prucka, S.K.; Hjalt, T. Fate Maps of Neural Crest and Mesoderm in the Mammalian Eye. Investig. Opthalmology Vis. Sci. 2005, 46, 4200–4208. [Google Scholar] [CrossRef]
- Trost, A.; Schroedl, F.; Lange, S.; Rivera, F.J.; Tempfer, H.; Korntner, S.; Stolt, C.C.; Wegner, M.; Bogner, B.; Kaser-Eichberger, A.; et al. Neural Crest Origin of Retinal and Choroidal Pericytes. Investig. Opthalmology Vis. Sci. 2013, 54, 7910–7921. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Melero-Martin, J.M.; De Obaldia, M.E.; Kang, S.-Y.; Khan, Z.A.; Yuan, L.; Oettgen, P.; Bischoff, J. Engineering Robust and Functional Vascular Networks In Vivo With Human Adult and Cord Blood–Derived Progenitor Cells. Circ. Res. 2008, 103, 194–202. [Google Scholar] [CrossRef] [Green Version]
- Velden, D.L.; Houthuijzen, J.M.; Roodhart, J.M.; Werkhoven, E.; Voest, E.E. Detection of endogenously circulating mesenchymal stem cells in human cancer patients. Int. J. Cancer 2018, 143, 2516–2524. [Google Scholar] [CrossRef] [Green Version]
- Trost, A.; Lange, S.; Schroedl, F.; Bruckner, D.; Motloch, K.A.; Bogner, B.; Kaser-Eichberger, A.; Strohmaier, C.; Runge, C.; Aigner, L.; et al. Brain and Retinal Pericytes: Origin, Function and Role. Front. Cell Neurosci. 2016, 10, 20. [Google Scholar] [CrossRef] [Green Version]
- Guimarães-Camboa, N.; Cattaneo, P.; Sun, Y.; Moore-Morris, T.; Gu, Y.; Dalton, N.D.; Rockenstein, E.; Masliah, E.; Peterson, K.L.; Stallcup, W.B.; et al. Pericytes of Multiple Organs Do Not Behave as Mesenchymal Stem Cells In Vivo. Cell Stem Cell 2017, 20, 345–359.e5. [Google Scholar] [CrossRef] [Green Version]
- Volz, K.S.; Jacobs, A.H.; Chen, H.I.; Poduri, A.; McKay, A.S.; Riordan, D.P.; Kofler, N.; Kitajewski, J.; Weissman, I.; Red-Horse, K. Pericytes are progenitors for coronary artery smooth muscle. Elife 2015, 4, e10036. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Li, W.; Lei, F.; Li, X. The regulatory role of micro RNA s in angiogenesis-related diseases. J. Cell Mol. Med. 2018, 22, 4568–4587. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeng, J.; Xiong, Y.; Li, G.; Liu, M.; He, T.; Tang, Y.; Chen, Y.; Cai, L.; Jiang, R.; Tao, J. MiR-21 is Overexpressed in Response to High Glucose and Protects Endothelial Cells from Apoptosis. Exp. Clin. Endocrinol. Diabetes 2013, 121, 425–430. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Malik, A.B.; Rehman, J. Endothelial progenitor cells and vascular repair. Curr. Opin. Hematol. 2014, 21, 224–228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, H.; Wang, B.; Jiang, C.; Li, R.; Zhao, J. Endothelial progenitor cell-derived exosomes facilitate vascular endothelial cell repair through shuttling miR-21-5p to modulate Thrombospondin-1 expression. Clin. Sci. 2019, 133, 1629–1644. [Google Scholar] [CrossRef]
- Medina, R.J.; O’Neill, C.L.; Humphreys, M.W.; Gardiner, T.A.; Stitt, A.W. Outgrowth endothelial cells: Characterization and their potential for reversing ischemic retinopathy. Investig. Ophthalmol. Vis. Sci. 2010, 51, 5906–5913. [Google Scholar] [CrossRef] [Green Version]
- Quiroz, J.; Yazdanyar, A. Animal models of diabetic retinopathy. Ann. Transl. Med. 2021, 9, 1272. [Google Scholar] [CrossRef]
- Olivares, A.M.; Althoff, K.; Chen, G.F.; Wu, S.; Morrisson, M.A.; DeAngelis, M.M.; Haider, N. Animal Models of Diabetic Retinopathy. Curr. Diabetes Rep. 2017, 17, 93. [Google Scholar] [CrossRef] [Green Version]
- Cai, X.; McGinnis, J.F. Diabetic Retinopathy: Animal Models, Therapies, and Perspectives. J. Diabetes Res. 2016, 2016, 3789217. [Google Scholar] [CrossRef] [Green Version]
- Lai, A.K.W.; Lo, A.C.Y. Animal Models of Diabetic Retinopathy: Summary and Comparison. J. Diabetes Res. 2013, 2013, 106594. [Google Scholar] [CrossRef]
- Jiang, X.; Yang, L.; Luo, Y. Animal Models of Diabetic Retinopathy. Curr. Eye Res. 2015, 40, 761–771. [Google Scholar] [CrossRef] [PubMed]
- Hailey, D.W.; Rambold, A.S.; Satpute-Krishnan, P.; Mitra, K.; Sougrat, R.; Kim, P.K.; Lippincott-Schwartz, J. Mitochondria Supply Membranes for Autophagosome Biogenesis during Starvation. Cell 2010, 141, 656–667. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Evans, C.S.; Holzbaur, E.L. Degradation of engulfed mitochondria is rate-limiting in Optineurin-mediated mitophagy in neurons. Elife 2020, 9, e50260. [Google Scholar] [CrossRef] [PubMed]
- Yuan, A.; Rao, M.V.; Veeranna; Nixon, R.A. Neurofilaments and Neurofilament Proteins in Health and Disease. Cold Spring Harb. Perspect. Biol. 2017, 9, a018309. [Google Scholar] [CrossRef] [Green Version]
- Garcia-Barros, M.; Paris, F.; Cordon-Cardo, C.; Lyden, D.; Rafii, S.; Haimovitz-Friedman, A.; Fuks, Z.; Kolesnick, R. Tumor Response to Radiotherapy Regulated by Endothelial Cell Apoptosis. Science 2003, 300, 1155–1159. [Google Scholar] [CrossRef] [Green Version]
- Moding, E.J.; Castle, K.D.; Perez, B.A.; Oh, P.; Min, H.D.; Norris, H.; Ma, Y.; Cardona, D.M.; Lee, C.-L.; Kirsch, D.G. Tumor cells, but not endothelial cells, mediate eradication of primary sarcomas by stereotactic body radiation therapy. Sci. Transl. Med. 2015, 7, 278ra34. [Google Scholar] [CrossRef] [Green Version]
- Chaib, S.; Tchkonia, T.; Kirkland, J.L. Cellular senescence and senolytics: The path to the clinic. Nat. Med. 2022, 28, 1556–1568. [Google Scholar] [CrossRef]
- Grosse, L.; Wagner, N.; Emelyanov, A.; Molina, C.; Lacas-Gervais, S.; Wagner, K.-D.; Bulavin, D.V. Defined p16High Senescent Cell Types Are Indispensable for Mouse Healthspan. Cell Metab. 2020, 32, 87–99.e6. [Google Scholar] [CrossRef]
- Ghozy, S.; Reda, A.; Varney, J.; Elhawary, A.S.; Shah, J.; Murry, K.; Sobeeh, M.G.; Nayak, S.S.; Azzam, A.Y.; Brinjikji, W.; et al. Neuroprotection in Acute Ischemic Stroke: A Battle Against the Biology of Nature. Front. Neurol. 2022, 13, 870141. [Google Scholar] [CrossRef]
- Chamorro, Á.; Dirnagl, U.; Urra, X.; Planas, A.M. Neuroprotection in acute stroke: Targeting excitotoxicity, oxidative and nitrosative stress, and inflammation. Lancet Neurol. 2016, 15, 869–881. [Google Scholar] [CrossRef]
- El-Remessy, A.B.; Tawfik, H.E.; Matragoon, S.; Pillai, B.; Caldwell, R.B. Peroxynitrite Mediates Diabetes-Induced Endothelial Dysfunction: Possible Role of Rho Kinase Activation. Exp. Diabetes Res. 2010, 2010, 247861. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Al-Gayyar, M.M.; Abdelsaid, M.A.; Matragoon, S.; Pillai, B.A.; El-Remessy, A.B. Neurovascular Protective Effect of FeTPPs in N-Methyl-D-Aspartate Model: Similarities to Diabetes. Am. J. Pathol. 2010, 177, 1187–1197. [Google Scholar] [CrossRef]
- Ali, T.K.; Matragoon, S.; Pillai, B.A.; Liou, G.I.; El-Remessy, A.B. Peroxynitrite Mediates Retinal Neurodegeneration by Inhibiting Nerve Growth Factor Survival Signaling in Experimental and Human Diabetes. Diabetes 2008, 57, 889–898. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, W.; Fan, Z.; Han, Y.; Lu, S.; Zhang, D.; Bai, X.; Xu, W.; Li, J.; Wang, H. Curcumin attenuates peroxynitrite-induced neurotoxicity in spiral ganglion neurons. Neurotoxicology 2011, 32, 150–157. [Google Scholar] [CrossRef]
- Goel, A.; Aggarwal, B.B. Curcumin, the Golden Spice from Indian Saffron, Is a Chemosensitizer and Radiosensitizer for Tumors and Chemoprotector and Radioprotector for Normal Organs. Nutr. Cancer 2010, 62, 919–930. [Google Scholar] [CrossRef] [PubMed]
- Zoi, V.; Galani, V.; Tsekeris, P.; Kyritsis, A.P.; Alexiou, G.A. Radiosensitization and Radioprotection by Curcumin in Glioblastoma and Other Cancers. Biomedicines 2022, 10, 312. [Google Scholar] [CrossRef]
- Yildirim, B.A.; Çetin, E.; Topkan, E.; Ozyigit, G.; Cengiz, M.; Surucu, S.; Usubutun, A.; Akyol, F. Prevention of radiation-induced retinopathy with amifostine in wistar albino rats. Retina 2015, 35, 1458–1464. [Google Scholar] [CrossRef]
- Brizel, D.M. Does amifostine have a role in chemoradiation treatment? Lancet Oncol. 2003, 4, 378–380. [Google Scholar] [CrossRef]
- Oronsky, B.; Scribner, C.; Aggarwal, R.; Cabrales, P. RRx-001 protects normal tissues but not tumors via Nrf2 induction and Bcl-2 inhibition. J. Cancer Res. Clin. Oncol. 2019, 145, 2045–2050. [Google Scholar] [CrossRef]
- Vomund, S.; Schäfer, A.; Parnham, M.J.; Brüne, B.; Von Knethen, A. Nrf2, the Master Regulator of Anti-Oxidative Responses. Int. J. Mol. Sci. 2017, 18, 2772. [Google Scholar] [CrossRef] [Green Version]
- Ning, S.; Bednarski, M.; Oronsky, B.; Scicinski, J.; Saul, G.; Knox, S.J. Dinitroazetidines Are a Novel Class of Anticancer Agents and Hypoxia-Activated Radiation Sensitizers Developed from Highly Energetic Materials. Cancer Res. 2012, 72, 2600–2608. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rajala, R.V.S. Aerobic Glycolysis in the Retina: Functional Roles of Pyruvate Kinase Isoforms. Front. Cell Dev. Biol. 2020, 8, 266. [Google Scholar] [CrossRef] [PubMed]
- Yu, D.-Y.; Cringle, S.J. Oxygen Distribution and Consumption within the Retina in Vascularised and Avascular Retinas and in Animal Models of Retinal Disease. Prog. Retin. Eye Res. 2001, 20, 175–208. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.-S.; Li, L.-Y.; Guan, Y.-D.; Yang, J.-M.; Cheng, Y. Anticancer strategies based on the metabolic profile of tumor cells: Therapeutic targeting of the Warburg effect. Acta Pharmacol. Sin. 2016, 37, 1013–1019. [Google Scholar] [CrossRef]
- Derkach, D.; Kehtari, T.; Renaud, M.; Heidari, M.; Lakshman, N.; Morshead, C.M. Metformin pretreatment rescues olfactory memory associated with subependymal zone neurogenesis in a juvenile model of cranial irradiation. Cell Rep. Med. 2021, 2, 100231. [Google Scholar] [CrossRef]
- Liu, Y.; Tang, G.; Zhang, Z.; Wang, Y.; Yang, G.-Y. Metformin promotes focal angiogenesis and neurogenesis in mice following middle cerebral artery occlusion. Neurosci. Lett. 2014, 579, 46–51. [Google Scholar] [CrossRef] [PubMed]
- Sambe, T.; Mason, R.P.; Dawoud, H.; Bhatt, D.L.; Malinski, T. Metformin treatment decreases nitroxidative stress, restores nitric oxide bioavailability and endothelial function beyond glucose control. Biomed. Pharmacother. 2018, 98, 149–156. [Google Scholar] [CrossRef]
- Monroe, A.T.; Bhandare, N.; Morris, C.G.; Mendenhall, W.M. Preventing radiation retinopathy with hyperfractionation. Int. J. Radiat. Oncol. 2005, 61, 856–864. [Google Scholar] [CrossRef]
- Wiegner, E.A.; Daly, M.E.; Murphy, J.D.; Abelson, J.; Chapman, C.H.; Chung, M.; Yu, Y.; Colevas, A.D.; Kaplan, M.J.; Fischbein, N.; et al. Intensity-Modulated Radiotherapy for Tumors of the Nasal Cavity and Paranasal Sinuses: Clinical Outcomes and Patterns of Failure. Int. J. Radiat. Oncol. 2012, 83, 243–251. [Google Scholar] [CrossRef]
- Askoxylakis, V.; Hegenbarth, P.; Timke, C.; Saleh-Ebrahimi, L.; Debus, J.; Röder, F.; Huber, P.E. Intensity modulated radiation therapy (IMRT) for sinonasal tumors: A single center long-term clinical analysis. Radiat. Oncol. 2016, 11, 17. [Google Scholar] [CrossRef] [Green Version]
- Mothersill, C.; Seymour, C.B. Bystander and Delayed Effects after Fractionated Radiation Exposure. Radiat. Res. 2002, 158, 626–633. [Google Scholar] [CrossRef] [PubMed]
- Mothersill, C.; Moriarty, M.; Seymour, C. Radiotherapy and the potential exploitation of bystander effects. Int. J. Radiat. Oncol. 2004, 58, 575–579. [Google Scholar] [CrossRef] [PubMed]










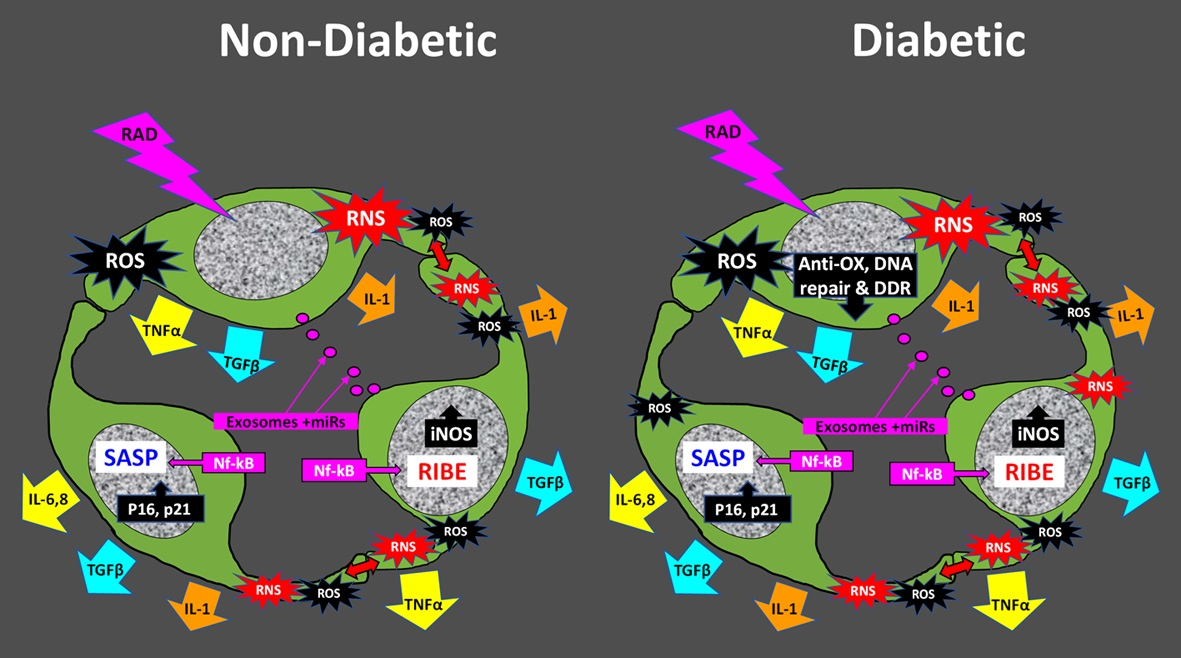{kind=link}
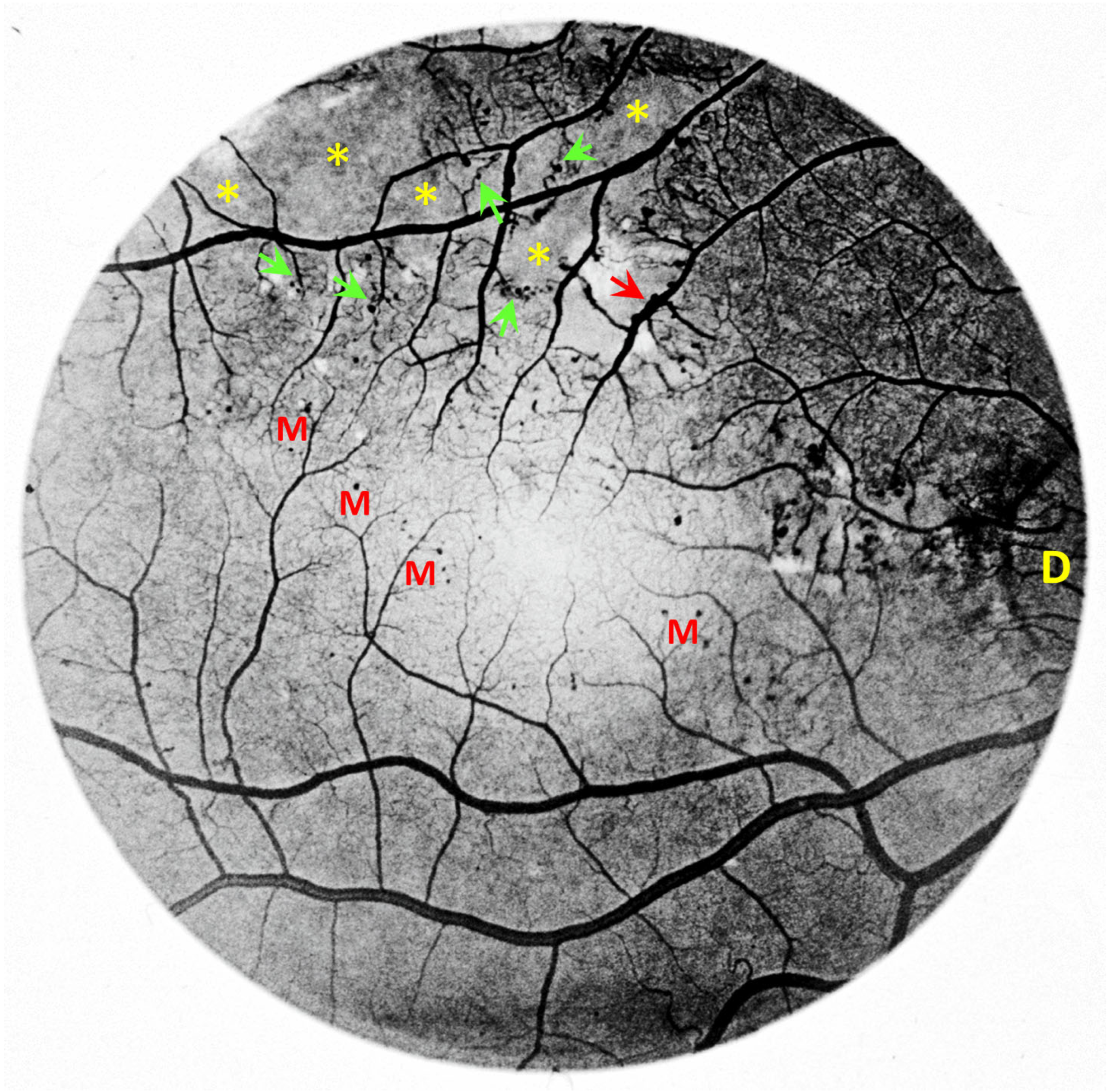{kind=link}
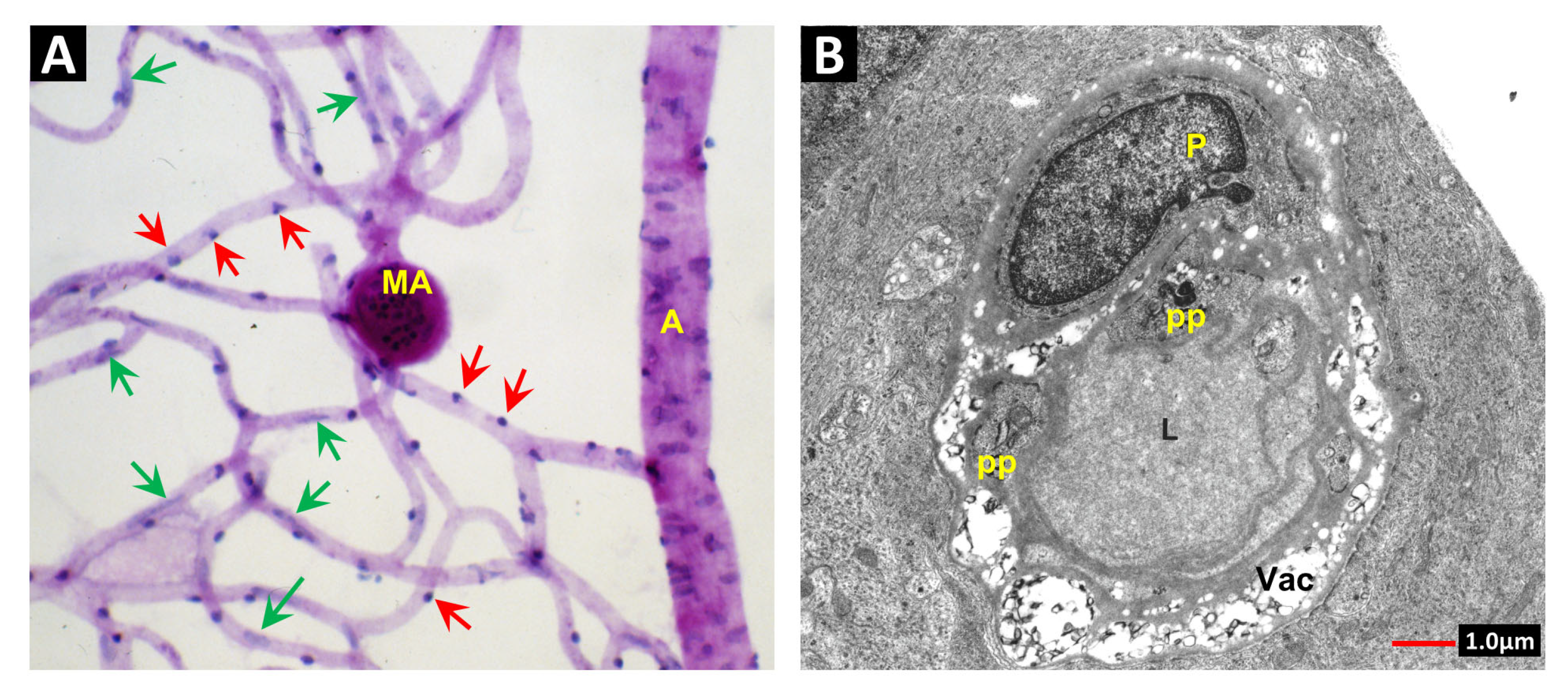{kind=link}
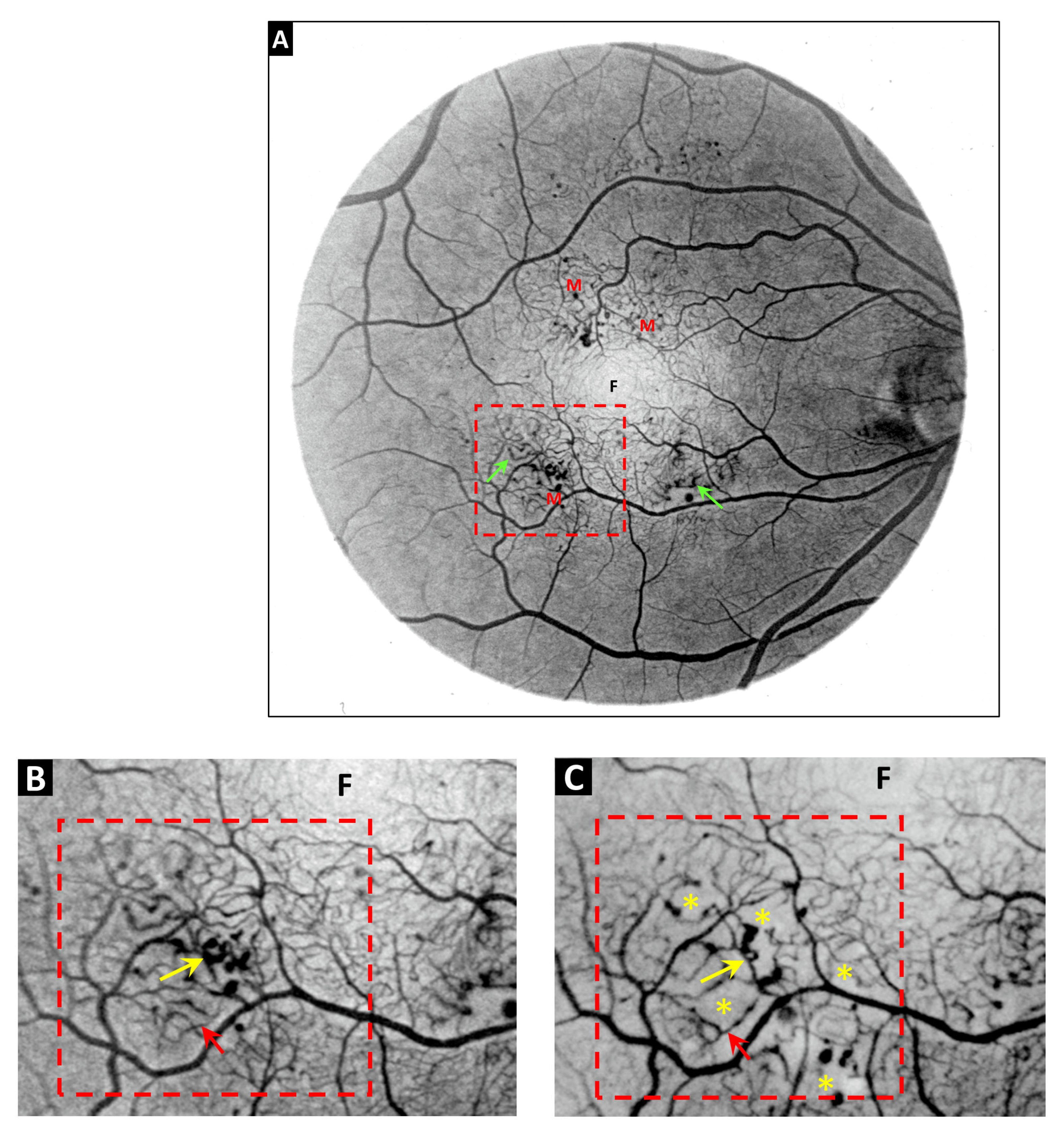{kind=link}
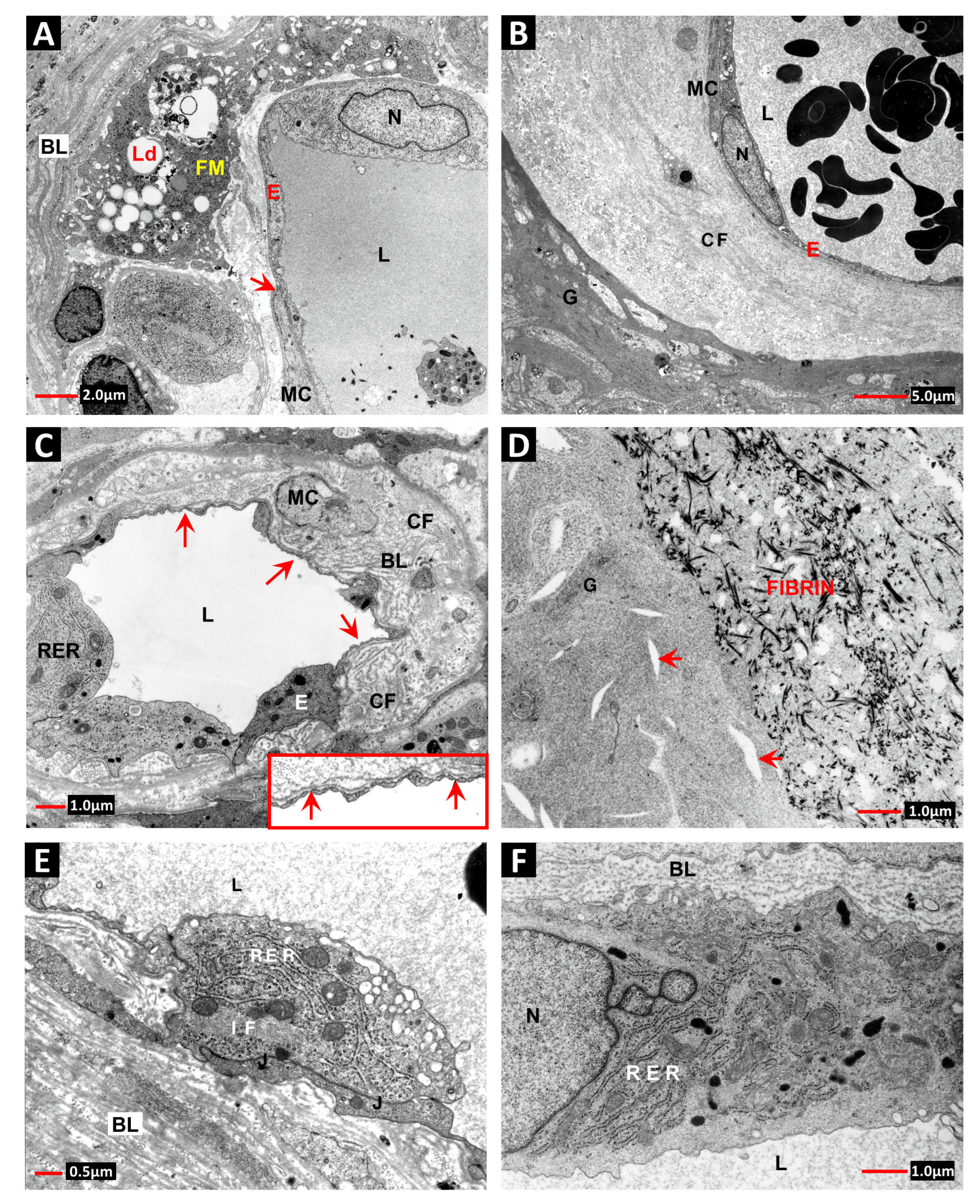{kind=link}
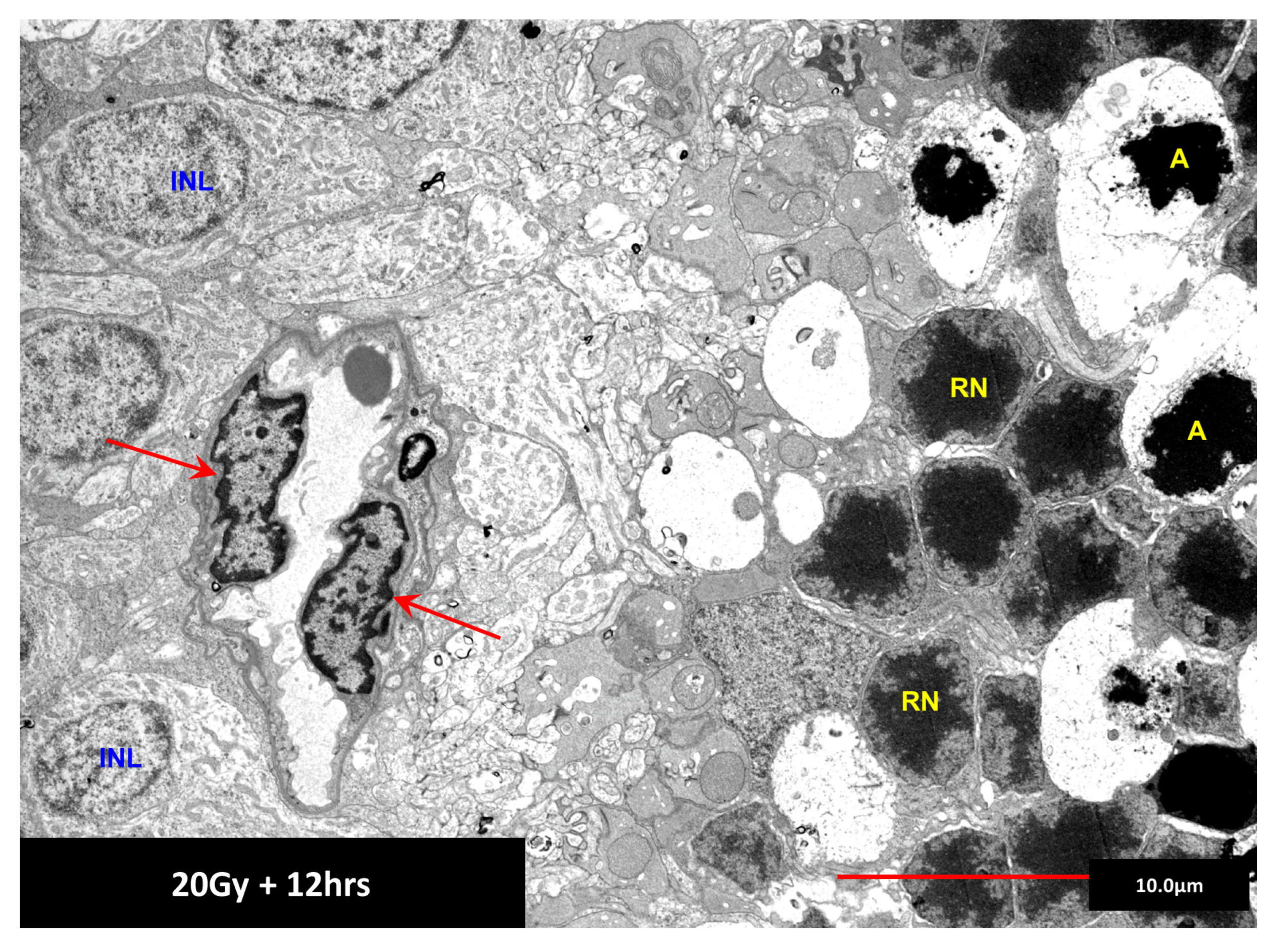{kind=link}
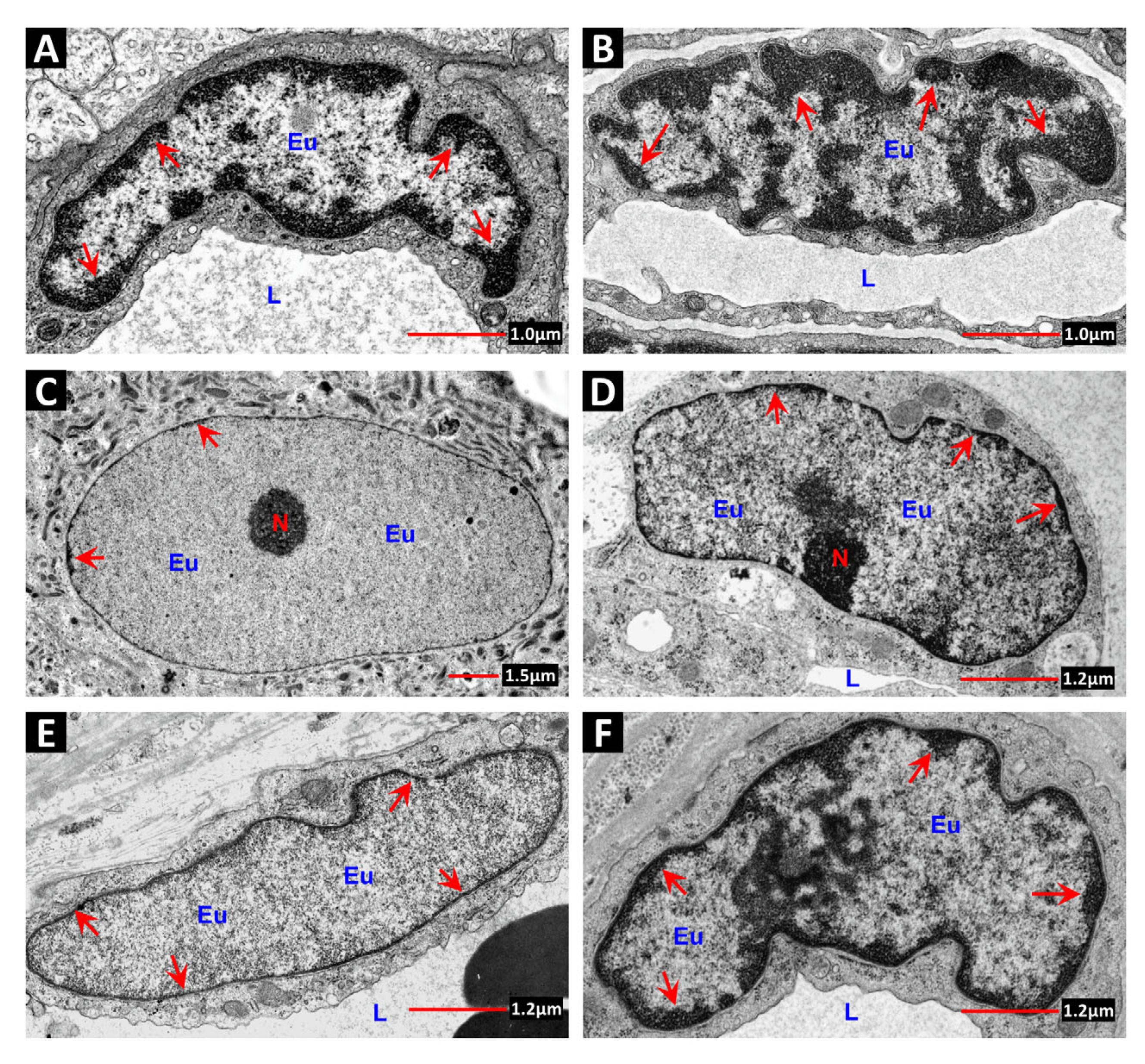{kind=link}
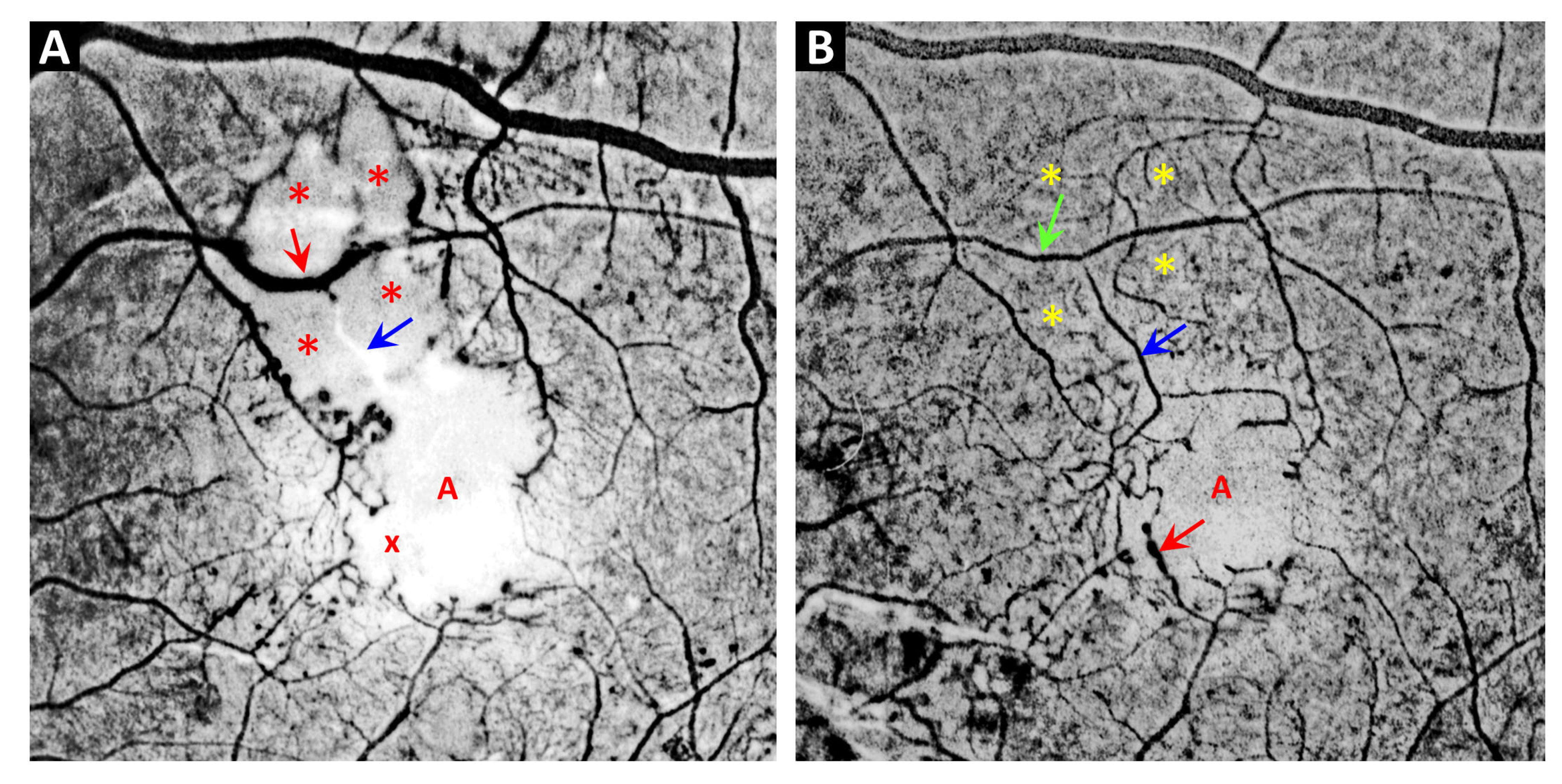{kind=link}
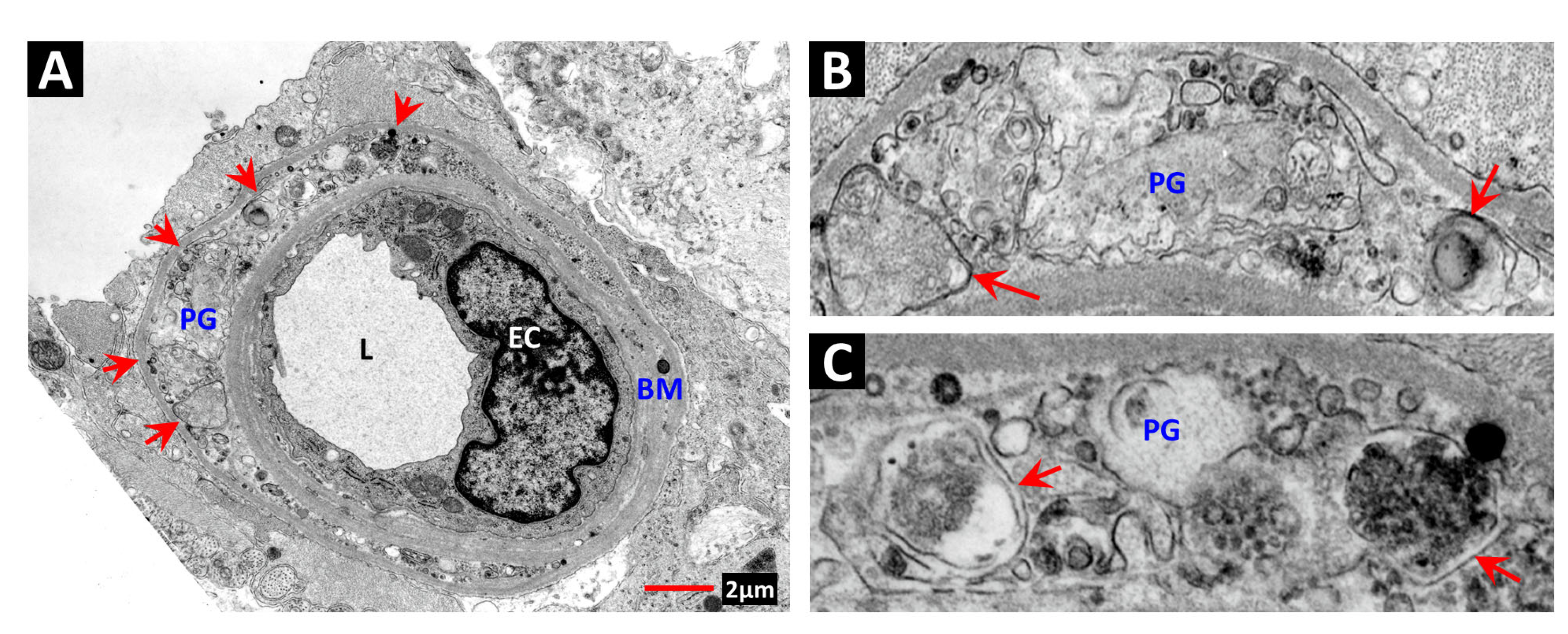{kind=link}
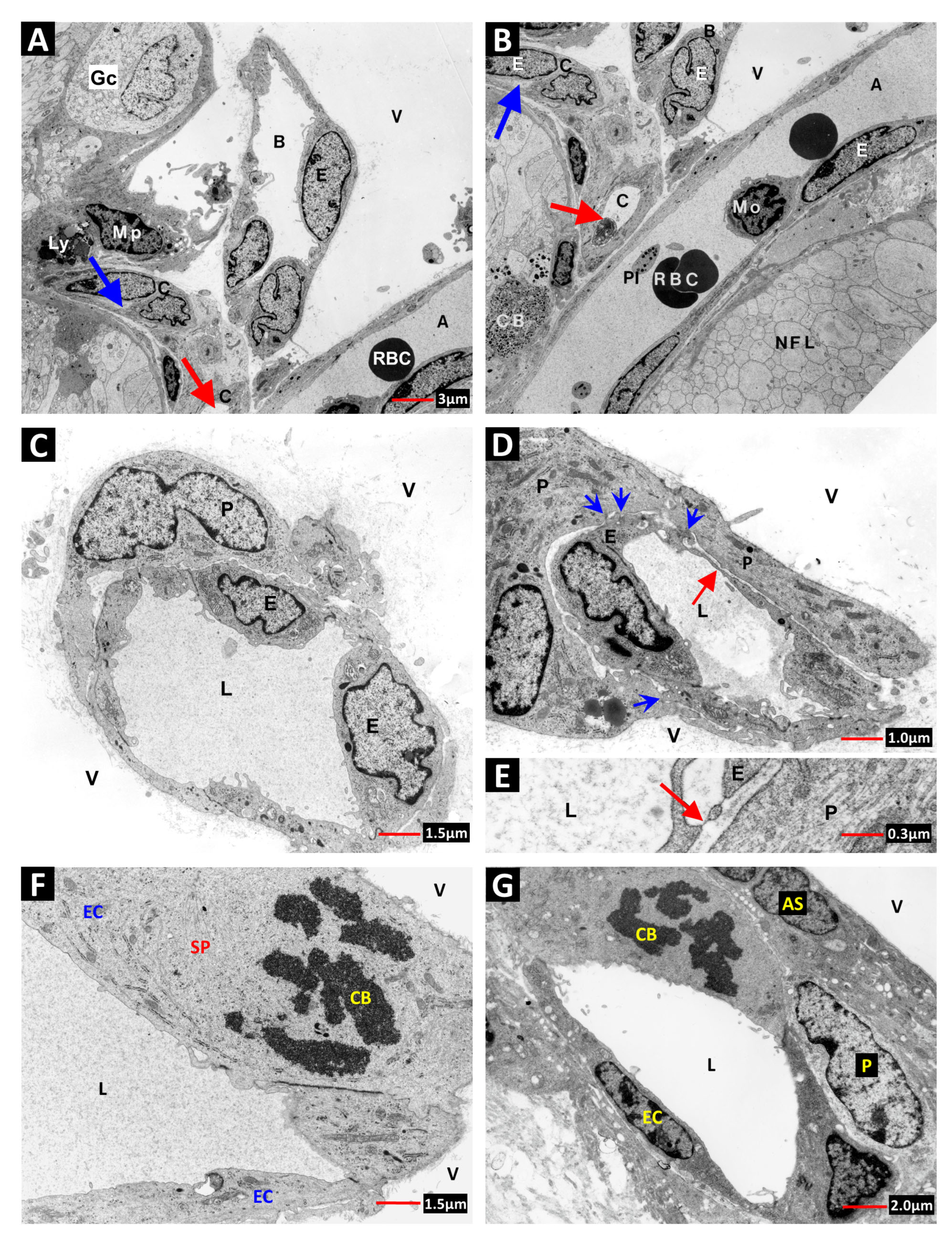{kind=link}
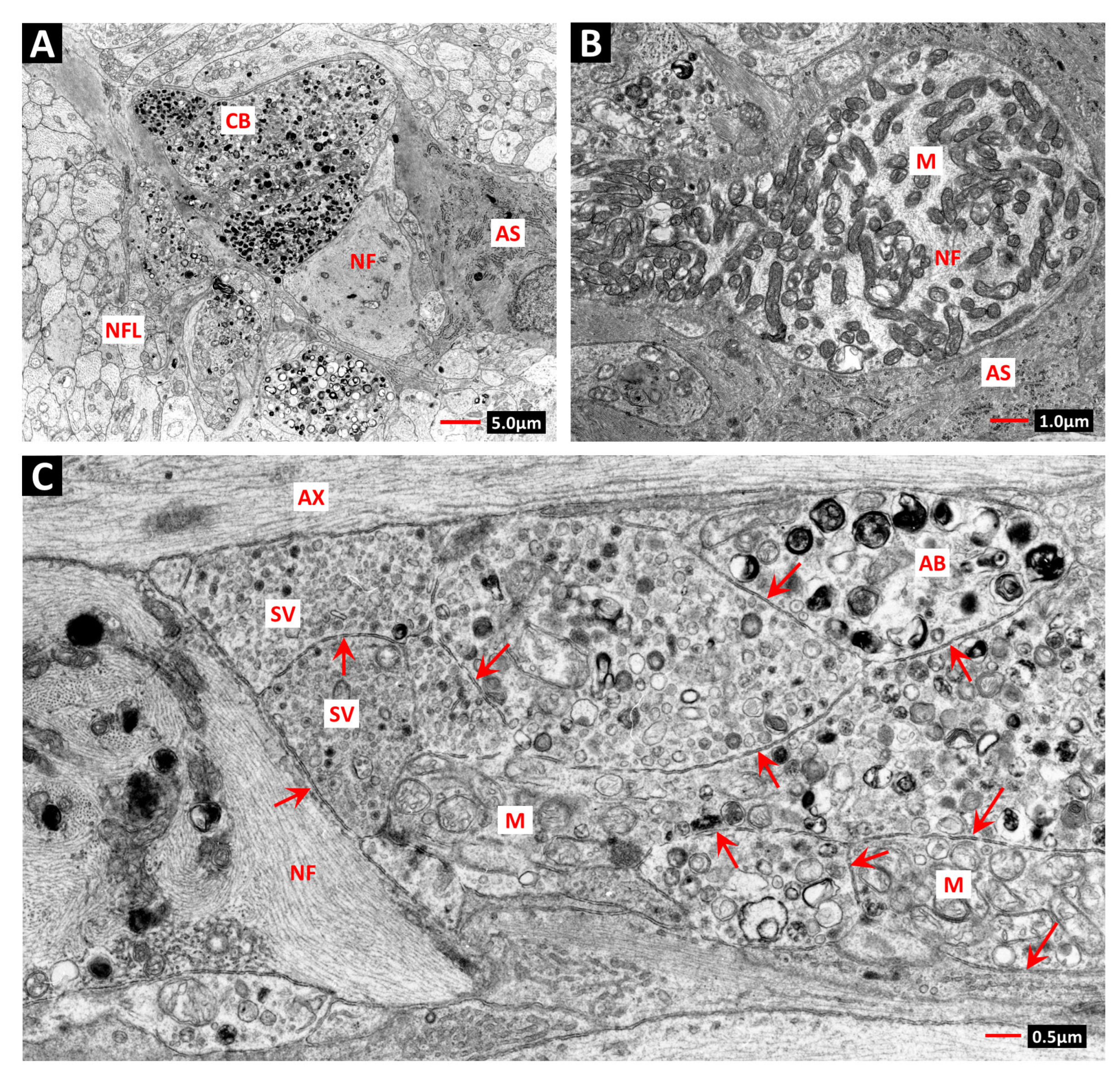{kind=link}
| Vascular Pathology | Diabetic Retinopathy | Radiation Retinopathy |
|---|---|---|
| Loss of pericytes or vascular smooth muscle cells in vessels with surviving endothelial cells | Yes | No |
| Loss of endothelial cells in vessels with surviving pericytes or vascular smooth muscle cells | No | Yes |
| Generalized thickening of capillary basement membranes | Yes | No |
| Vessels with fenestrated endothelial cells | Only in end-stage disease | Yes |
| Vascular occlusion and inner retinal ischaemia | Yes | Yes |
| Vascular recovery | No—Space-filling of BM tubes by glial cells is permanent | Limited recovery |
| Vascular remodelling | IRMA | Telangiectasia |
| Telangiectatic vessels with expanded pseudoadventitia and fenestrated endothelial cells with euchromatic nuclei | No | Yes |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gardiner, T.A.; Archer, D.B.; Silvestri, G.; Amoaku, W.M. Radiation and Diabetic Retinopathy: A Dark Synergy. Int. J. Transl. Med. 2023, 3, 120-159. https://doi.org/10.3390/ijtm3010011
Gardiner TA, Archer DB, Silvestri G, Amoaku WM. Radiation and Diabetic Retinopathy: A Dark Synergy. International Journal of Translational Medicine. 2023; 3(1):120-159. https://doi.org/10.3390/ijtm3010011
Chicago/Turabian StyleGardiner, Tom A., Desmond B. Archer, Giuliana Silvestri, and Winfried M. Amoaku. 2023. "Radiation and Diabetic Retinopathy: A Dark Synergy" International Journal of Translational Medicine 3, no. 1: 120-159. https://doi.org/10.3390/ijtm3010011