
DNA Damage and the Gut Microbiome: From Mechanisms to Disease Outcomes

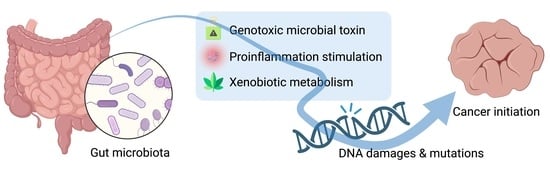
Abstract
:
1. Introduction
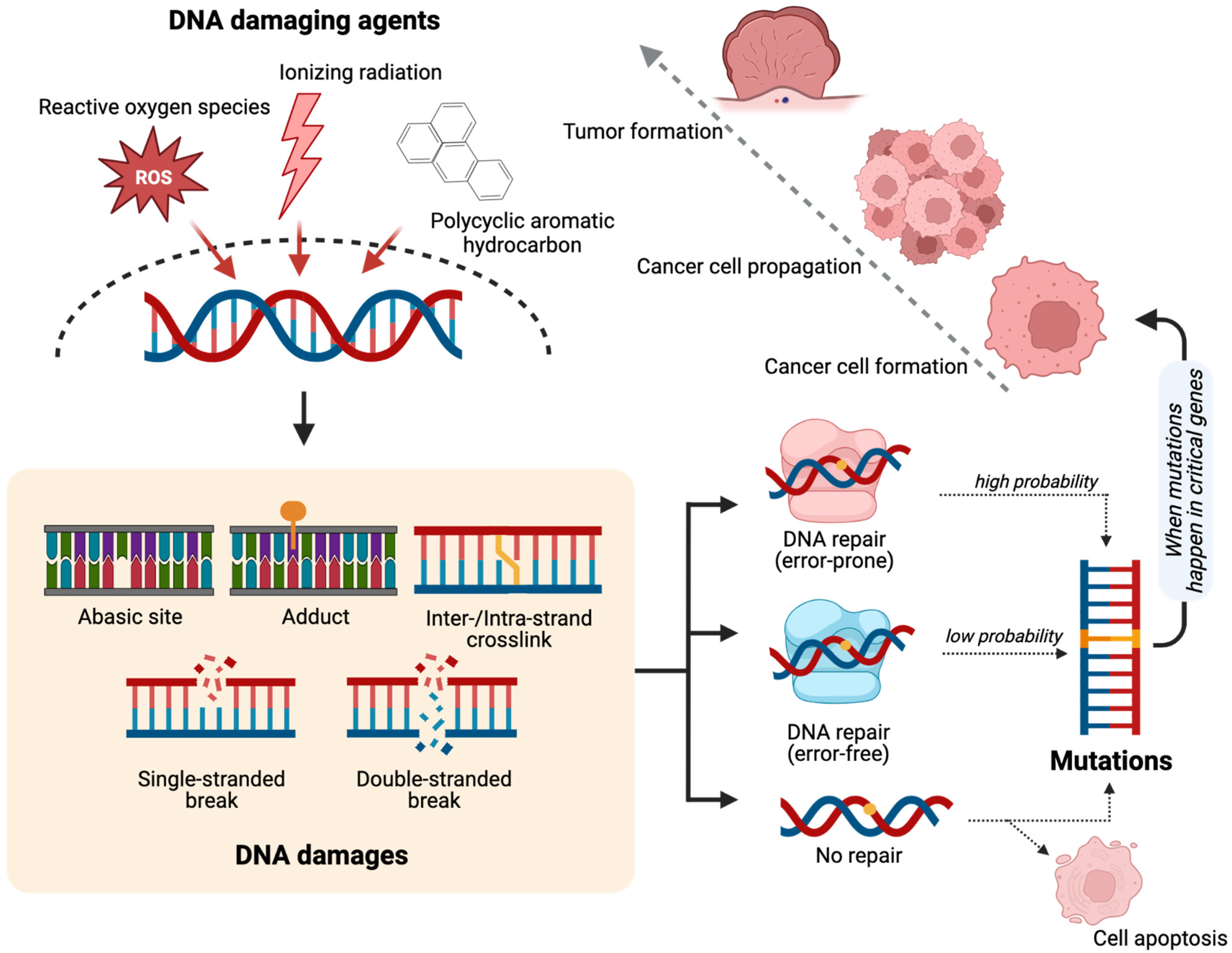
2. DNA Damage and Cancer Development
3. DNA Damage Attributed to the Gut Microbiome
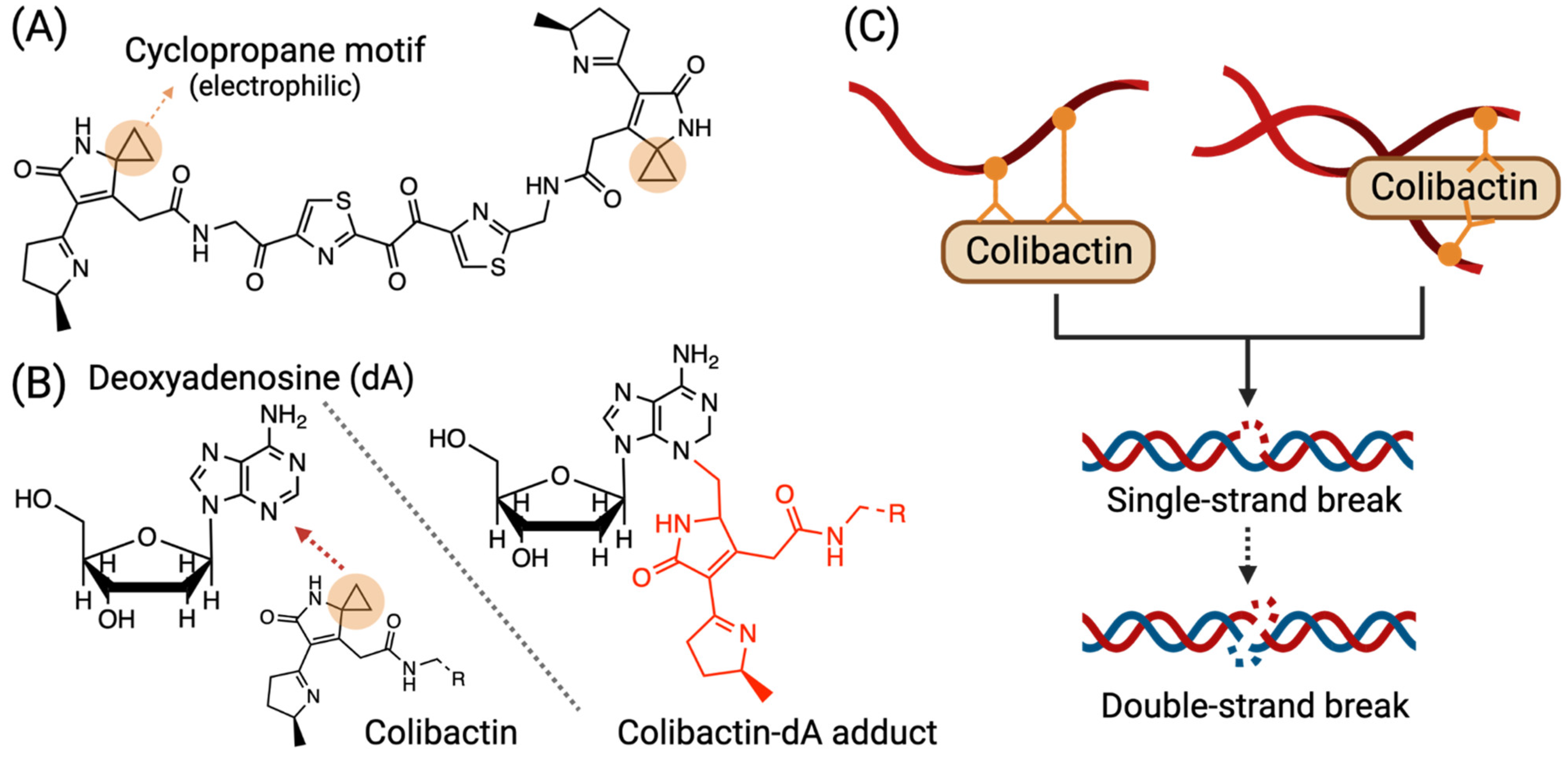
3.1. Colibactin-Derived DNA Damage
3.2. Other Toxins: Cytolethal Distending Toxins and Typhoid Toxin
3.3. Indirect Pathways and Systemic Effects
4. Genotoxic Endogenous Processes Modulated by the Gut Microbiome
4.1. Bile Acids and Lipid Metabolism
4.2. Proinflammation and Inflammation
4.2.1. Helicobacter Pylori
4.2.2. Bacterial Lipopolysaccharides and Other Microbial Products
4.3. Xenobiotic Biotransformation

{kind=link}
{kind=link}
{kind=link}
| Class | Name | PubChem CID | Use | Specific Gut Microbiome Species | Mechanism | Reference |
|---|---|---|---|---|---|---|
| Nitro-PAHs | 2-nitrofluorene | 11831 | By-product of combustion | NA | CONV-R mice experienced higher total DNA adduct levels than GF mice in all tissues collected. | [106] |
| NA | SPF mice and HFA mice had higher total DNA adduct levels in local (e.g., colon epithelium) and distant (e.g., liver) tissues. | [107] | ||||
| 2-acetylaminofluorene | By-product of combustion | NA | CONV-R mice experienced higher total DNA adduct levels than GF mice in all tissues collected. | [106] | ||
| 6-nitrobenzo[a]pyrene | 44374 | Engine emission | NA | Microbiome reduced 6- Nitrobenzo[a] pyrene to 6-nitrosobenzo[a]pyrene (PCID 119358) and 6-aminobenzo[a]pyrene (PCID 23911), whereby 6-nitrosobenzo[a]pyrene showed direct mutagenicity. | [102,103] | |
| 1-nitropyrene | 21694 | By-product of combustion | NA | Specific DNA adducts were detected only in CONV-R but not in ABT mice. | [108] | |
| P. magnus | P. magnus metabolized sample had higher genotoxicity. | [104] | ||||
| 2-Amino-1-methyl-6-phenylimidazo[4,5-b]pyridine | 1530 | Known mutagen found in cooked foods and in cigarette smoke. | L. rhamnosus | CONV-R mice additionally fed with L. rhanmosus had lower total DNA adduct levels in the colon tissues compared to control CONV-R mice. | [105] | |
| 3-methyl-3H-imidazo[4,5-f]quinolin-2-amine | 53462 | Known mutagen found in cooked foods and in cigarette smoke. | NA | SPF mice and HFA mice had higher total DNA adduct levels in local (e.g., colon epithelium) and distant (e.g., liver) tissues. | [107] | |
| 2-Amino-9H-pyrido[2,3-b]indole | 62805 | Known mutagen found in cooked foods and in cigarette smoke. | S. faecalis, C. butyricum, B. mesentericus | HFA mice additionally administered with the probiotic mixture (Sf, Cb, Bm) had lower total DNA adduct level than the control HFA mice. | [109] | |
| MelQx | 62275 | Known mutagen found in cooked foods and in cigarette smoke. | E. hallii, L. reuteri, L. rossiae | The three bacteria tested were able to convert MelQx to a new microbial metabolite (MelQx-M1) with lower mutagenicity. | [110,111] | |
| Dinitrotoluenes | 2-nitrotoluene | 6944 | Production of dyes, pesticides, and rubber chemicals. | NA | DNA repair response was only observed in inoculated animal rather than GF animal. | [112] |
| Toxin | Aflatoxin B1 | 186907 | Mutagen produced by specific molds, particularly Aspergillus spp. | L. rhamnosus, P. freudenreichii | Healthy young men (n = 90) with potential exposure to Aflatoxin B1 were assigned to the control group or probiotic-administered group. The probiotic-administered group had lower Aflatoxin B1-induced DNA adduct. | [113] |
5. Gut Microbiome and Cancer Development: From Disease Associations to Mechanistic Understanding
5.1. Colorectal Cancer
5.2. Gastric Cancer
5.3. Extra-Gastrointestinal Cancer
6. Missing Pieces and Future Direction
7. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lu, K.; Hsiao, Y.-C.; Liu, C.-W.; Schoeny, R.; Gentry, R.; Starr, T.B. A Review of Stable Isotope Labeling and Mass Spectrometry Methods to Distinguish Exogenous from Endogenous DNA Adducts and Improve Dose–Response Assessments. Chem. Res. Toxicol. 2021, 35, 7–29. [Google Scholar] [CrossRef] [PubMed]
- Nagai, H.; Kim, Y.H. Cancer prevention from the perspective of global cancer burden patterns. J. Thorac. Dis. 2017, 9, 448–451. [Google Scholar] [CrossRef] [PubMed]
- Yu, R.; Lai, Y.; Hartwell, H.J.; Moeller, B.C.; Doyle-Eisele, M.; Kracko, D.; Bodnar, W.M.; Starr, T.B.; Swenberg, J.A. Formation, Accumulation, and Hydrolysis of Endogenous and Exogenous Formaldehyde-Induced DNA Damage. Toxicol. Sci. Off. J. Soc. Toxicol. 2015, 146, 170–182. [Google Scholar] [CrossRef] [PubMed]
- Pontel, L.B.; Rosado, I.V.; Burgos-Barragan, G.; Garaycoechea, J.I.; Yu, R.; Arends, M.J.; Chandrasekaran, G.; Broecker, V.; Wei, W.; Liu, L.; et al. Endogenous Formaldehyde Is a Hematopoietic Stem Cell Genotoxin and Metabolic Carcinogen. Mol. Cell 2015, 60, 177–188. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.S.; Groopman, J.D. DNA damage by mycotoxins. Mutat. Res. 1999, 424, 167–181. [Google Scholar] [CrossRef] [PubMed]
- Thursby, E.; Juge, N. Introduction to the human gut microbiota. Biochem. J. 2017, 474, 1823–1836. [Google Scholar] [CrossRef]
- Fan, Y.; Pedersen, O. Gut microbiota in human metabolic health and disease. Nat. Rev. Microbiol. 2021, 19, 55–71. [Google Scholar] [CrossRef]
- Sender, R.; Fuchs, S.; Milo, R. Revised Estimates for the Number of Human and Bacteria Cells in the Body. PLoS Biol. 2016, 14, e1002533. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.Y.; Tsolis, R.M.; Bäumler, A.J. The microbiome and gut homeostasis. Science 2022, 377, eabp9960. [Google Scholar] [CrossRef]
- Petersen, C.; Round, J.L. Defining dysbiosis and its influence on host immunity and disease. Cell Microbiol. 2014, 16, 1024–1033. [Google Scholar] [CrossRef]
- Rogers, A.W.L.; Tsolis, R.M.; Bäumler, A.J. Salmonella versus the Microbiome. Microbiol. Mol. Biol. Rev. 2021, 85, e00027-19. [Google Scholar] [CrossRef] [PubMed]
- Ahmadmehrabi, S.; Tang, W.H.W. Gut microbiome and its role in cardiovascular diseases. Curr. Opin. Cardiol. 2017, 32, 761–766. [Google Scholar] [CrossRef]
- Wang, Z.; Klipfell, E.; Bennett, B.J.; Koeth, R.; Levison, B.S.; Dugar, B.; Feldstein, A.E.; Britt, E.B.; Fu, X.; Chung, Y.-M.; et al. Gut flora metabolism of phosphatidylcholine promotes cardiovascular disease. Nature 2011, 472, 57–63. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Li, X.; Yang, F.; Zhao, R.; Pan, X.; Liang, J.; Tian, L.; Li, X.; Liu, L.; Xing, Y.; et al. Gut Microbiota-Dependent Marker TMAO in Promoting Cardiovascular Disease: Inflammation Mechanism, Clinical Prognostic, and Potential as a Therapeutic Target. Front. Pharmacol. 2019, 10, 1360. [Google Scholar] [CrossRef]
- Kowalski, K.; Mulak, A. Brain-Gut-Microbiota Axis in Alzheimer’s Disease. J. Neurogastroenterol. Motil. 2019, 25, 48–60. [Google Scholar] [CrossRef] [PubMed]
- Jiang, C.; Li, G.; Huang, P.; Liu, Z.; Zhao, B. The Gut Microbiota and Alzheimer’s Disease. J. Alzheimer’s Dis. 2017, 58, 1–15. [Google Scholar] [CrossRef]
- Hsiao, Y.-C.; Liu, C.-W.; Chi, L.; Yang, Y.; Lu, K. Effects of Gut Microbiome on Carcinogenic DNA Damage. Chem. Res. Toxicol. 2020, 33, 2130–2138. [Google Scholar] [CrossRef]
- Martin, L.J. DNA damage and repair: Relevance to mechanisms of neurodegeneration. J. Neuropathol. Exp. Neurol. 2008, 67, 377–387. [Google Scholar] [CrossRef]
- Jackson, S.P.; Bartek, J. The DNA-damage response in human biology and disease. Nature 2009, 461, 1071–1078. [Google Scholar] [CrossRef]
- Abbotts, R.; Wilson, D.M., 3rd. Coordination of DNA single strand break repair. Free Radic. Biol. Med. 2017, 107, 228–244. [Google Scholar] [CrossRef]
- Vítor, A.C.; Huertas, P.; Legube, G.; de Almeida, S.F. Studying DNA Double-Strand Break Repair: An Ever-Growing Toolbox. Front. Mol. Biosci. 2020, 7, 24. [Google Scholar] [CrossRef]
- Yu, T.-W.; Anderson, D. Reactive oxygen species-induced DNA damage and its modification: A chemical investigation. Mutat. Res. /Fundam. Mol. Mech. Mutagen. 1997, 379, 201–210. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, S.; Anai, H.; Hanada, K. Mechanisms of interstrand DNA crosslink repair and human disorders. Genes Environ. 2016, 38, 9. [Google Scholar] [CrossRef] [PubMed]
- Wilson, M.R.; Jiang, Y.; Villalta, P.W.; Stornetta, A.; Boudreau, P.D.; Carrá, A.; Brennan, C.A.; Chun, E.; Ngo, L.; Samson, L.D.; et al. The human gut bacterial genotoxin colibactin alkylates DNA. Science 2019, 363, eaar7785. [Google Scholar] [CrossRef] [PubMed]
- Tsukanov, V.V.; Smirnova, O.V.; Kasparov, E.V.; Sinyakov, A.A.; Vasyutin, A.V.; Tonkikh, J.L.; Cherepnin, M.A. Dynamics of Oxidative Stress in Helicobacter pylori-Positive Patients with Atrophic Body Gastritis and Various Stages of Gastric Cancer. Diagnostics 2022, 12, 1203. [Google Scholar] [CrossRef]
- Kidane, D.; Murphy, D.L.; Sweasy, J.B. Accumulation of abasic sites induces genomic instability in normal human gastric epithelial cells during Helicobacter pylori infection. Oncogenesis 2014, 3, e128. [Google Scholar] [CrossRef]
- Thakur, B.K.; Malaisé, Y.; Martin, A. Unveiling the Mutational Mechanism of the Bacterial Genotoxin Colibactin in Colorectal Cancer. Mol. Cell 2019, 74, 227–229. [Google Scholar] [CrossRef]
- Bossuet-Greif, N.; Vignard, J.; Taieb, F.; Mirey, G.; Dubois, D.; Petit, C.; Oswald, E.; Nougayrède, J.P. The Colibactin Genotoxin Generates DNA Interstrand Cross-Links in Infected Cells. mBio 2018, 9, e02393-17. [Google Scholar] [CrossRef]
- Xue, M.; Wernke, K.M.; Herzon, S.B. Depurination of Colibactin-Derived Interstrand Cross-Links. Biochemistry 2020, 59, 892–900. [Google Scholar] [CrossRef]
- Lopez Chiloeches, M.; Bergonzini, A.; Frisan, T. Bacterial Toxins Are a Never-Ending Source of Surprises: From Natural Born Killers to Negotiators. Toxins 2021, 13, 426. [Google Scholar] [CrossRef]
- Bezine, E.; Vignard, J.; Mirey, G. The cytolethal distending toxin effects on Mammalian cells: A DNA damage perspective. Cells 2014, 3, 592–615. [Google Scholar] [CrossRef] [PubMed]
- Elwell, C.A.; Dreyfus, L.A. DNase I homologous residues in CdtB are critical for cytolethal distending toxin-mediated cell cycle arrest. Mol. Microbiol. 2000, 37, 952–963. [Google Scholar] [CrossRef] [PubMed]
- Mao, X.; DiRienzo, J.M. Functional studies of the recombinant subunits of a cytolethal distending holotoxin. Cell Microbiol. 2002, 4, 245–255. [Google Scholar] [CrossRef] [PubMed]
- Ibler, A.E.M.; ElGhazaly, M.; Naylor, K.L.; Bulgakova, N.A.; F. El-Khamisy, S.; Humphreys, D. Typhoid toxin exhausts the RPA response to DNA replication stress driving senescence and Salmonella infection. Nat. Commun. 2019, 10, 4040. [Google Scholar] [CrossRef]
- Li, Z.-R.; Li, J.; Cai, W.; Lai, J.Y.H.; McKinnie, S.M.K.; Zhang, W.-P.; Moore, B.S.; Zhang, W.; Qian, P.-Y. Macrocyclic colibactin induces DNA double-strand breaks via copper-mediated oxidative cleavage. Nat. Chem. 2019, 11, 880–889. [Google Scholar] [CrossRef]
- Fahrer, J.; Huelsenbeck, J.; Jaurich, H.; Dörsam, B.; Frisan, T.; Eich, M.; Roos, W.P.; Kaina, B.; Fritz, G. Cytolethal distending toxin (CDT) is a radiomimetic agent and induces persistent levels of DNA double-strand breaks in human fibroblasts. DNA Repair 2014, 18, 31–43. [Google Scholar] [CrossRef] [PubMed]
- Putze, J.; Hennequin, C.; Nougayrède, J.P.; Zhang, W.; Homburg, S.; Karch, H.; Bringer, M.A.; Fayolle, C.; Carniel, E.; Rabsch, W.; et al. Genetic structure and distribution of the colibactin genomic island among members of the family Enterobacteriaceae. Infect. Immun. 2009, 77, 4696–4703. [Google Scholar] [CrossRef]
- Auvray, F.; Perrat, A.; Arimizu, Y.; Chagneau, C.V.; Bossuet-Greif, N.; Massip, C.; Brugère, H.; Nougayrède, J.P.; Hayashi, T.; Branchu, P.; et al. Insights into the acquisition of the pks island and production of colibactin in the Escherichia coli population. Microb. Genom. 2021, 7, 000579. [Google Scholar] [CrossRef]
- Vizcaino, M.I.; Crawford, J.M. The colibactin warhead crosslinks DNA. Nat. Chem. 2015, 7, 411–417. [Google Scholar] [CrossRef]
- Xue, M.; Shine, E.; Wang, W.; Crawford, J.M.; Herzon, S.B. Characterization of Natural Colibactin–Nucleobase Adducts by Tandem Mass Spectrometry and Isotopic Labeling. Support for DNA Alkylation by Cyclopropane Ring Opening. Biochemistry 2018, 57, 6391–6394. [Google Scholar] [CrossRef]
- Wernke, K.M.; Xue, M.; Tirla, A.; Kim, C.S.; Crawford, J.M.; Herzon, S.B. Structure and bioactivity of colibactin. Bioorg. Med. Chem. Lett. 2020, 30, 127280. [Google Scholar] [CrossRef]
- Jinadasa, R.N.; Bloom, S.E.; Weiss, R.S.; Duhamel, G.E. Cytolethal distending toxin: A conserved bacterial genotoxin that blocks cell cycle progression, leading to apoptosis of a broad range of mammalian cell lineages. Microbiology 2011, 157, 1851–1875. [Google Scholar] [CrossRef]
- Cortes-Bratti, X.; Frisan, T.; Thelestam, M. The cytolethal distending toxins induce DNA damage and cell cycle arrest. Toxicon 2001, 39, 1729–1736. [Google Scholar] [CrossRef] [PubMed]
- Jiao, X.; Smith, S.; Stack, G.; Liang, Q.; Bradley, A.; Kellam, P.; Galán, J.E. Generation and Characterization of Typhoid Toxin-Neutralizing Human Monoclonal Antibodies. Infect. Immun. 2020, 88, e00292-20. [Google Scholar] [CrossRef] [PubMed]
- Song, J.; Gao, X.; Galán, J.E. Structure and function of the Salmonella Typhi chimaeric A(2)B(5) typhoid toxin. Nature 2013, 499, 350–354. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Rivera, L.D.; Bowen, B.M.; den Bakker, H.C.; Duhamel, G.E.; Wiedmann, M. Characterization of the cytolethal distending toxin (typhoid toxin) in non-typhoidal Salmonella serovars. Gut Pathog. 2015, 7, 19. [Google Scholar] [CrossRef]
- Chang, S.-J.; Jin, S.C.; Jiao, X.; Galán, J.E. Unique features in the intracellular transport of typhoid toxin revealed by a genome-wide screen. PLoS Pathog. 2019, 15, e1007704. [Google Scholar] [CrossRef]
- Iftekhar, A.; Berger, H.; Bouznad, N.; Heuberger, J.; Boccellato, F.; Dobrindt, U.; Hermeking, H.; Sigal, M.; Meyer, T.F. Genomic aberrations after short-term exposure to colibactin-producing E. coli transform primary colon epithelial cells. Nat. Commun. 2021, 12, 1003. [Google Scholar] [CrossRef]
- Damek-Poprawa, M.; Haris, M.; Volgina, A.; Korostoff, J.; DiRienzo, J.M. Cytolethal distending toxin damages the oral epithelium of gingival explants. J. Dent. Res. 2011, 90, 874–879. [Google Scholar] [CrossRef]
- Graillot, V.; Dormoy, I.; Dupuy, J.; Shay, J.W.; Huc, L.; Mirey, G.; Vignard, J. Genotoxicity of Cytolethal Distending Toxin (CDT) on Isogenic Human Colorectal Cell Lines: Potential Promoting Effects for Colorectal Carcinogenesis. Front. Cell. Infect. Microbiol. 2016, 6, 34. [Google Scholar] [CrossRef] [Green Version]
- Fowler, C.C.; Galán, J.E. Decoding a Salmonella Typhi Regulatory Network that Controls Typhoid Toxin Expression within Human Cells. Cell Host Microbe 2018, 23, 65–76.e66. [Google Scholar] [CrossRef] [PubMed]
- Butt, J.; Epplein, M. Helicobacter pylori and colorectal cancer-A bacterium going abroad? PLoS Pathog 2019, 15, e1007861. [Google Scholar] [CrossRef] [PubMed]
- Jackson, L.; Britton, J.; Lewis, S.A.; McKeever, T.M.; Atherton, J.; Fullerton, D.; Fogarty, A.W. A population-based epidemiologic study of Helicobacter pylori infection and its association with systemic inflammation. Helicobacter 2009, 14, 108–113. [Google Scholar] [CrossRef]
- Lamb, A.; Chen, L.F. Role of the Helicobacter pylori-induced inflammatory response in the development of gastric cancer. J. Cell Biochem. 2013, 114, 491–497. [Google Scholar] [CrossRef]
- Zhao, Q.; Song, C.; Wang, K.; Li, D.; Yang, Y.; Liu, D.; Wang, L.; Zhou, N.; Xie, Y. Prevalence of Helicobacter pylori babA, oipA, sabA, and homB genes in isolates from Chinese patients with different gastroduodenal diseases. Med. Microbiol. Immunol. 2020, 209, 565–577. [Google Scholar] [CrossRef]
- Sugimoto, M.; Ohno, T.; Graham, D.Y.; Yamaoka, Y. Helicobacter pylori outer membrane proteins on gastric mucosal interleukin 6 and 11 expression in Mongolian gerbils. J. Gastroenterol. Hepatol. 2011, 26, 1677–1684. [Google Scholar] [CrossRef]
- Yamaoka, Y.; Kwon, D.H.; Graham, D.Y. A M(r) 34,000 proinflammatory outer membrane protein (oipA) of Helicobacter pylori. Proc. Natl. Acad. Sci. USA 2000, 97, 7533–7538. [Google Scholar] [CrossRef] [PubMed]
- Ranneh, Y.; Ali, F.; Akim, A.M.; Hamid, H.A.; Khazaai, H.; Fadel, A. Crosstalk between reactive oxygen species and pro-inflammatory markers in developing various chronic diseases: A review. Appl. Biol. Chem. 2017, 60, 327–338. [Google Scholar] [CrossRef]
- Kumar, R.; Hemminki, K. Separation of 7-methyl- and 7-(2-hydroxyethyl)-guanine adducts in human DNA samples using a combination of TLC and HPLC. Carcinogenesis 1996, 17, 485–492. [Google Scholar] [CrossRef]
- Nilsson, R.; Liu, N.-A. Nuclear DNA damages generated by reactive oxygen molecules (ROS) under oxidative stress and their relevance to human cancers, including ionizing radiation-induced neoplasia part I: Physical, chemical and molecular biology aspects. Radiat. Med. Prot. 2020, 1, 140–152. [Google Scholar] [CrossRef]
- Swenberg, J.A.; Lu, K.; Moeller, B.C.; Gao, L.; Upton, P.B.; Nakamura, J.; Starr, T.B. Endogenous versus exogenous DNA adducts: Their role in carcinogenesis, epidemiology, and risk assessment. Toxicol. Sci. Off. J. Soc. Toxicol. 2011, 120 (Suppl. S1), S130–S145. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.J.; Wu, E. The role of gut microbiota in immune homeostasis and autoimmunity. Gut Microbes 2012, 3, 4–14. [Google Scholar] [CrossRef]
- Jiao, Y.; Wu, L.; Huntington, N.D.; Zhang, X. Crosstalk Between Gut Microbiota and Innate Immunity and Its Implication in Autoimmune Diseases. Front. Immunol. 2020, 11, 282. [Google Scholar] [CrossRef] [PubMed]
- Pourrajab, B.; Fatahi, S.; Sohouli, M.H.; Găman, M.A.; Shidfar, F. The effects of probiotic/synbiotic supplementation compared to placebo on biomarkers of oxidative stress in adults: A systematic review and meta-analysis of randomized controlled trials. Crit. Rev. Food Sci. Nutr. 2022, 62, 490–507. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Wang, B.; Xu, H.; Tang, L.; Li, Y.; Gong, L.; Wang, Y.; Li, W. Probiotic Bacillus Attenuates Oxidative Stress- Induced Intestinal Injury via p38-Mediated Autophagy. Front. Microbiol. 2019, 10, 2185. [Google Scholar] [CrossRef]
- Lin, W.-Y.; Lin, J.-H.; Kuo, Y.-W.; Chiang, P.-F.R.; Ho, H.-H. Probiotics and their Metabolites Reduce Oxidative Stress in Middle-Aged Mice. Curr. Microbiol. 2022, 79, 104. [Google Scholar] [CrossRef]
- Prete, R.; Long, S.L.; Gallardo, A.L.; Gahan, C.G.; Corsetti, A.; Joyce, S.A. Beneficial bile acid metabolism from Lactobacillus plantarum of food origin. Sci. Rep. 2020, 10, 1165. [Google Scholar] [CrossRef]
- Begley, M.; Hill, C.; Gahan Cormac, G.M. Bile Salt Hydrolase Activity in Probiotics. Appl. Environ. Microbiol. 2006, 72, 1729–1738. [Google Scholar] [CrossRef]
- Ajouz, H.; Mukherji, D.; Shamseddine, A. Secondary bile acids: An underrecognized cause of colon cancer. World J. Surg. Oncol. 2014, 12, 164. [Google Scholar] [CrossRef]
- Zeng, H.; Umar, S.; Rust, B.; Lazarova, D.; Bordonaro, M. Secondary Bile Acids and Short Chain Fatty Acids in the Colon: A Focus on Colonic Microbiome, Cell Proliferation, Inflammation, and Cancer. Int. J. Mol. Sci. 2019, 20, 1214. [Google Scholar] [CrossRef] [Green Version]
- Dalile, B.; Van Oudenhove, L.; Vervliet, B.; Verbeke, K. The role of short-chain fatty acids in microbiota-gut-brain communication. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 461–478. [Google Scholar] [CrossRef] [PubMed]
- Silva, Y.P.; Bernardi, A.; Frozza, R.L. The Role of Short-Chain Fatty Acids From Gut Microbiota in Gut-Brain Communication. Front. Endocrinol. Lausanne 2020, 11, 25. [Google Scholar] [CrossRef] [PubMed]
- Heinken, A.; Ravcheev, D.A.; Baldini, F.; Heirendt, L.; Fleming, R.M.T.; Thiele, I. Systematic assessment of secondary bile acid metabolism in gut microbes reveals distinct metabolic capabilities in inflammatory bowel disease. Microbiome 2019, 7, 75. [Google Scholar] [CrossRef] [PubMed]
- Zeng, H.; Claycombe, K.J.; Reindl, K.M. Butyrate and deoxycholic acid play common and distinct roles in HCT116 human colon cell proliferation. J. Nutr. Biochem. 2015, 26, 1022–1028. [Google Scholar] [CrossRef]
- Dossa, A.Y.; Escobar, O.; Golden, J.; Frey, M.R.; Ford, H.R.; Gayer, C.P. Bile acids regulate intestinal cell proliferation by modulating EGFR and FXR signaling. Am. J. Physiol. Gastrointest. Liver. Physiol. 2016, 310, G81–G92. [Google Scholar] [CrossRef]
- Lajczak-McGinley, N.K.; Porru, E.; Fallon, C.M.; Smyth, J.; Curley, C.; McCarron, P.A.; Tambuwala, M.M.; Roda, A.; Keely, S.J. The secondary bile acids, ursodeoxycholic acid and lithocholic acid, protect against intestinal inflammation by inhibition of epithelial apoptosis. Physiol. Rep. 2020, 8, e14456. [Google Scholar] [CrossRef]
- Chattopadhyay, I.; Gundamaraju, R.; Jha, N.K.; Gupta, P.K.; Dey, A.; Mandal, C.C.; Ford, B.M. Interplay between Dysbiosis of Gut Microbiome, Lipid Metabolism, and Tumorigenesis: Can Gut Dysbiosis Stand as a Prognostic Marker in Cancer? Dis. Markers 2022, 2022, 2941248. [Google Scholar] [CrossRef]
- Liu, Y.; Zhang, S.; Zhou, W.; Hu, D.; Xu, H.; Ji, G. Secondary Bile Acids and Tumorigenesis in Colorectal Cancer. Front. Oncol. 2022, 12, 813745. [Google Scholar] [CrossRef]
- Hylemon, P.B.; Zhou, H.; Pandak, W.M.; Ren, S.; Gil, G.; Dent, P. Bile acids as regulatory molecules. J. Lipid. Res. 2009, 50, 1509–1520. [Google Scholar] [CrossRef]
- Powolny, A.; Xu, J.; Loo, G. Deoxycholate induces DNA damage and apoptosis in human colon epithelial cells expressing either mutant or wild-type p53. Int. J. Biochem. Cell Biol. 2001, 33, 193–203. [Google Scholar] [CrossRef] [Green Version]
- Tatishchev, S.F.; VanBeek, C.; Wang, H.L. Helicobacter pylori infection and colorectal carcinoma: Is there a causal association? J. Gastrointest. Oncol. 2012, 3, 380–385. [Google Scholar]
- Wroblewski, L.E.; Peek, R.M., Jr.; Wilson, K.T. Helicobacter pylori and gastric cancer: Factors that modulate disease risk. Clin. Microbiol. Rev. 2010, 23, 713–739. [Google Scholar] [CrossRef]
- Chaturvedi, R.; Asim, M.; Romero-Gallo, J.; Barry, D.P.; Hoge, S.; de Sablet, T.; Delgado, A.G.; Wroblewski, L.E.; Piazuelo, M.B.; Yan, F.; et al. Spermine oxidase mediates the gastric cancer risk associated with Helicobacter pylori CagA. Gastroenterology 2011, 141, 1696–1708.e7082. [Google Scholar] [CrossRef] [PubMed]
- Yong, X.; Tang, B.; Li, B.-S.; Xie, R.; Hu, C.-J.; Luo, G.; Qin, Y.; Dong, H.; Yang, S.-M. Helicobacter pylori virulence factor CagA promotes tumorigenesis of gastric cancer via multiple signaling pathways. Cell Commun. Signal. 2015, 13, 30. [Google Scholar] [CrossRef] [PubMed]
- Jones, K.R.; Whitmire, J.M.; Merrell, D.S. A Tale of Two Toxins: Helicobacter Pylori CagA and VacA Modulate Host Pathways that Impact Disease. Front. Microbiol. 2010, 1, 115. [Google Scholar] [CrossRef] [PubMed]
- Parsonnet, J.; Friedman, G.D.; Orentreich, N.; Vogelman, H. Risk for gastric cancer in people with CagA positive or CagA negative Helicobacter pylori infection. Gut 1997, 40, 297–301. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.Q.; Zheng, G.F.; Sumanac, K.; Irvine, E.J.; Hunt, R.H. Meta-analysis of the relationship between <em>cagA</em> seropositivity and gastric cancer. Gastroenterology 2003, 125, 1636–1644. [Google Scholar] [CrossRef]
- Stephens, M.; von der Weid, P.Y. Lipopolysaccharides modulate intestinal epithelial permeability and inflammation in a species-specific manner. Gut Microbes 2020, 11, 421–432. [Google Scholar] [CrossRef]
- Ngkelo, A.; Meja, K.; Yeadon, M.; Adcock, I.; Kirkham, P.A. LPS induced inflammatory responses in human peripheral blood mononuclear cells is mediated through NOX4 and Giα dependent PI-3kinase signalling. J. Inflamm. 2012, 9, 1. [Google Scholar] [CrossRef] [PubMed]
- Guo, S.; Al-Sadi, R.; Said, H.M.; Ma, T.Y. Lipopolysaccharide Causes an Increase in Intestinal Tight Junction Permeability in Vitro and in Vivo by Inducing Enterocyte Membrane Expression and Localization of TLR-4 and CD14. Am. J. Pathol. 2013, 182, 375–387. [Google Scholar] [CrossRef]
- Candelli, M.; Franza, L.; Pignataro, G.; Ojetti, V.; Covino, M.; Piccioni, A.; Gasbarrini, A.; Franceschi, F. Interaction between Lipopolysaccharide and Gut Microbiota in Inflammatory Bowel Diseases. Int. J. Mol. Sci. 2021, 22, 6242. [Google Scholar] [CrossRef] [PubMed]
- Salguero, M.V.; Al-Obaide, M.A.I.; Singh, R.; Siepmann, T.; Vasylyeva, T.L. Dysbiosis of Gram-negative gut microbiota and the associated serum lipopolysaccharide exacerbates inflammation in type 2 diabetic patients with chronic kidney disease. Exp. Ther. Med. 2019, 18, 3461–3469. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Chen, W.D.; Wang, Y.D. The Relationship Between Gut Microbiota and Inflammatory Diseases: The Role of Macrophages. Front. Microbiol. 2020, 11, 1065. [Google Scholar] [CrossRef]
- Goodwin, A.C.; Destefano Shields, C.E.; Wu, S.; Huso, D.L.; Wu, X.; Murray-Stewart, T.R.; Hacker-Prietz, A.; Rabizadeh, S.; Woster, P.M.; Sears, C.L.; et al. Polyamine catabolism contributes to enterotoxigenic Bacteroides fragilis-induced colon tumorigenesis. Proc. Natl. Acad. Sci. USA 2011, 108, 15354–15359. [Google Scholar] [CrossRef] [PubMed]
- Weersma, R.K.; Zhernakova, A.; Fu, J. Interaction between drugs and the gut microbiome. Gut 2020, 69, 1510. [Google Scholar] [CrossRef] [PubMed]
- Falony, G.; Joossens, M.; Vieira-Silva, S.; Wang, J.; Darzi, Y.; Faust, K.; Kurilshikov, A.; Bonder, M.J.; Valles-Colomer, M.; Vandeputte, D.; et al. Population-level analysis of gut microbiome variation. Science 2016, 352, 560–564. [Google Scholar] [CrossRef] [PubMed]
- Koppel, N.; Maini Rekdal, V.; Balskus, E.P. Chemical transformation of xenobiotics by the human gut microbiota. Science 2017, 356, eaag2770. [Google Scholar] [CrossRef] [PubMed]
- Pollet, R.M.; D’Agostino, E.H.; Walton, W.G.; Xu, Y.; Little, M.S.; Biernat, K.A.; Pellock, S.J.; Patterson, L.M.; Creekmore, B.C.; Isenberg, H.N.; et al. An Atlas of β-Glucuronidases in the Human Intestinal Microbiome. Structure 2017, 25, 967–977.e965. [Google Scholar] [CrossRef] [PubMed]
- Dashnyam, P.; Mudududdla, R.; Hsieh, T.-J.; Lin, T.-C.; Lin, H.-Y.; Chen, P.-Y.; Hsu, C.-Y.; Lin, C.-H. β-Glucuronidases of opportunistic bacteria are the major contributors to xenobiotic-induced toxicity in the gut. Sci. Rep. 2018, 8, 16372. [Google Scholar] [CrossRef]
- Takasuna, K.; Hagiwara, T.; Hirohashi, M.; Kato, M.; Nomura, M.; Nagai, E.; Yokoi, T.; Kamataki, T. Involvement of beta-glucuronidase in intestinal microflora in the intestinal toxicity of the antitumor camptothecin derivative irinotecan hydrochloride (CPT-11) in rats. Cancer Res. 1996, 56, 3752–3757. [Google Scholar]
- Wallace, B.D.; Wang, H.; Lane, K.T.; Scott, J.E.; Orans, J.; Koo, J.S.; Venkatesh, M.; Jobin, C.; Yeh, L.-A.; Mani, S.; et al. Alleviating Cancer Drug Toxicity by Inhibiting a Bacterial Enzyme. Science 2010, 330, 831–835. [Google Scholar] [CrossRef] [PubMed]
- Fu, P.P.; Cerniglia, C.E.; Richardson, K.E.; Heflich, R.H. Nitroreduction of 6-nitrobenzo[a]pyrene: A potential activation pathway in humans. Mutat. Res. Lett. 1988, 209, 123–129. [Google Scholar] [CrossRef] [PubMed]
- Zhan, D.J.; Chiu, L.H.; Von Tungeln, L.S.; Herreno-Saenz, D.; Cheng, E.; Evans, F.E.; Heflich, R.H.; Fu, P.P. Characterization of DNA adducts in Chinese hamster ovary cells treated with mutagenic doses of 1- and 3-nitrosobenzo[a]pyrene and the trans-7,8-diol-anti-9,10-epoxides of 1- and 3-nitrobenzo[a]pyrene. Mutat. Res. 1997, 379, 43–52. [Google Scholar] [CrossRef] [PubMed]
- Kataoka, K.; Kinouchi, T.; Akimoto, S.; Ohnishi, Y. Bioactivation of cysteine conjugates of 1-nitropyrene oxides by cysteine conjugate beta-lyase purified from Peptostreptococcus magnus. Appl. Environ. Microbiol. 1995, 61, 3781–3787. [Google Scholar] [CrossRef] [PubMed]
- Luca, D.; Milena, V.; Francesca, T.; Ermanno, F.; Giovanni, C. Protective Effects of Probiotic Lactobacillus rhamnosus IMC501 in Mice Treated with PhIP. J. Microbiol. Biotechnol. 2014, 24, 371–378. [Google Scholar] [CrossRef]
- Möller, L.; Zeisig, M.; Midtvedt, T.; Gustafsson, J.A. Intestinal microflora enhances formation of DNA adducts following administration of 2-NF and 2-AAF. Carcinogenesis 1994, 15, 857–861. [Google Scholar] [CrossRef]
- Hirayama, K.; Baranczewski, P.; Åkerlund, J.-E.; Midtvedt, T.; Möller, L.; Rafter, J. Effects of human intestinal flora on mutagenicity of and DNA adduct formation from food and environmental mutagens. Carcinogenesis 2000, 21, 2105–2111. [Google Scholar] [CrossRef]
- Kinouchi, T.; Kataoka, K.; Miyanishi, K.; Akimoto, S.; Ohnishi, Y. Role of intestinal microflora in metabolism of glutathione conjugates of 1-nitropyrene 4,5-oxide and 1-nitropyrene 9,10-oxide. Tohoku J. Exp. Med. 1992, 168, 119–122. [Google Scholar] [CrossRef]
- Horie, H.; Zeisig, M.; Hirayama, K.; Midtvedt, T.; Möller, L.; Rafter, J. Probiotic mixture decreases DNA adduct formation in colonic epithelium induced by the food mutagen 2-amino-9H-pyrido [2,3-b]indole in a human-flora associated mouse model. Eur. J. Cancer Prev. 2003, 12, 101–107. [Google Scholar] [CrossRef]
- Zhang, J.; Empl, M.T.; Schwab, C.; Fekry, M.I.; Engels, C.; Schneider, M.; Lacroix, C.; Steinberg, P.; Sturla, S.J. Gut Microbial Transformation of the Dietary Imidazoquinoxaline Mutagen MelQx Reduces Its Cytotoxic and Mutagenic Potency. Toxicol. Sci. 2017, 159, 266–276. [Google Scholar] [CrossRef]
- Zhang, J.; Empl, M.T.; Schneider, M.; Schröder, B.; Stadnicka-Michalak, J.; Breves, G.; Steinberg, P.; Sturla, S.J. Gut microbial transformation of the dietary mutagen MeIQx may reduce exposure levels without altering intestinal transport. Toxicol. Vitr. 2019, 59, 238–245. [Google Scholar] [CrossRef] [PubMed]
- Doolittle, D.J.; Sherrill, J.M.; Butterworth, B.E. Influence of Intestinal Bacteria, Sex of the Animal, and Position of the Nitro Group on the Hepatic Genotoxicity of Nitrotoluene Isomers in Vivo1. Cancer Res. 1983, 43, 2836–2842. [Google Scholar] [PubMed]
- El-Nezami, H.S.; Polychronaki, N.N.; Ma, J.; Zhu, H.; Ling, W.; Salminen, E.K.; Juvonen, R.O.; Salminen, S.J.; Poussa, T.; Mykkänen, H.M. Probiotic supplementation reduces a biomarker for increased risk of liver cancer in young men from Southern China. Am. J. Clin. Nutr. 2006, 83, 1199–1203. [Google Scholar] [CrossRef] [PubMed]
- Saffarian, A.; Mulet, C.; Regnault, B.; Amiot, A.; Tran-Van-Nhieu, J.; Ravel, J.; Sobhani, I.; Sansonetti, P.J.; Pédron, T. Crypt- and Mucosa-Associated Core Microbiotas in Humans and Their Alteration in Colon Cancer Patients. mBio 2019, 10, e01315–e01319. [Google Scholar] [CrossRef] [PubMed]
- Wu, N.; Yang, X.; Zhang, R.; Li, J.; Xiao, X.; Hu, Y.; Chen, Y.; Yang, F.; Lu, N.; Wang, Z.; et al. Dysbiosis signature of fecal microbiota in colorectal cancer patients. Microb. Ecol. 2013, 66, 462–470. [Google Scholar] [CrossRef]
- Bonnet, M.; Buc, E.; Sauvanet, P.; Darcha, C.; Dubois, D.; Pereira, B.; Déchelotte, P.; Bonnet, R.; Pezet, D.; Darfeuille-Michaud, A. Colonization of the Human Gut by E. coli and Colorectal Cancer Risk. Clin. Cancer Res. 2014, 20, 859–867. [Google Scholar] [CrossRef]
- Vipperla, K.; O’Keefe, S.J. Diet, microbiota, and dysbiosis: A ‘recipe’ for colorectal cancer. Food Funct. 2016, 7, 1731–1740. [Google Scholar] [CrossRef]
- Jahani-Sherafat, S.; Alebouyeh, M.; Moghim, S.; Ahmadi Amoli, H.; Ghasemian-Safaei, H. Role of gut microbiota in the pathogenesis of colorectal cancer; a review article. Gastroenterol. Hepatol. Bed. Bench. 2018, 11, 101–109. [Google Scholar]
- Genua, F.; Raghunathan, V.; Jenab, M.; Gallagher, W.M.; Hughes, D.J. The Role of Gut Barrier Dysfunction and Microbiome Dysbiosis in Colorectal Cancer Development. Front. Oncol. 2021, 11, 626349. [Google Scholar] [CrossRef]
- Bongers, G.; Pacer, M.E.; Geraldino, T.H.; Chen, L.; He, Z.; Hashimoto, D.; Furtado, G.C.; Ochando, J.; Kelley, K.A.; Clemente, J.C.; et al. Interplay of host microbiota, genetic perturbations, and inflammation promotes local development of intestinal neoplasms in mice. J. Exp. Med. 2014, 211, 457–472. [Google Scholar] [CrossRef]
- Hu, B.; Elinav, E.; Huber, S.; Strowig, T.; Hao, L.; Hafemann, A.; Jin, C.; Wunderlich, C.; Wunderlich, T.; Eisenbarth, S.C.; et al. Microbiota-induced activation of epithelial IL-6 signaling links inflammasome-driven inflammation with transmissible cancer. Proc. Natl. Acad. Sci. USA 2013, 110, 9862–9867. [Google Scholar] [CrossRef] [PubMed]
- Uemura, N.; Okamoto, S.; Yamamoto, S.; Matsumura, N.; Yamaguchi, S.; Yamakido, M.; Taniyama, K.; Sasaki, N.; Schlemper, R.J. Helicobacter pylori infection and the development of gastric cancer. N. Engl. J. Med. 2001, 345, 784–789. [Google Scholar] [CrossRef] [PubMed]
- Mera, R.; Fontham, E.T.; Bravo, L.E.; Bravo, J.C.; Piazuelo, M.B.; Camargo, M.C.; Correa, P. Long term follow up of patients treated for Helicobacter pylori infection. Gut 2005, 54, 1536–1540. [Google Scholar] [CrossRef] [PubMed]
- Wong, B.C.; Lam, S.K.; Wong, W.M.; Chen, J.S.; Zheng, T.T.; Feng, R.E.; Lai, K.C.; Hu, W.H.; Yuen, S.T.; Leung, S.Y.; et al. Helicobacter pylori eradication to prevent gastric cancer in a high-risk region of China: A randomized controlled trial. JAMA 2004, 291, 187–194. [Google Scholar] [CrossRef] [PubMed]
- Gunathilake, M.N.; Lee, J.; Choi, I.J.; Kim, Y.-I.; Ahn, Y.; Park, C.; Kim, J. Association between the relative abundance of gastric microbiota and the risk of gastric cancer: A case-control study. Sci. Rep. 2019, 9, 13589. [Google Scholar] [CrossRef]
- Yang, J.; Zhou, X.; Liu, X.; Ling, Z.; Ji, F. Role of the Gastric Microbiome in Gastric Cancer: From Carcinogenesis to Treatment. Front. Microbiol. 2021, 12, 641322. [Google Scholar] [CrossRef]
- Yoshimoto, S.; Loo, T.M.; Atarashi, K.; Kanda, H.; Sato, S.; Oyadomari, S.; Iwakura, Y.; Oshima, K.; Morita, H.; Hattori, M.; et al. Obesity-induced gut microbial metabolite promotes liver cancer through senescence secretome. Nature 2013, 499, 97–101. [Google Scholar] [CrossRef]
- Dapito, D.H.; Mencin, A.; Gwak, G.Y.; Pradere, J.P.; Jang, M.K.; Mederacke, I.; Caviglia, J.M.; Khiabanian, H.; Adeyemi, A.; Bataller, R.; et al. Promotion of hepatocellular carcinoma by the intestinal microbiota and TLR4. Cancer Cell 2012, 21, 504–516. [Google Scholar] [CrossRef]
- Xuan, C.; Shamonki, J.M.; Chung, A.; Dinome, M.L.; Chung, M.; Sieling, P.A.; Lee, D.J. Microbial dysbiosis is associated with human breast cancer. PLoS ONE 2014, 9, e83744. [Google Scholar] [CrossRef]
- Chen, J.; Douglass, J.; Prasath, V.; Neace, M.; Atrchian, S.; Manjili, M.H.; Shokouhi, S.; Habibi, M. The microbiome and breast cancer: A review. Breast Cancer Res. Treat. 2019, 178, 493–496. [Google Scholar] [CrossRef]
- Marshall, B.J.; Warren, J.R. Unidentified curved bacilli in the stomach of patients with gastritis and peptic ulceration. Lancet 1984, 1, 1311–1315. [Google Scholar] [CrossRef]
- Marshall, B.J.; Armstrong, J.A.; McGechie, D.B.; Glancy, R.J. Attempt to fulfil Koch’s postulates for pyloric Campylobacter. Med. J. Aust. 1985, 142, 436–439. [Google Scholar] [CrossRef] [PubMed]
- Plaza-Díaz, J.; Álvarez-Mercado, A.I.; Ruiz-Marín, C.M.; Reina-Pérez, I.; Pérez-Alonso, A.J.; Sánchez-Andujar, M.B.; Torné, P.; Gallart-Aragón, T.; Sánchez-Barrón, M.T.; Reyes Lartategui, S.; et al. Association of breast and gut microbiota dysbiosis and the risk of breast cancer: A case-control clinical study. BMC Cancer 2019, 19, 495. [Google Scholar] [CrossRef] [Green Version]
- Pérez, J.C. Fungi of the human gut microbiota: Roles and significance. Int. J. Med. Microbiol. 2021, 311, 151490. [Google Scholar] [CrossRef] [PubMed]
- Raimondi, S.; Amaretti, A.; Gozzoli, C.; Simone, M.; Righini, L.; Candeliere, F.; Brun, P.; Ardizzoni, A.; Colombari, B.; Paulone, S.; et al. Longitudinal Survey of Fungi in the Human Gut: ITS Profiling, Phenotyping, and Colonization. Front. Microbiol. 2019, 10, 1575. [Google Scholar] [CrossRef] [PubMed]
- Huffnagle, G.B.; Noverr, M.C. The emerging world of the fungal microbiome. Trends Microbiol. 2013, 21, 334–341. [Google Scholar] [CrossRef] [Green Version]


| Type | Definition | Example DDAs | Mechanism | Repair Pathway | Repair Error | Reference | |
|---|---|---|---|---|---|---|---|
| Exogenous | Endogenous | ||||||
| Abasic site | Loss of a purine or pyrimidine base in a DNA sequence. | Ionizing radiation | ROS, DNA glycosylases | DDAs attack and break the glycosidic linkages between the deoxyribose and the nitrogenous base of a nucleotide. | BER (major) and NER (minor) | More error free | |
| Adduct | DNA nucleotides covalently bound to substances that add a functional group to the DNA’s primary structure. | PAHs, formaldehyde, aflatoxin | ROS, endogenous alkylating agents (e.g., formaldehyde) | Generally, the electrophilic sites of DDA attack the nucleophilic sites of the nucleotide and form the covalent bond. | Structurally dependent, including DR, BER, NER, MMR. | Structurally dependent bulky adducts generally lead to error-prone repairs. | |
| Deamination | Removal of an amino group from a nucleotide. | NA * | MT, nitric oxide | (1) DDAs cause deamination, such as deaminating dC to dU. (2) Misincorporation of dUMP instead of dTMP during replication. | BER | More error free | |
| Single-strand break | Discontinuities in one strand of the DNA’s double helix. | Ionizing radiation | ROS | DDAs cause cleavage, thus discontinuity, in one strand of the DNA duplex. | SSBR, HR, BER | More error prone | [20] |
| Double-strand break | Discontinuities in both strands of the DNA’s double helix. | Ionizing radiation, bleomycin, neocarzinostatin | Colibactin, hydrogen peroxide | DDAs cause cleavage, thus discontinuity, in both strands of the DNA duplex. | DSBR, NHEJ, HR | Majorly error prone | [21,22] |
| Intra- and inter-strand crosslink | Two nucleotides in the same (intra-) or different (inter-) strands of DNA were reacted to form a covalent bond. | Nitrogen mustards, cisplatin, psoralens | Nitrous acid, aldehydes (e.g., malondialdehyde) | DDAs often have two independently reactive groups that bind with two nucleotide residues of DNA to form a crosslink. | NER, HR, BER | Majorly error prone | [23] |
| DNA Damage | Specific Gut Microbiome Species | Mechanism | Reference |
|---|---|---|---|
| DNA adduct | pks+ Enterobacteriaceae spp. | Some specific bacteria that harbor the pks genomic island (pks+) synthesize various colibactins, which can conjugate to DNA and form a colibactin–DNA adduct. | [24] |
| H. pylori | H. pylori disrupts intracellular processes in the gut epithelium that cause inflammation, and the host responds by involving immune cells through their release of cytokines, forming reactive oxygen/nitrogen species (ROS and RNS), which can eventually attack DNA to form adducts, such as 8-oxo-dG. | [25,26] | |
| (Not applicable) | DNA adducts related to oxidative stress (i.e., 8-oxo-dG) are lower in the small intestine of SPF mice than in GF mice. 5-Cl-dC, a DNA adduct attributed to neutrophil activity, is higher in colon and small intestine of GF mice than SPF mice. Lipid-peroxidation-induced DNA adduct, N2-ε-dG, is higher in the liver of SPF mice than in GF mice. | [17] | |
| DNA crosslinking | pks+ Enterobacteriaceae spp. | pks+ bacteria induce colibactin–DNA adduct and can then form DNA inter-strand crosslinks. | [27,28,29] |
| DNA single-strand break | pks+ Enterobacteriaceae spp. | DNA inter-strand crosslinks formed by colibactin can be depurinated, subsequently leading to single-strand breaks. | |
| E. coli, C. jejuni, and others | CDT is produced by some pathogenic Gram-negative bacteria. Most members of CDTs hold similar structures, sequence homology, and endonuclease activities of DNase I, which can induce single-strand breaks (nicks) in DNA. | [30,31,32,33] | |
| S. typhi, S. enterica, and other Salmonella species | TT have been identified in several Salmonella spp. TT released from bacteria possess endonuclease activities similar to CDT, which can introduce single-strand breaks. | [30,34] | |
| DNA double-strand break | pks+ Enterobacteriaceae spp. | When colibactins introduce accumulating single-strand breaks, and two closed nicks face each other on opposite strands, a DSB can be created. | [29] |
| Some species of colibactins (e.g., colibactin-645) from pks+ bacteria, under certain situations (e.g., presence of Cu (II)), induce DNA double-strand breaks. | [27,35] | ||
| E. coli, C. jejuni, and others | Highly concentrated CDT accumulates single-strand breaks, and when two closed nicks face each other on opposite strands, a DSB can be created. | [30,31,36] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hsiao, Y.-C.; Liu, C.-W.; Yang, Y.; Feng, J.; Zhao, H.; Lu, K. DNA Damage and the Gut Microbiome: From Mechanisms to Disease Outcomes. DNA 2023, 3, 13-32. https://doi.org/10.3390/dna3010002
Hsiao Y-C, Liu C-W, Yang Y, Feng J, Zhao H, Lu K. DNA Damage and the Gut Microbiome: From Mechanisms to Disease Outcomes. DNA. 2023; 3(1):13-32. https://doi.org/10.3390/dna3010002
Chicago/Turabian StyleHsiao, Yun-Chung, Chih-Wei Liu, Yifei Yang, Jiahao Feng, Haoduo Zhao, and Kun Lu. 2023. "DNA Damage and the Gut Microbiome: From Mechanisms to Disease Outcomes" DNA 3, no. 1: 13-32. https://doi.org/10.3390/dna3010002